
Abstract
Ferroptosis is a recently characterized cell death phenotype resulting from iron-catalyzed peroxidation of polyunsaturated fatty acid phospholipids. Increased dysfunctional iron metabolism is thought to lead to increased levels of iron and ferroptosis, which in turn leads to cell and organismal death at least in the nematode Caenorhabditis elegans. Drugs that block lipid peroxidation or scavenge intracellular iron extend healthspan and lifespan in C. elegans independently of other mechanisms such as the daf-1/daf-16 (insulin/insulin-like growth factor 1 [IGF-1]) pathway, but unlike many aging mechanisms do not alter temporal scaling across the life cycle of C. elegans, but rather act at specific late points in the organism's life history, temporarily blocking execution of critical dysfunction that results in listless worms. As such, inhibition of ferroptosis may be a means to extend healthspan and treat frailty and possibly neurodegenerative diseases that have a reported role for iron dyshomeostasis. However, a significant effort to understand ferroptosis in the context of mammalian and human biology is necessary. For example, some tumors block ferroptosis to survive. The constraints of balancing iron metabolism are significant and will require careful consideration in any drug development program.
What Is Ferroptosis?
Ferroptosis is a recently described cell death phenotype originally found to be induced in HRAS+ tumor cell lines by erastin an inhibitor of the cystine/glutamate antiporter, which reduced intracellular cysteine and thus glutathione (GSH) levels 1,2 (Fig. 1). More recently Dixon et al. 3 noted the characteristic features of this phenotype linked to the relative abundance of iron (catalytic ferrous [Fe2+]) in comparison with sulfur (sulfhydryls). Thus, either