
Abstract
Liver cells are easily damaged by oxidative stress during progression both in liver development and throughout adult life, resulting in tissue pathology that ranges from simple hepatitis to nonalcoholic fatty liver disease. In this study, we determined the attenuation of oxidative stress in liver cells with pretreatment of rosmarinic acid (RA), which is an antioxidant agent from Rosmarinus officinalis. The human liver cell line L02 was damaged by hydrogen peroxide (H2O2). In the RA treatment group, the viability of L02 cells increased and the intracellular reactive oxygen species levels decreased compared with the H2O2-induced damage group. Analysis of flow cytometry revealed that the percentage of G2/M cell cycle arrest and cell apoptosis decreased in the RA treatment group. This alteration was associated with activation of a G2/M DNA damage and oxidative stress apoptotic signal. Furthermore, we determined the redox-sensitive protein expression of mitogen-activated protein kinases (MAPKs), quinone acceptor oxidoreductase 1 (NQO1), and nuclear factor E2-related factor 2 (Nrf2), and the expression of both MAPKs and Nrf2 was activated in the RA group. Results showed that the relevant protein expression of MAPKs and Nrf2 was activated in the RA group. Thus, RA protected L02 cells from oxidative damage through suppressing cell cycle arrest and cell apoptosis with the activation of MAPK and Nrf2 signaling pathways.
Introduction
A diverse set of toxic, metabolic, and inflammatory elements causes liver damage and disease. Cell death mainly occurs by apoptosis or necrosis when the liver suffers damage and then resulting in tissue pathology such as nonalcoholic fatty liver disease (NAFLD). NAFLD ranges from simple steatosis, nonalcoholic steatohepatitis, fibrosis, liver cirrhosis to end-stage hepatocellular carcinoma (HCC). 1 In these liver pathologies, HCC is the most ordinary (70%–90%) histologic type of hepar cancer. 2 Besides, NAFLD has been estimated as the primary cause of liver interrelated morbidity and even an indication for liver transplantation projected. 3 However, despite its high prevalence and severity, currently there is no pharmacological agent for the treatment of NAFLD.
A “Two-Hit” hypothesis has been widely proposed to explain the pathogenesis of NAFLD. The first hit involves an imbalance in the fatty acid and triglyceride metabolism in hepatocytes resulting in lipid accretion in the liver. The second hit includes oxidative stress and lipid peroxidation resulting in NASA with an increase in cytokine production and inflammation production. 4 In the first hit, as the main store of lipids in the liver, the triglycerides are synthesized from the free fatty acids (FFA). The excessive FFA can also promote hepatocyte apoptosis through increased production of reactive oxygen species (ROS). 5 The increase of ROS production leads to antioxidative defense decline in NAFLD. Thus, oxidative stress has been considered a key factor in the pathophysiology of NAFLD. 6 Hydrogen peroxide (H2O2) is a vital cause of oxidative damage for its longer half-life than the other ROS and it can easily transform into the hydroxyl radical, one of the most destructive free radicals. 7 Furthermore, it is generated from almost all sources of oxidative stress in many kinds of cells and tissues. In the liver, the augmented hepatic oxidative stress caused by H2O2 increased the progression of NAFLD. 8 H2O2 has been reported to trigger apoptosis in hepatocytes. 9,10 However, the cellular and molecular mechanisms under oxidative stress are not fully understood.
There are reports that oxidative stress is intimately involved in the regulation of mitogen-activated protein kinases (MAPKs) and nuclear factor erythroid2-related factor 2 (Nrf2) signaling pathways. 7 The close connection between oxidative stress and MAPKs and Nrf2 signaling prompted us to investigate the potential mechanism in liver pathology. The expression of MAPK, Nrf2, Keap1, and NQO1 was alternated in mouse liver after the mequindox-induced oxidative stress. 11 Redox-sensitive MAPKs are all serine/threonine kinases regulated by the proline residue, and MAPKs include the c-Jun N-terminal kinase (JNK), the p38 kinase (p38), and the extracellular signal-related kinases (ERK1/2). 12 Meanwhile, Nrf2 has been recognized as an important player in regulating the cellular antioxidant response. Nfr2 belongs to the cap “n” collar basic leucine zipper family. Nrf2 interacts with the Kelch-like ECH-associated protein 1 (Keap1) in the cytosol before exposure to oxidants and electrophiles. However, while under oxidative stress, Nrf2 is released and translated from the cytosol to nucleus due to modification of Cys residues of CRLKeap1. 13
More recently, dietary antioxidants have been proposed as therapeutic agents to counteract oxidative stress-induced liver damage such as NAFLD. 14 As a phenolic compound, rosmarinic acid (RA) extracted from Rosmarinus officinalis exhibits good antioxidant properties (Fig. 1). Certainly, some studies indicated that RA has several other biological activities, including antimemory deficits, 15 antifibrosis, 16 anticancer, 17 antiradiation, 18 antiaging, 19 anti-inflammation, 20 anticonvulsant. 21 Today, much is known about the chemistry and antioxidant potential of RA as a result of many studies. Reports have showed RA enhanced the activity of antioxidant enzymes in aging mice, 22 and ameliorated acute liver damage in CCL4-intoxicated mice through ameliorating oxidative stress by activation of the Nrf2 and heme oxygenase-1 (HO-1) expression. 23 The current study was conducted to investigate the cytoprotective effect of RA against H2O2-induced oxidative stress and further elucidated its new potential mechanism of signal transduction on L02 cells.
