
Abstract
Myocardial infarction (MI) remains the leading cause of mortality and morbidity worldwide. It is caused by a thrombotic occlusion of coronary vessel/s that leads to cardiomyocyte death. As a response, inflammatory and fibrotic responses are initiated to replace the necrotic tissue and remodel the heart. However, in most cases, these responses are excessively activated, which accentuates the injury and causes adverse cardiac remodeling, often leading to heart failure. This is highly attributed to the dysregulated repair mechanism brought by reduced regenerative capacity of the adult heart, chronic inflammation, and other patient factors, such as comorbidities, diet, and lifestyle. Because of the negative consequences of excessive inflammation and fibrosis in post-MI responses, inhibiting factors associated with these processes are one of the major approaches in MI management. Several therapies have been developed to broadly and/or selectively inhibit inflammation- and fibrosis-associated proteins over the past decades and have shown promise in addressing post-MI complications. However, challenges (e.g., off-targets, problems with drug delivery, dosage, route, and cost) and efficacy of these interventions in the clinical setting remain. Hence, alternative approaches to optimally alleviate these post-MI processes are still much needed. In this review, we discuss the possible use of plasmapheresis, a technique that involves extracorporeal replacement of blood plasma, as a treatment for MI. We provide an overview of the inflammatory and fibrotic responses after MI and focus on how plasmapheresis can be an approach to target these pathways.
Introduction
Myocardial infarction (MI) contributes to the global health burden and results in high morbidity and mortality worldwide. MI occurs when there is an abrupt reduction of blood flow in the myocardium, causing necrosis, ultimately leading to heart failure. Immediate restoration of blood flow to rescue the ischemic tissue and reduce the infarct size, usually through thrombolysis, percutaneous coronary intervention, and coronary artery bypass grafting, has been the main clinical intervention for MI. Aside from these, patients receive pharmacological interventions such as aspirin, statins, beta-blockers, angiotensin-converting enzyme inhibitors (ACEis), and angiotensin receptor blockers (ARBs) with the goal to reduce infarct size and improve ventricular remodeling. However, studies have shown that these therapies have limited efficacy in reducing MI recurrence and 10-year mortality risk, and rehospitalization remains high. 1 –4 Thus, new strategies for treating MI are in high demand.
The severity of inflammatory and fibrotic response post-MI are major therapeutic targets, as these processes play critical roles in determining the extent of infarct and subsequent left ventricle remodeling. Moreover, improving cardiac regeneration post-MI has been of great interest because the restricted regeneration capacity of adult cardiomyocytes has been shown to contribute to the elaborated fibrosis post-MI. 5 However, amid the many attempts to target inflammation, fibrosis, and regeneration after MI, no therapy was shown to address all these factors. Moreover, off-target events, high cost, and complexity of the treatments summarize the key challenges of these therapies. 1,6
Recent studies reported the involvement of systemic responses in the clinical outcomes of MI. 7,8 For example, MI is associated with elevated levels of circulating inflammatory factors, increased activation of peripheral immune cells and age-associated pathways, dysregulated immune system, and generalized increase in atherosclerotic inflammation. 7,9 Moreover, systemic effects of MI were also found to be linked with the release of paracrine factors that cause inflammation and senescence in distal organs, such as the spleen, kidney, and liver. 10
Here, we evaluate the potential use of therapeutic plasma exchange (TPE), also referred to as plasmapheresis, as a treatment for MI. It involves systemic removal or exchange of blood elements and was recently found to reduce systemic inflammation and fibrosis, while promoting multitissue rejuvenation using age-neutral saline as replacement fluid in murine models. 11,12 This review describes the molecular responses, which are elicited by MI, and the potential of plasmapheresis to attenuate these pathogenic systemic changes via dilution of the factors in blood.
Inflammatory Response After MI
Following an acute myocardial infarction episode, ischemic cellular damage triggers a series of inflammatory reactions to clear debris and facilitate healing. 13,14 The inflammatory response consists of proinflammatory and anti-inflammatory phases, which should be in balance for productive wound healing (Fig. 1).