excess iron or sulfur deficiency contributes to ferroptosis. Central to the mechanism is iron-catalyzed peroxidation of polyunsaturated fatty acid phospholipids. GSH plays a central role as a ligand for cytosolic Fe2+ iron 4 and as the key substrate of glutathione peroxidase-4, which eliminates membrane lipid peroxides that contribute to ferroptotic cell death. 5 –7
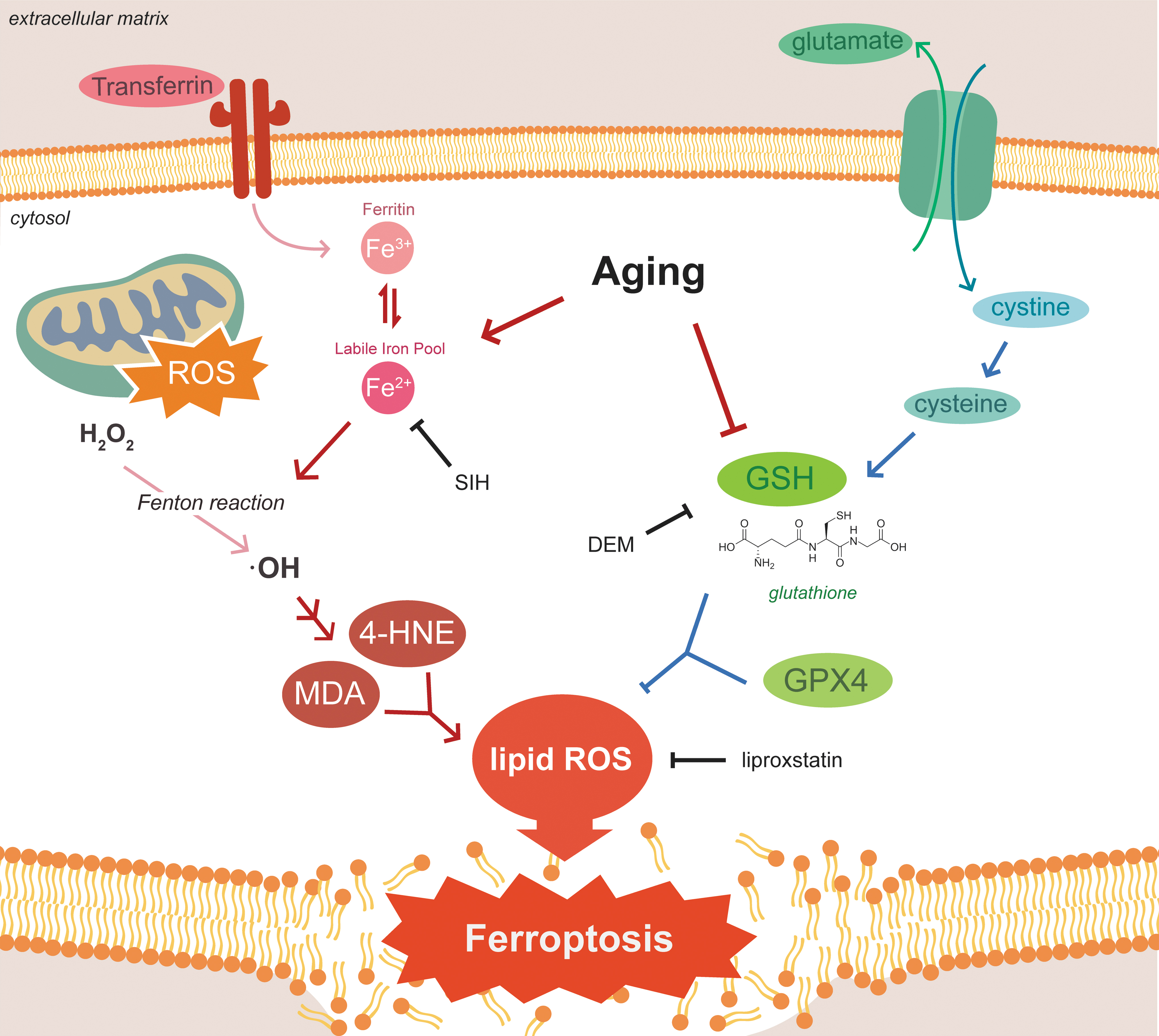
Pathogenesis of ferroptosis. Aging is associated with lower levels of reduced GSH and increased ferritin and pool of labile iron. Jenkins et al. 17 demonstrated that lowering of GSH with DEM was accompanied by ∼50% mortality of Caenorhabditis elegans worms that was mitigated by Lip-1 a known (although not very potent) inhibitor of ferroptosis through inactivation of lipid peroxide radicals and SIH, a lipophilic acylhydrazone that scavenges intracellular iron for extracellular clearance. Relatively stable and diffusible toxic lipid peroxidation end-products 4-HNE and MDA may mediate the paracrine propagation of ferroptosis to neighboring cells. •OH, hydroxyl radical; 4-HNE, 4-hydroxynonenal; DEM, diethyl maleate; Fe2+, ferrous; Fe3+, ferric; GPX4, glutathione peroxidase-4; GSH, glutathione; H2O2, , hydrogen peroxide; Lip-1, liproxstatin; MDA, malondialdhyde; ROS, reactive oxygen species; SIH, salicylaldehyde isonicotinyl hydrazone.
The histopathological hallmarks of ferroptosis are dysmorphic small mitochondria (reduced crista, condensed membranes, and ruptured outer membranes) and none of the hallmarks of other mechanisms of cell death, including apoptosis or autophagic cell death. Furthermore, knockdown of necroptosis mediators receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and receptor-interacting serine/threonine-protein kinase 3 (RIPK3) does NOT protect cells.