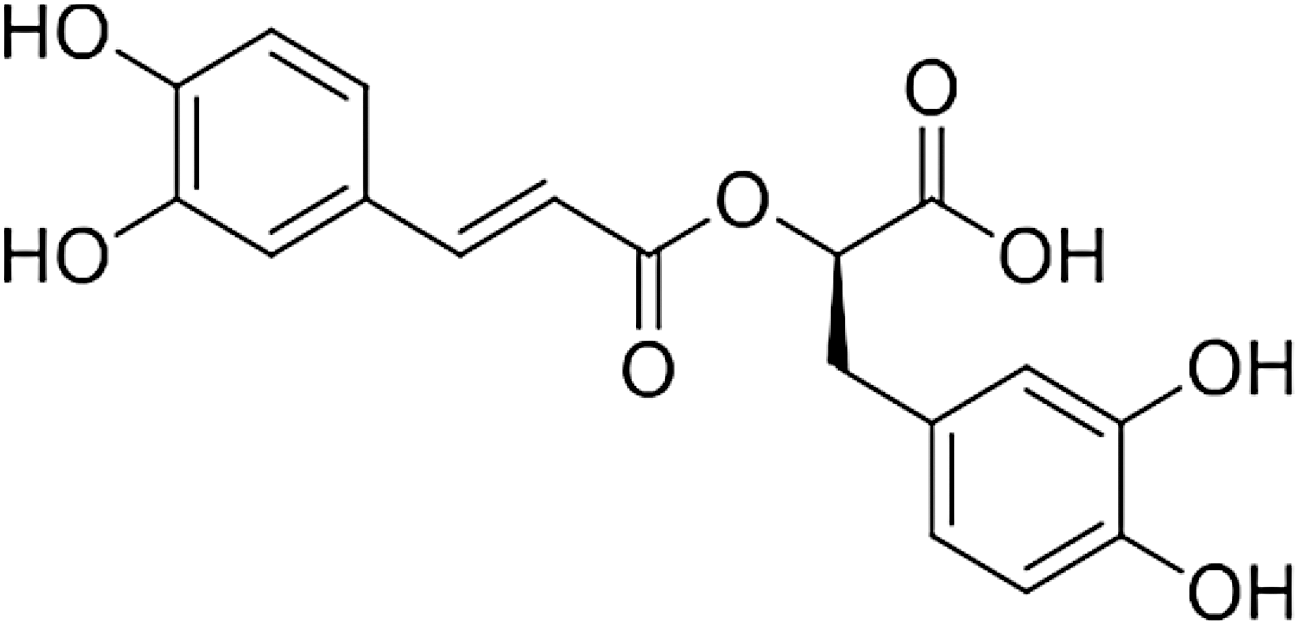
Chemical structure of rosmarinic acid.
Materials and Methods
Rosmarinic acid and l-ascorbic acid (Vit.C [VC]) were purchased from Sigma-Aldrich (St. Louis, MO, USA). H2O2 (hydrogen peroxide) was from Guangzhou Chemical Reagent Factory. CCK-8 kit, ROS assay kit, cell cycle and annexin V-FITC apoptosis detection kit, RIPA, PMSF, primary antibody dilution, second antibody dilution, and BCA protein assay kit were all from Beyotime Biotechnology (Shanghai, China). The nuclear/cytosol fractionation kit was bought from Sangon Biotech (Shanghai, China). The primary antibodies for p-JNK, JNK, p-p38, p38, p-ERK1/2, ERK1/2, Nrf2, NQO1, laminB1, and β-actin, and the horseradish peroxidase (HRP)-linked second antibody were from Cell Signaling Technology (Boston, MA, USA).
Cell culture
The hepatic cell line L02 was obtained from the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 μg/mL streptomycin, 100 U/mL penicillin, and 2 mM l-glutamine at 95% air with 5% CO2 in a humidified incubator. The culture medium was replaced with fresh medium every 2 to 3 days.
Cell viability assay
Cell viability was measured by the CCK-8 kit. L02 cells were seeded on 96-well plates (Corning) at a density of 2 × 104 cells per well in 100 μL medium with three replicate wells. After 12 hours of incubation, the cells were pretreated with DMEM containing RA (0.25, 1, and 5 μM) or VC (100 μM) for 24 hours, and then stimulated with H2O2 (400 μM) for 6 hours. The RA and VC were washed away with phosphate-buffered saline (PBS) once, before stimulation with H2O2. Then, 10 μL of CCK-8 solution was added into each well and the optical densities at 450 nm were measured using an enzyme-labeled instrument (Thermo) after incubation at the cell culture incubator for 1 hour.
DCFH-DA fluorescence assay
Measurement of intracellular ROS was performed by the ROS assay kit. L02 cells were seeded on 96-well plates with 2 × 104 cells per well in 100 μL medium and 6-well plates with 3 × 105 cells per well in 2 mL medium with three replicate wells, respectively. After 12 hours of incubation, the cells were pretreated with DMEM containing RA (0.25, 1, and 5 μM) and VC (100 μM) for 24 hours, and then stimulated with H2O2 (400 μM) for 6 hours. The RA and VC were washed away with PBS once, before stimulation with H2O2. Then, the cells were incubated with 10 μM DCFH-DA for 30 minutes at 37°C in the dark. The green fluorescence of DCF in 96-well plates and 6-well plates was detected by Molecular Devices (Sutter Instrument Company, MD) and flow cytometry (CytoFLEX; Beckman Coulter), respectively, with an excitation wavelength of 485 nm and an emission wavelength of 525 nm.