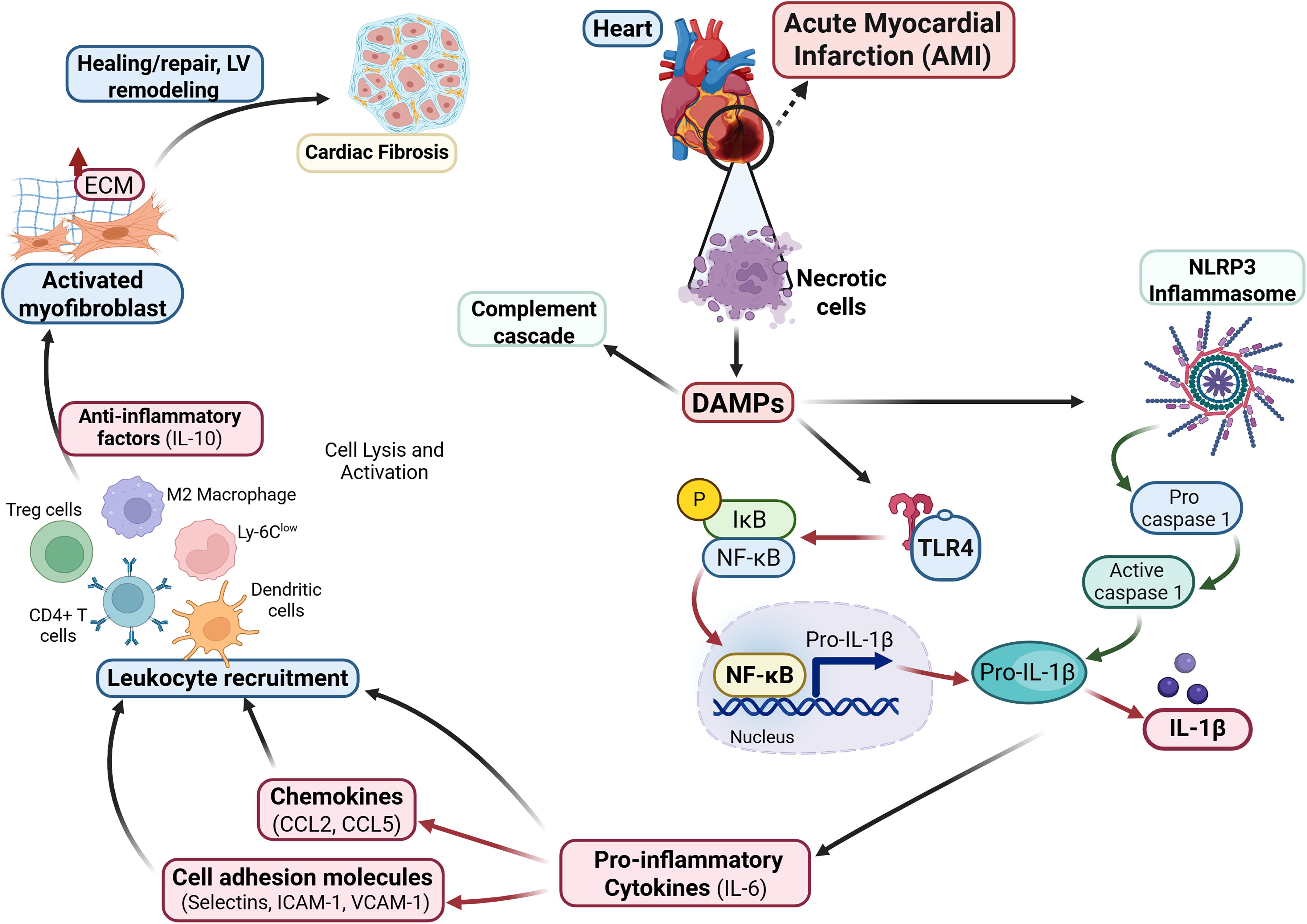
Overview of the inflammatory response following an acute myocardial infarction (AMI). AMI triggers intense initial proinflammatory responses, such as elevation of inflammatory mediators and complement cascades, and recruitment of inflammatory cells via DAMPs/TLR4/IL-1 signaling. Inflammasomes activate caspase-1, which initiate the release of IL-1β, a known proinflammatory factor that amplifies the secretion of other proinflammatory cytokines. These responses lead to leukocyte infiltration in the injured area to clear necrotic cells. To modulate these responses, CD4+ T cells and dendritic cells are recruited in the injured area and initiate the anti-inflammatory responses. Overlap between inflammation and regeneration leads to minimal fibrosis and effective wound healing. IL-1, interleukin-1; DAMPs, damage-associated molecular patterns; TLR, toll-like receptor.