Loss of GSH and neuroferritinopathy play a role in the ferroptosis recognized during neuronal aging and age-related neurodegenerative diseases. 8,9 Iron storage protein ferritin increases during aging, 10 especially in elderly individuals with high neocortical amyloid-β load. 11 A recent prospective study found an association of elevated ferritin levels with cardiovascular and all-cause mortality. 12 Masaldan et al. 13 found that radiation-induced senescent cells exhibited impaired ferritinophagy and inhibition of ferroptosis, suggesting that their resistance to cell death goes beyond apoptosis. Ferroptosis may play a damaging role in stroke, 14 neurodegenerative diseases, 13 cardiomyopathy 15 and traumatic brain injury. 16
Evidence Connecting Ferroptosis to Aging in Caenorhabditis elegans
Jenkins et al. 17 hypothesized that elevation of Fe2+ iron and depletion of GSH during life primes cells of aged animals for ferroptosis. In Caenorhabditis elegans GSH levels drop ∼50% from day 1 aged juveniles to old worms aged 10 days and both total iron and areal density of iron increase with age as measured by synchrotron-based X-ray fluorescence microscopy. 18,19
To investigate mechanisms, Jenkins et al. 17 used a pharmacological approach to manipulate iron and GSH levels in C. elegans worms of various ages. GSH was acutely depleted using direct chemical conjugation by diethyl maleate (DEM). DEM (>5 mM) treatment of day 4 (adult, at end of reproductive phase) C. elegans worms caused a ∼50% depletion of GSH in 24 hours. which was accompanied by ∼50% mortality. Liproxstatin (Lip-1, 200 μM) is a known (although not very potent) inhibitor of ferroptosis through inactivation of lipid peroxide radicals. 3,20 Salicylaldehyde isonicotinyl hydrazone (SIH, 250 μM) is a lipophilic acylhydrazone that scavenges intracellular iron for extracellular clearance. 21 Lip-1 and SIH both rescued the DEM intoxicated 4-day-old worms with Lip-1 inhibition of ferroptosis occurring downstream of GSH depletion consistent with results found in cell culture models. 20,22
Ferroptosis can “propagate” in a paracrine manner outward from affected cells. Although details of this mechanism are unclear, relatively stable and diffusible toxic lipid peroxidation end-products 4-hydroxynonenal and malondialdhyde may mediate this effect. 23 Jenkins et al. 17 utilized propidium iodide (PI), a fluorescent intercalating agent that binds to DNA and stains dead cells (cannot cross membranes of live cells) to follow ferroptosis of individual cells. Interestingly, PI-positive dying/dead cells did NOT accumulate with aging. However, animals treated with DEM exhibited a >10-fold increase in PI-positive cells that was reduced to background by subsequent reduction in the labile iron pool by SIH or curtailment of damaged peroxidized membranes by Lip-1.
Fe2+ iron in the labile iron pool catalyzes the Fenton reaction to generate hydroxyl radicals from hydrogen peroxide that feed the catalytic increase in lipid peroxidation characteristic of ferroptosis and aging (Fig. 1). Direct in vivo ascertainment of Fe2+ versus ferric iron can be measured by X-ray absorption near edge structure (XANES) spectroscopy using fluorescence detection. 18 Ganio et al. 18 and James et al. 19 utilized XANES to document an increase in Fe+2 with aging. Jenkins et al. 17 confirmed the age-associated increase in the labile iron pool and showed that Lip-1 and SIH treatment normalized values to those of young worms. These interventions provided a significant increase in median lifespans, ∼70% with Lip-1 and ∼100% with SIH. However, the lifespan curves of Lip-1 and SIH demonstrated distinct effects. SIH (the iron scavenger) extended mid-life with a squaring of the survival curve at end of life, whereas Lip-1, the membrane peroxidation blocker, altered late life survival. More on this in “Modulating Ferroptosis to Benefit Lifespan Not Due to Temporal Scaling.”
Modulating Ferroptosis to Benefit Lifespan Not Due to Temporal Scaling
Stroustrup et al. 24 suggested that most longevity interventions in C. elegans (e.g., dietary restriction, temperature modulation, genetic modulation [e.g., daf-1/daf-16 {insulin/insulin-like growth factor 1 (IGF-1) pathway}, hif-1 {hypoxia-inducible factor}, and hsf-1 {heat shock factor}]) alter lifespan by an apparent stretching or shrinking of time, denoted “temporal scaling.” In this paradigm, an intervention extends lifespan by altering all physiological determinants of risk of death to the same extent throughout adult life. Thus, temporal scaling results when the risk of death is modulated by the intervention acting solely on the rate constant of a single stochastic process. The shapes of the Kaplan–Meir survival curves generated by SIH and Lip-1 modulation of ferroptosis point to a mechanism inconsistent with a temporal scaling model. SIH reduced mid-life risk of death, whereas Lip-1 markedly reduced the mortality rate in postreproductive late-life. Additional studies showed that modulation of ferroptosis could be demonstrated on top of a temporal scaling intervention such as temperature. Furthermore, although iron accumulation may contribute to many processes that can modulate the rate of aging, inhibition of ferroptosis reduces frailty (late life survival) rather than modulating the global rate of aging. Perhaps the most parsimonious explanation is that iron dyshomeostasis and ferroptosis are not fundamental to mechanisms that underlie the gradual dysfunction observed with and possibly underlying aging, but act toward the end of life as “executioners” to rapidly damage and degrade function.