Cell cycle assay
Cell cycle was measured by the cell cycle and apoptosis analysis kit. L02 cells were seeded on six-well plates at a density of 3 × 105 cells per well in 2 mL medium with three replicate wells. After 12 hours of incubation, the cells were pretreated with DMEM containing RA (0.25, 1, and 5 μM) and VC (100 μM) for 24 hours, and then stimulated with H2O2 (400 μM) for 6 hours. The RA and VC were washed away with PBS once, before stimulation with H2O2. Then the cells were harvested by trypsin digestion, washed with ice bath precooling PBS and fixed with ice bath precooling 70% ethyl alcohol at 4°C overnight. Then the cells were further incubated with propidium iodide (PI) staining solution for 30 minutes at 37°C in the dark. The red fluorescence of PI was detected by flow cytometry (CytoFLEX; Beckman Coulter) with an excitation wavelength of 488 nm and an emission wavelength of 630 nm.
Cell apoptosis assay
Cell apoptosis was identified using the annexin V-FITC apoptosis detection kit. L02 cells were seeded on six-well plates at a density of 3 × 105 cells per well in 2 mL medium with three replicate wells. After 12 hours of incubation, the cells were pretreated with DMEM containing RA (0.25, 1, and 5 μM) and VC (100 μM) for 24 hours, and then stimulated with H2O2 (400 μM) for 6 hours. The RA and VC were washed away with PBS once, before stimulation with H2O2. Then, cells were harvested, washed, and incubated in 195 μL of annexin V-FITC binding buffer, and then added to 5 μL of annexin V-FITC and 10 μL of PI for 20 minutes of incubation at room temperature in the dark. The percentage of apoptotic cells was detected using flow cytometry (Beckman Coulter, CytoFLEX) with an excitation wavelength of 488 nm and an emission wavelength of 530 nm.
Western blotting assay
L02 cells were seeded on six-well plates at a density of 3 × 105 cells per well in 2 mL medium with three replicate wells. After 12 hours of incubation, cells were pretreated with DMEM containing RA (0.25, 1, and 5 μM) and VC (100 μM) for 24 hours, and then stimulated with H2O2 (400 μM) for 6 hours. The RA and VC were washed away with PBS once, before stimulation with H2O2. Then, cells were lysed with radioimmunoprecipitation assay (RIPA) buffer containing 1 mM phenylmethanesulfonyl fluoride (PMSF) for the total protein extraction in the nuclear and cytosol fractionation. The cells were lysed with buffer A, buffer B, buffer C from the nuclear/cytosol fractionation kit for the nuclear and cytosol fractionation protein extraction, respectively. Protein concentrations were quantified using bicinchoninic acid (BCA) hydrate disodium salt protein assay kit. The boiled proteins were separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto the polyvinylidene fluoride (PVDF; Millipore) membrane using Western Semi-dry Transfer System (Bio-Rad). After being blocked with 5% skim milk in Tris-buffered saline Tween (TBST) buffer for 3 hours at room temperature, the membranes were incubated with primary antibodies for p-JNK, JNK, p-p38, p38, p-ERK, ERK, and β-actin (dilution 1:3000) overnight at 4°C, followed by a secondary antibody coupled to HRP (dilution 1:5000) for 1 hour at room temperature. Finally, the blots were developed using the enhanced chemiluminescent (ECL) detection system (Tanon, China).
Statistical analysis
All experiments were performed independently three times. Statistical analysis was performed by using PRISM 5.0 (GraphPad software). Group differences were assessed by the Student's t-test or ANOVA multiple comparisons. p Values of less than 0.05 were considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
The protective effect of RA on the viability of H2O2-induced L02 cells
Exposure of L02 cells to various concentrations of H2O2 for 6 hours resulted in a dose-dependent descending in cell viability (Fig. 2A). Figure 2A shows that H2O2 of 400 μM could cause a 40.06% ± 5.59% decrease in cell viability. Thus, 400 μM was selected as the oxidative damage concentration for later experimentations. While the exposure of L02 cells to H2O2 (400 μM) for 6 hours significantly decreased cell viability, the RA-containing medium in addition significantly increased cell viability (Fig. 2B). The antioxidant VC (100 μM) also increased the viability of H2O2-induced L02 cells, and it was used as positive control (Fig. 2B). These results suggested that RA protected L02 cells from H2O2-induced oxidative damage.