The proinflammatory responses (e.g., recruitment of immune cells, activation of complement cascades, and release of endogenous alarm signals called damage-associated molecular patterns) take place after MI. 9,15 The release of cytokines (IL-1β, IL-6) and chemokines (CCL2 and CCL5), which are the main drivers of post-MI inflammation, mediate the recruitment of leukocytes; their roles after MI have been extensively reviewed. 16 –18 Briefly, IL‐1β signaling stimulates chemokine synthesis and promotes leukocyte infiltration in the infarct area to clear dead cells and debris, as well as to delay myofibroblast conversion, 19 and suppress the synthesis of α‐smooth muscle actin. 20,21 Meanwhile, IL-6, aside from its role in leukocyte infiltration, contributes to the development of atherosclerotic plaque, cardiac hypertrophy, and cardiomyocyte loss. 22 Chemokines (CC and CXC) have been implicated in neutrophil, monocyte, fibroblast, and vascular cell recruitment, and work together with cytokines to modulate inflammatory responses post-MI. 23,24
To limit the extent of inflammatory response, anti-inflammatory signals are activated in the infarct area by recruiting macrophage subsets and Tregs, CD4+ T cells, and dendritic cells. They release anti-inflammatory factors, such as IL-10, which trigger the differentiation of cardiac fibroblasts to myofibroblasts and secretion of extracellular matrix (ECM) proteins, eventually leading to cardiac fibrosis. 15
The balance between the inflammatory and anti-inflammatory mediators is required for productive cardiac repair and for preventing adverse cardiac remodeling. 25 In animal models, defects in these regulatory mechanisms result in prolonged and expanded inflammatory responses, which in turn prolong the fibrotic response and aggravate collagen deposition. 26,27
Fibrotic Response after MI
Cardiac fibrosis is an important part of the reparative process following MI. It occurs acutely following inflammation and is initiated by immune cell migration and ECM deposition around the infarcted area. 28,29 This mainly results in replacement fibrosis, wherein an expanding interstitium replaces the necrosed cellular tissue.
The process involves complex interactions of immune cells, endothelial cells, cardiomyocytes, ECM components, and a wide array of molecular mediators. Some of the key factors associated in this post-MI fibrosis are interleukins IL-1β, IL-6, tumor necrosis factor (TNF)-α, and matrix metalloproteinases, which recruit macrophages to clear the cellular debris and degrade the extracellular matrix, while transforming growth factor (TGF)-β activates fibroblasts (Fig. 2).
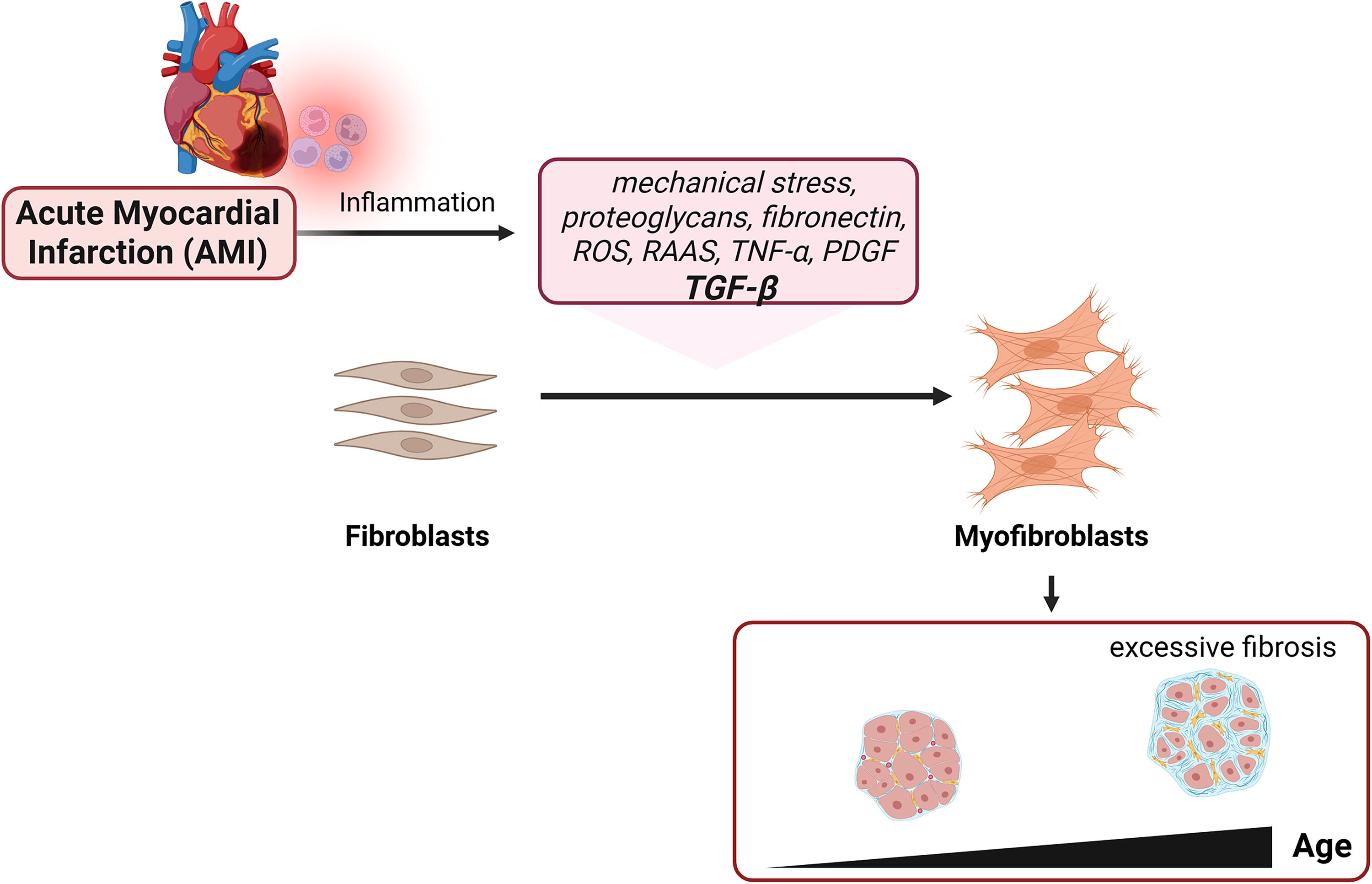
Overview of the fibrotic response following an acute myocardial infarction (AMI). AMI triggers fibrotic response, which is initiated by inflammation and TGF-β activation. This leads to myofibroblast proliferation to produce and organize collagen and other extracellular matrices (ECM), forming scar tissue. In young individuals, this response often leads to normal scarring. But with age, excessive myofibroblast activation occurs, which causes excessive fibrosis.