Inhibition of Ferroptosis Improves Healthspan and Frailty
Interventions that augment the lifespans of worms (and other organisms) are often detrimental to healthspan and fitness. 25,26 For example, with dietary restriction the “cost” of longevity may be a smaller size, reduced fertility, and paucity of movement. 27 Interestingly, the SIH-treated animals grew substantially larger than controls (control 1440 ± 123 μm vs. SIH 1696 ± 64 μm). A surrogate measure of “healthspan” in C. elegans is the steady decline in motility. 28 Treatment with SIH or Lip-1 markedly improved the maximum velocity of aging worms as well as the distance traveled. Thus, modulation of ferroptosis improves “frailty” among aging C. elegans worms.
Medical Implications
Modulation of healthspan by interventions that modulate ferroptosis may provide medical benefit. Finding a “squaring” of the late-life survival curve with an improvement in motility and movement velocity in worms supports this contention. However, do these findings translate to mammals and ultimately to humans? Frailty is a significant quality of life issue impacting healthspan affecting >10% of the elderly, 29 characterized by decreased strength, endurance, functional reserve, unexplained weight loss, easy exhaustion, reduced appetite and increased probability of infection, morbidity, and death. 30 –32 A “Frailty Index” measures the accumulated effects of aging, including social, physical, and cognitive impairments. 33 Afflicted patients are currently managed with nutritional support (e.g., calories and vitamin D), physical therapy regimens (both aerobic and resistance training), and optimized medication management (attention to drug–drug interactions and dose attenuation). More effective interventions are needed. Iron has been reported to increase with aging in rats and people. 34 –37 So is there more than a correlation? Perhaps modulating accumulation of iron and reducing resulting ferroptosis resulting from increased iron is worthy of investigation as a possible therapeutic intervention, although much work would need to be done to establish the iron/ferroptosis/frailty connection in mammals and humans.
Beyond accumulation with age, dyshomeostasis of iron plays a key role in several important neurodegenerative diseases among them Alzheimer's disease, Lewy Body disease, and frontotemporal dementia. Iron contributes to aggregation and pathogenicity of beta-amyloid, 38 tau, 39 alpha synuclein, 40 as well as TDP43. 41,42 A reduction of excess iron or inhibition of ferroptosis are potential points of therapeutic intervention that need to be investigated further. Similarly, ferroptosis plays a role in cardiac dysfunction associated with cardiomyopathy and can be alleviated by its inhibition. 15 However, the particular underlying mechanisms of toxic accumulation of iron may differ in these different pathologies.
Beyond chelation to control reduce potentially toxic excess iron or scavenging of lipid peroxides to prevent ferroptosis, activation of 5′ adenosine monophosphate-activated protein kinase (AMPK) by energy stress (caloric restriction) and presumably drugs such as metformin inhibits ferroptosis, which may explain some of the apparent antiaging benefits of metformin and caloric restriction. Moreover, caloric restriction may be a relatively safe way to attenuate ferroptosis, having been under intensive study as a way to extend healthspan and lifespan.
The general problem with interventions for iron dyshomeostasis is that iron metabolism needs to be finely tuned, as it is detrimental to have excess or insufficient iron or ferroptosis. For example, contrary to what might be needed to inhibit frailty or neurodegeneration, restoring or stimulating ferroptosis function in cancer cells is a promising new therapeutic approach to kill tumors.
Ultimately, the significance of ferroptosis as a target for antiaging therapies depends on the validity of the hypothesis that altered iron metabolism associated with aging (which needs to be carefully maintained for good health), is a potential key target for the development of new therapeutics for frailty, neurodegenerative diseases, cardiomyopathy and in general for late stage effects of aging. The caveat is that inhibition of ferroptosis presents potential problems with promoting the survival of cancer or precancerous cells. We expect significant attempts to develop drugs that modulate iron metabolism and ferroptosis, which ultimately may have profound effects on healthspan.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.