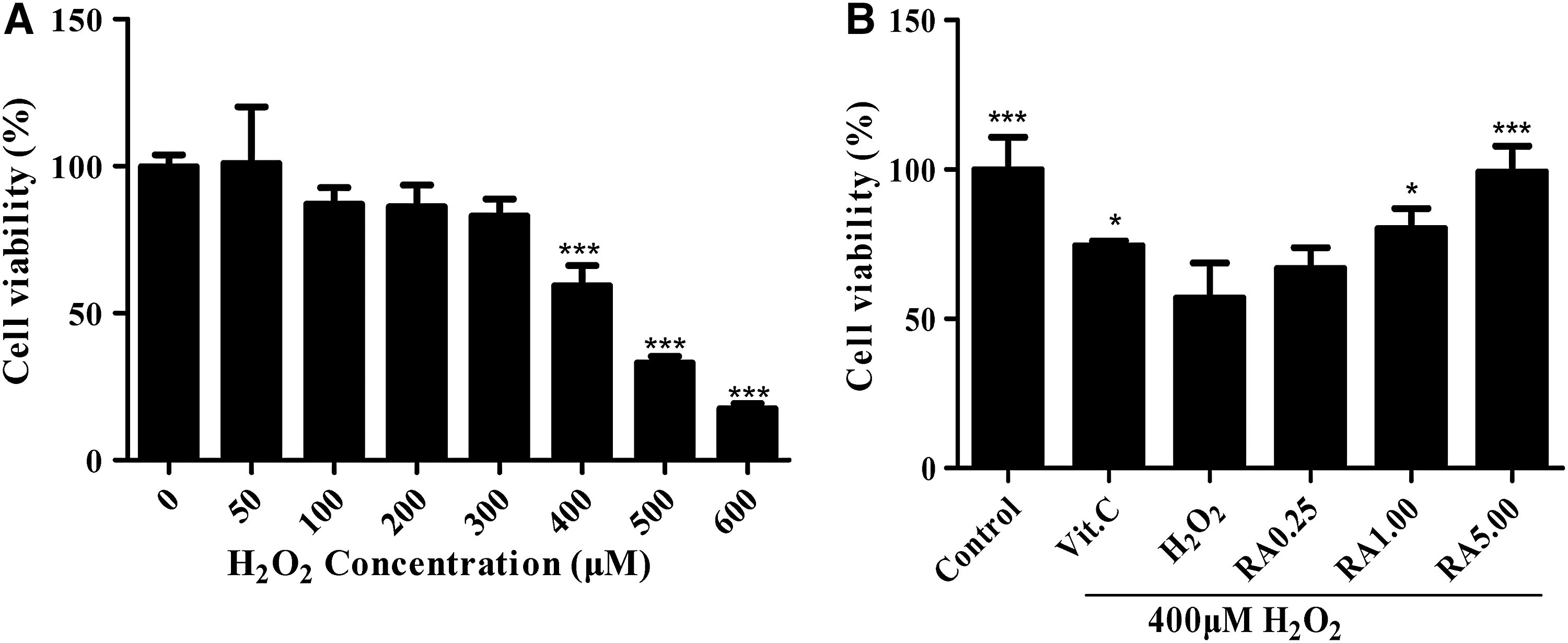
Cell viabilities of L02 cells were assayed by CCK-8.
RA reduced H2O2-induced ROS production
H2O2-induced L02 cells robustly augmented the ROS production, while RA-containing serum (5.00 μM) drastically reduced ROS levels in the damaged cells, as shown in Figure 3A–C. The antioxidant VC (100 μM) also showed the significant effects of inhibiting H2O2-induced ROS production, and it was used as positive control (Fig. 3A–C). These results showed that RA protected L02 cells by decreasing the ROS production. The potential cytoprotective effect might be the involvement of the induction of superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), total glutathione (GSH), and malondialdehyde (MDA). 24
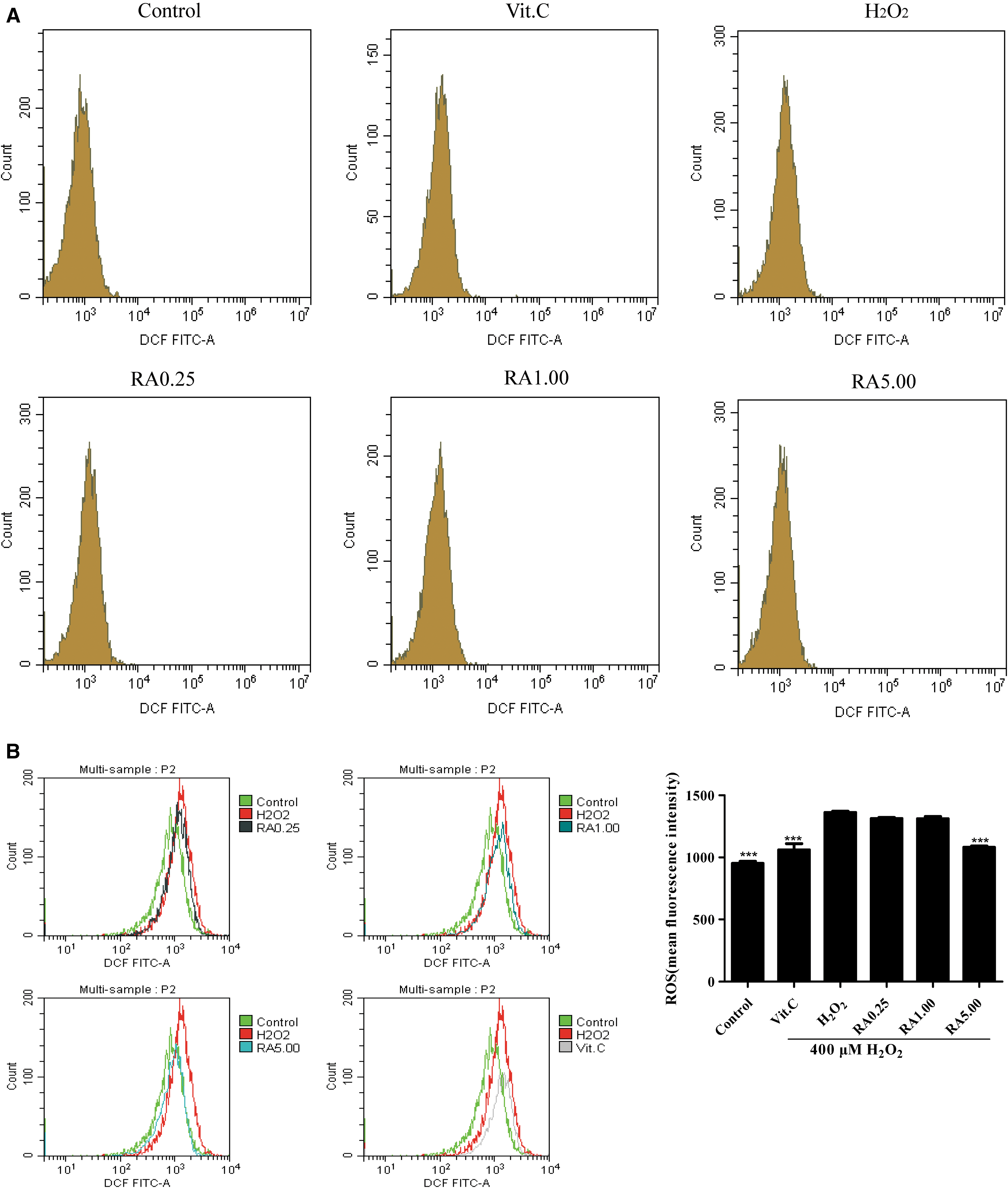
The ROS production in L02 cells was assayed in each group.

RA inhibited cell G2/M phase arrest progression and cell apoptosis caused by oxidative stress