Post-MI fibrosis is needed for the survival of an organism. The deposited ECM provides tensile strength to the injured myocardium, maintains structural integrity, and provides a scaffold for future remodeling. Without fibrosis, the injured myocardium is more prone to cardiac aneurysm and rupture. 26,30 –32 Moreover, the repaired scaffold maintains the function of the heart conduit by retaining the blood passageway, serving as a temporary bridge for the cellular gaps in the myocardium. 33 Fibrosis is also a migratory medium for the inflammatory and reparative cells to migrate toward the affected areas.
Despite these clear benefits and roles in the reparative process, myocardial fibrosis, if excessive, causes pathological cardiac remodeling with subsequent adverse outcomes. Because of the lack of inherent contractility and inherent stiffness, scar tissue can interfere with the effective blood pumping. 30,31 This results in a loss in pumping efficiency and filling capacity, leading to systolic and diastolic dysfunction. The highly dense and fibrous tissue also hampers the migration and formation of cellular structures that are acutely needed during the remodeling process. A lack of tissue remodeling can seriously affect the reparative process by interfering with gas and nutrient exchange and the establishment of cell niches that are important for the immediate regeneration of the surrounding areas.
Ultimately, excessive fibrosis is deemed a major cause of heart failure, arrhythmias, and other adverse sequelae that increase patients’ risk for hospitalization and mortality after MI. This has led to interest in targeting fibrosis as a potential therapy over the past decades. 34,35 Yet, currently, most therapies targeting the activity of fibroblasts are still in early preclinical stages. 35
The importance of post-MI processes for cardiac health highlights the need to further investigate the modulation of cardiac fibrosis after MI with the goal of modifying disease via appropriate repair response while clinically minimizing the extent of defective scar formation and adverse remodeling.
Therapeutic Targeting of Inflammation and Fibrosis After MI
Given the negative consequences of excessive and prolonged inflammation and fibrosis after MI, several strategies have aimed to attenuate the initial proinflammatory response, enhance the anti-inflammatory response, and reduce the fibrotic response (Table 1). 20
Examples of Broad-Range and Targeted Therapies for MI Patients
ACEi, angiotensin-converting enzyme inhibitors; ARBs, angiotensin receptor blockers; CCL, chemokine C-C motif ligand; COX, cyclooxygenase; ECG, electrocardiogram; MI, myocardial infarction; IL, interleukin; LV, left ventricle; MAPK, mitogen-activated protein kinase; NSAID, nonsteroidal anti‐inflammatory drugs; PCI, percutaneous coronary intervention; PDGF, platelet-derived growth factors; STEMI, ST-elevation myocardial infarction; TGF, transforming growth factor.