To further investigate the protective effect of RA against oxidative stress in L02 cells, cell cycle was performed. As shown in Figure 4A and C, the percentage of G2/M cell cycle arrest was 19.55% ± 0.69% in the H2O2-induced damage group, while only 3.53% ± 0.98% in the normal control group. The result suggested a G2/M cell cycle arrest in the H2O2-induced damage group compared with the normal control group. In the RA treatment group, the G2/M phase was partially blocked to 12.46% ± 3.19%, 11.33% ± 3.98%, 8.6% ± 0.95% (at 0.25, 1.00, and 5.00 μM), respectively. Also, the percentage of cells in S phase increased by 15.66% ± 1.21%, 18.10% ± 3.97%, and 19.13% ± 1.92%, while the percentage of cells in G0/G1 phase decreased by 8.59% ± 3.69%, 9.88% ± 0.30%, and 8.10% ± 1.55% compared with the H2O2-induced damage group, which was in association with the changed percentage of the G2/M cell cycle arrest. The antioxidant VC (100 μM) showed a similarity to the effect of RA, and it was used as positive control. The results suggested that RA might decrease the percentage of the G2/M cell cycle arrest against oxidative damage in L02 cells.

Cell cycle arrest and cell apoptosis in L02 cells were assayed in each group.
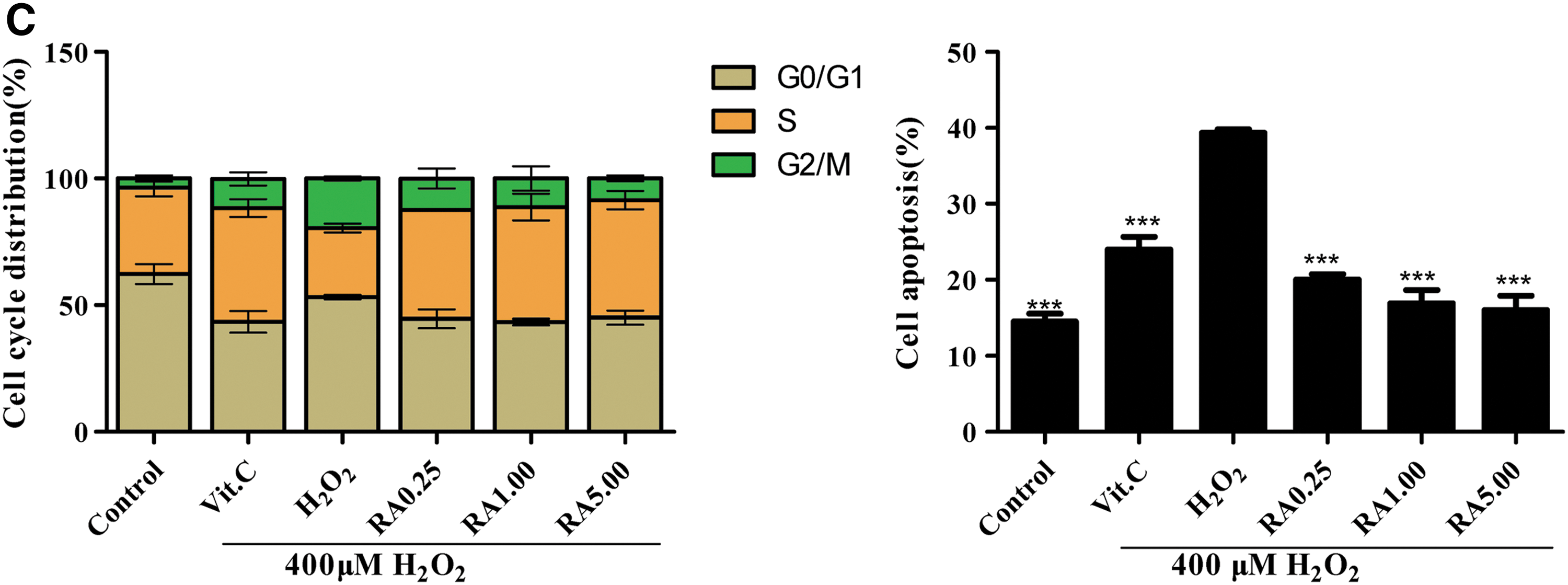
To determine whether RA reduced H2O2-induced cell apoptosis, cell apoptosis was performed. The percentage of apoptotic cells significantly increased in the H2O2-induced oxidative damage group, compared with the normal control group (39.36% ± 0.37% vs. 14.46% ± 0.87%, Fig. 5B, C). However, pretreatment of L02 cells with RA (at 0.25, 1.00, and 5.00 μM) reduced the proportion of apoptotic cells to 20.0% ± 0.58%, 16.89% ± 1.45%, and 16.03% ± 1.52%, respectively. The antioxidant VC (100 μM) also distinctly reduced cell apoptosis, and it was used as positive control (Fig. 4B, C). The results suggested that RA might decrease cell apoptosis against oxidative damage in L02 cells.
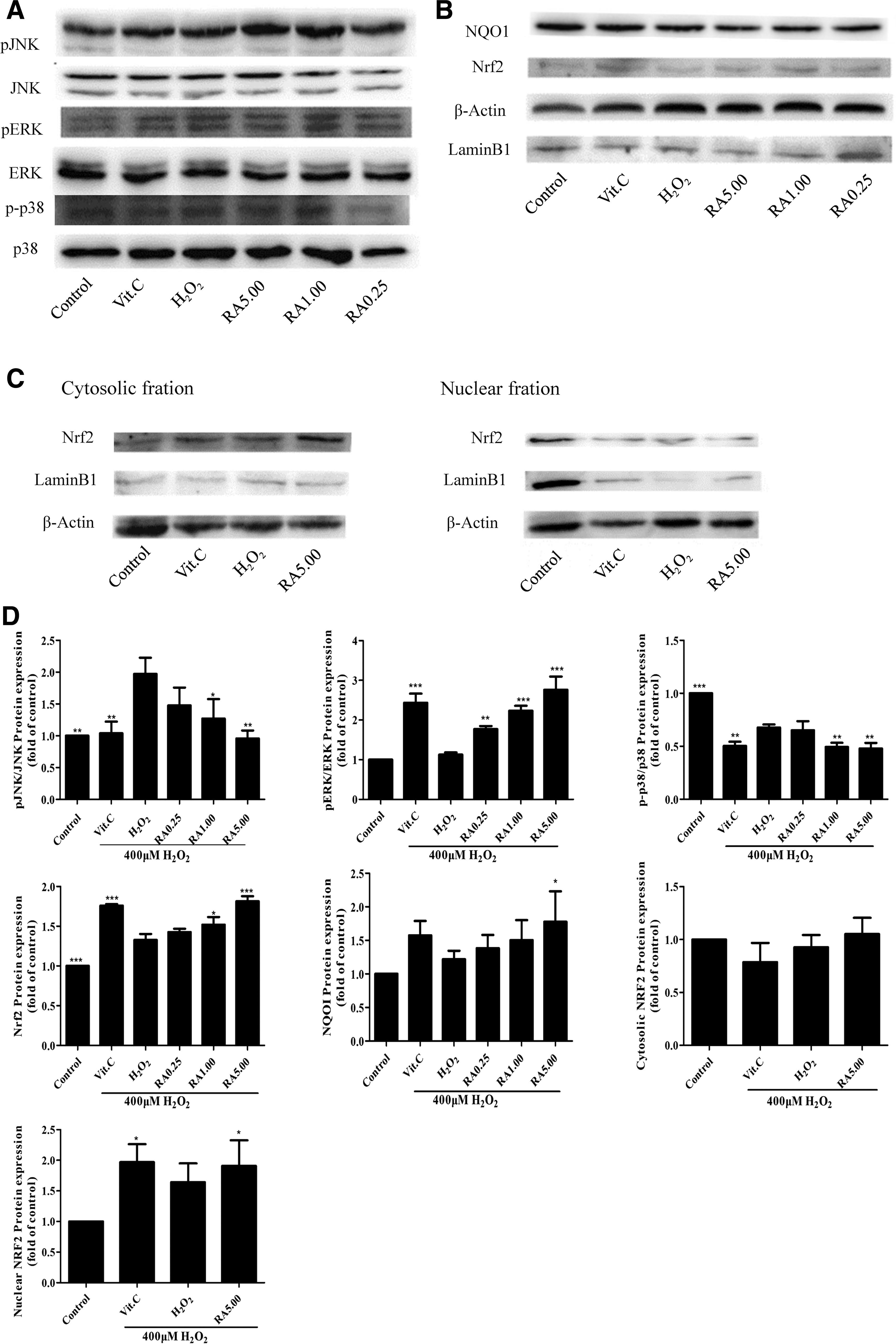
Western blotting analyses were conducted in each group.
RA attenuated oxidative damage by the activation of MAPK and Nrf2 signaling pathways
To further determine whether RA attenuated oxidative damage in L02 cells by inducing various signaling pathways, the expression of some oxidative stress-related major genes (e.g., MAPK, Nrf2, NQO1) was evaluated using Western blotting. Exposure to H2O2 significantly increased the phosphorylation of JNK, while RA and VC could reverse the JNK protein expression (Fig. 5A, D). In Figure 5A and D, the p38 protein expression was downregulated in both the RA treatment group and the H2O2-induced damage group, but the p38 protein expression decreased more in the RA treatment group than the H2O2-induced damage group to protect L02 cells from oxidative stress. The ERK1/2 protein expression was upregulated in both the RA treatment group and the H2O2-induced damage group, but the ERK1/2 protein expression increased more in the RA treatment group than the H2O2-induced damage group to protect L02 cells from oxidative stress.
The expression of Nrf2 and NQO1 was significantly induced in the RA (5 μM) treatment group than the H2O2-induced oxidative damage group (Fig. 5B, D). To further investigate the activation role of RA on Nrf2, we examined whether RA facilitated Nrf2 translocation from the cytoplasm to the nucleus. LaminB1 was used as reference protein in the nucleus, and β-actin was used as reference protein in the cytoplasm. As shown in Figure 5C and D, Nrf2 protein expression was induced in the nuclear fraction and cytosolic fraction, and the nuclear fraction increased more than the cytosolic fraction relatively in the RA treatment group, which indicated that RA could activate Nrf2 through upregulating its phosphorylation and facilitating its nuclear translocation. The antioxidant VC (100 μM) also activated the MAPK and Nrf2 signaling pathways, and it was used as positive control. As a result, RA showed a cellular protection response to oxidative stress by regulation of the MAPK and Nrf2 signaling pathways.
Discussion
NAFLD remains a high prevalence and rising incidence worldwide. ROS generate in microsomes, peroxisomes, and mitochondria during liver damage. Oxidant formulation has been suggested to be interrelated in the pathogenesis and progression of NAFLD, and several reports highlighted the efficacy of antioxidants for its treatment. For example, the antioxidant supplementation provided a significant attenuation of insulin resistance in overweight patients with liver injury. 25 Also, antioxidant interventions, which include vitamin E, vitamin D, coenzyme Q, polyphenols, polyunsaturated fatty acids, and preprobiotics, have enhanced antioxidant defense in NAFLD. 26 In particular, RA represents a highly effective and reliable property for the treatment of liver damage than the clinical drug of silymarin and N-acetyl-l-cysteine (NAC) compounds. 23 In the present study, antioxidant intervention RA could protect L02 cells from oxidative stress-induced damage and increase cell viabilities.
Consistent with that ROS can promote pathologic polyploidization and cell cycle arrest in the liver cells, subsequently leading to cell death by apoptosis and necrosis. The previous report showed that hepatocytes from oxidative stress-induced NAFLD mice increased the G2/M phase progression, while the antioxidant-treated NAFLD hepatocytes returned to the normal cell division. 27 Besides, it has been recently described that in the research models of oxidative stress-induced liver damage, cell death by apoptosis is currently occurring. 28 Our results showed that RA decreased G2/M phase progression and subsequent cell death, which is in agreement that cell cycle arrest increases the likelihood of cell death during ROS accumulation. 29
Redox-sensitive MAPKs interact with ROS generation and cell apoptosis, 30 and meanwhile, transcription factor Nrf2 also plays an important role against oxidative stress. 31 As we know, oxidative stress could activate simple liver damage to the progress of NAFLD, and we further explored whether RA alleviated oxidative stress via the MAPK and Nrf2 signaling pathways in liver damage. The results suggested that RA would protect liver cells against damage by activation of MAPK and Nrf2 expression. MAPKs contribute to cell proliferation, survival, and apoptosis. In particular, the phosphorylation of p38 and JNK could induce apoptosis, while the phosphorylation of ERK1/2 protects oxidative stress-mediated cell damage. 32 Our research indicated that RA may alleviate liver cell oxidative damage by inhibiting JNK and p38 expression, and inducing ERK1/2 expression. Besides, MAPKs might regulate Nrf2 in hepatic apoptosis, which is in agreement that coptisineagainst AAPH (2,2′-azobis[2-methylpropionamidine] dihydrochloride)-induced oxidative stress. 33 Oxidative modification induces the Nrf2-Keap1 complex to suppress Nrf2 degradation to bind to antioxidant response elements (ARE) and facilitates nuclear translocation. Results in the study showed that RA could protect liver cells from oxidative damage by activating expression of Nrf2, facilitating nuclear translocation, and inducing its downstream target gene expression of NQO1.