Of key interest, most targets for MI are circulating factors, including but not limited to cytokines, chemokines, and miRNA. 63 To reiterate, these factors are vital in facilitating the healing process post-infarction. However, their excessive levels post-injury contribute to pathological remodeling. The dysregulated expression of these factors has been associated with aging, 64 lifestyle and diet, 65 comorbidities, and environmental factors. 66
Some of the most studied targets are the complement system, cytokines such as IL-1 β, IL-6, and TGF-β, and chemokines such as CCL2. These anti-inflammatory strategies have shown promise in animal models, being able to reduce infarct size and improve cardiac contractility and overall cardiac function. However, translation of these results to MI patients has been challenging (Table 1). Targeting the complement system through an antibody, pexelizumab, did not improve patients’ prognosis. 50 And while targeting IL-1 through anakinra, a recombinant interleukin‐1 receptor antagonist, has shown promise in reducing inflammation post-MI, 67 its effectiveness to reduce infarct size has shown variable results in clinical trials. 46 Similar limitations were noted for targeting inflammatory chemokines, such as CCL2, which showed fibrosis reduction and improved diastolic function post-MI in murine models, but limited reported clinical studies. 47,48
Targeting cell signaling pathways, which are responsive to MI, has also emerged as a clinical strategy in past decades. A network of pathways is activated post-MI to initiate the wound healing, and these are extensively reviewed. 68 This includes TGF-β/SMADs pathway, proangiogenic phosphoinositide-3 kinase/protein kinase B (PI3K/Akt) pathway, Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, and mitogen-activated protein kinases (MAPK) pathway, among others. Several molecular targets for these pathways have been applied post-MI (Table 1) and have shown benefits in addressing adverse cardiac remodeling. However, targeting these pathways has its limitations and challenges, following their complex role in other physiological functions and deleterious consequences of their skewing.
The limitations of these currently available strategies highlight the need to optimize and identify new therapeutic targets for MI management.
Plasmapheresis
The use of plasmapheresis in humans dates back to the 1950s when it was applied for the treatment of thrombotic thrombocytopenic purpura. 69 It involves the removal of plasma components (i.e., autoantibodies, immune complexes, cytokines, and toxins) in exchange for a replacement fluid such as saline with albumin or fresh frozen plasma (FFP). 70 To date, applications of TPE in the medical field have grown extensively and this procedure is currently use to treat several disease, for example, a first line treatment for myasthenia gravis, Guillain-Barre syndrome (GBS), chronic inflammatory demyelinating polyradiculoneuropathy, and chronic acquired demyelinating polyneuropathies, among others. 70 Table 2 enumerates the pathologies for which the therapeutic use of plasmapheresis is strongly indicated.
List of Diseases with Strong Recommendation (Grade 1A and 1B) for Therapeutic Plasmapheresis or Similar Techniques
DFPP, Double filtration plasmapheresis; IA, immunoadsorption; RBC exchange, red blood cell exchange; GRADE, Grading of Recommendations Assessment, Development, and Evaluation, 1 to 5, 1A being the highest; TPE, therapeutic plasma exchange. Adopted from Connelly-Smith et al. 70
Moreover, potential benefits of plasmapheresis in other pathologies are currently being investigated clinically. As of 2024, there are 263 ongoing clinical trials involving plasmapheresis. 71 Table 3 lists some clinical trials with published results that were performed the trials with healthy controls.
Comprehensive Table of Clinical Trials of Plasmapheresis from ClinicalTrials.gov with Results
*NCT# = National Clinical Trial number. Terms “Plasmapheresis”, “plasmapheresis device”, “plasma exchange”, and “plasma exchange with albumin” were used. Only studies with results available are shown. Only studies that include a control group are shown (phase I trials are excluded).
As discussed, curtailing excessive inflammation is a key element in treating MI, for which plasmapheresis can potentially provide an effective means to do so. To date, based on Table 3, none of the reported clinical trials investigated the use of plasmapheresis for MI cases. It has been used for heart transplants with the goal of limiting the circulating antibodies that interfere with organ transplants and lupus myocarditis with a similar goal of diluting the auto-reactive antibodies. 72 –74 Other examples, such as Wilson disease, which involves copper accumulation, leading to cardiac fibrosis, 75,76 are also treated with plasmapheresis to physically reduce the systemic toxins. 75,77,78
Interestingly, there are also several examples of inflammation-associated pathologies for which plasmapheresis has provided clear evidence of effectiveness. Here we provide some examples.