Conclusions
As previously reported, the upregulation of p38, JNK in MAPKs and the downregulation of ERK1/2 in MAPKs could induce hepatic apoptosis, while the upregulation of Nrf2 and its downstream target gene NQO1 protein expression could decrease hepatic apoptosis. 34,35 The inhibition of MAPK did not induce Nrf2 and HO-1 expression in the liver. 34,36,37 In summary, RA could decrease H2O2-induced oxidative damage to build a new redox homeostasis through decreasing cell cycle arrest and activating the MAPK and Nrf2 signaling pathways to suppress apoptosis (Fig. 6). These findings provided a new evidence to support RA as a potential candidate for the prevention or treatment of liver cell oxidative damage and related metabolic disorders such as NAFLD.
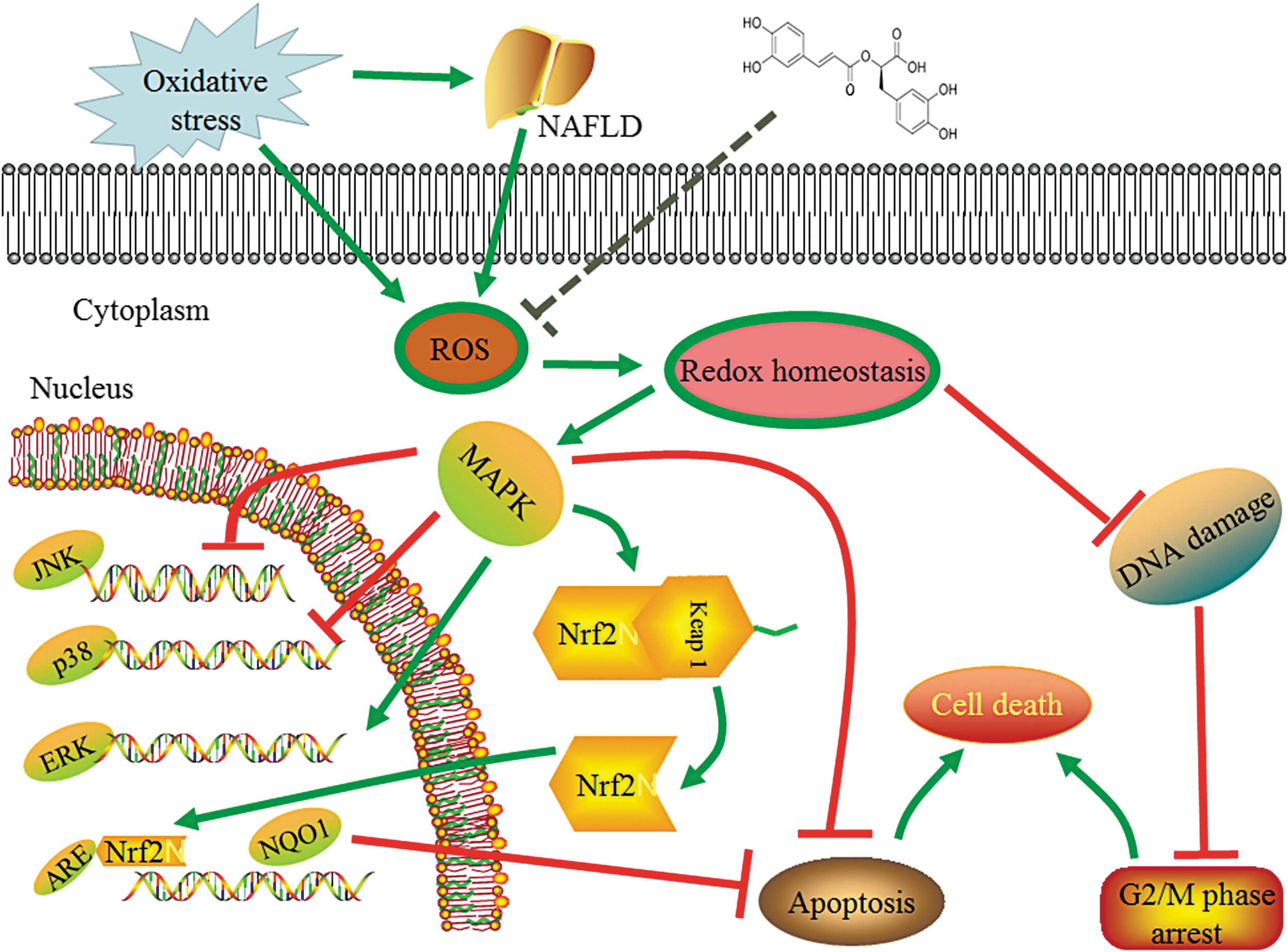
The proposed mechanisms of RA alleviating oxidative stress in L02 cells. ARE, antioxidant response elements; NAFLD, nonalcoholic fatty liver disease.
Footnotes
Acknowledgments
This work was supported by grants from the Special Funds of the Central Finance to Support the Development of Local Universities and Colleges, and Guangdong Province Department of Education (Grant 2015KGJHZ022).
Authors' Contributions
Y.D. was the principal investigator of the study and the writer of the original article, Z.Z. performed statistical analysis, Z.Y. and L.D. contributed to some experimental design, and H.H. and Z.H. contributed to the study design and helped in the review and editing of the original article.
Author Disclosure Statement
No competing financial interests exist.