COVID has been observed to cause a cytokine storm in the most severe cases. To treat this, plasmapheresis was approved as a potential therapy, 79 quickly showing its advantages while having minimal side effects. This has been discussed at length elsewhere 80 but it is interesting to note that plasmapheresis provided significant benefits on proinflammatory clearance by lowering cytokine levels, as well as, reducing mortality risk. 80,81
In acute disseminated encephalomyelitis (ADEM) and GBS, excessive inflammation was linked to brain and spinal cord damages, and TPE showed some success as second line therapy to eliminate autoantibodies, immune complexes, cytokines, and cellular debris. 82,83 Of note, the inflammatory response for ADEM and GBS involves T cell activation and overexpression of inflammatory cytokines (IFN-γ, TNF-α, IL-1β and IL-6). 84,85
Further, in atopic dermatitis, TPE and similar had some potential. 33,86 –88 In atopic dermatitis, there is a dysregulated immune response featuring enhanced type 2 inflammation, involving several cytokines such as IL-4, IL-5, IL-13, and IL-31 and chemokines. 89 These cytokines mediate several downstream processes that result in mast cell activation, IgE production, and skin barrier dysfunction. In a 2015 case report, the use of TPE was able to reduce plasma IgE and improve patient symptoms and quality of life. 87
Moreover, for the purposes of this review, plasmapheresis has been deemed useful in certain diseases revolving fibrosis. These include neonatal lupus, 90 graft-versus-host disease, 91,92 organ transplant, nephrogenic systemic fibrosis, 93 and systemic sclerosis. 94 These pathologies have an autoimmune basis, but these demonstrate how plasmapheresis can be applied to fibrosis-based complications.
Lastly, in preclinical studies, TPE improved the balance of circulating leukocytes and of systemic proteome in older individuals, and this procedure reduced fibrosis of skeletal muscle and liver in old mice. 12,95
Overall, plasmapheresis has been shown effective in modulating immune and inflammatory responses in a wide array of diseases. It is mainly used to eliminate immune complexes, cytokines, and toxins that contribute to the pathogenesis of various diseases. A single or a few procedures of plasmapheresis are relatively safe. The patient may suffer from dizziness, chills, faintness, nausea, blurry vision, feeling of coldness or breathlessness, or temporary low blood pressure. There have also been reports of other, more serious side effects, albeit with a lower incidence. These may include reduced immunity to disease, allergic reactions, blood clotting, infections, and shock. Still, compared to the anticytokine antibodies, exogenous receptors, and other comparable factors, TPE is expected to be less harmful for the overall organismal homeostasis.
Plasmapheresis as a Therapy for MI
After MI, immune cells (e.g., leukocytes, neutrophils, monocytes/macrophages), cytokines (e.g., IL-1β, IL6, TNF-α, TGF-β), and chemokines (e.g., CCL2) orchestrate heart inflammation and fibrosis, while key regulators of signal transduction pathways become highly elevated in plasma. 44,96 –99 At their healthy normal levels, these circulating factors and pathways are needed to initiate the repair mechanisms; however, their excessive levels amplify the inflammatory and fibrotic responses. Such dysregulation promotes pathological cardiac remodeling, which causes severe clinical complications. Hence, proper modulation of these factors and pathways is needed to achieve balance in proinflammatory, anti-inflammatory, and fibrotic responses, and plasmapheresis is known to restore the systemic balance to the determinants of signal transduction. 95
Through plasmapheresis, blood elements become systemically lowered. This includes reduction of immunological factors, such as cytokines (IL-1, IL-6), 100 pathogenic antibodies, 101 and complement components. 102,103 Our studies have explored the role of plasmapheresis in specific dampening of disease-associated signaling pathways, 95 and it is interesting to note that some of the mentioned factors, such as IL-1, IL-6, complement proteins, are associated with activation of inflammasome (NLRP3/Caspase-1) pathways, 104,105 MAPK, JAK/STAT3, and PI3K/AKT pathways, 106 and TGF-β pathways, 107 which are all associated with the pathogenesis of MI.
With these evidences, it is surmised that in the context of MI, plasmapheresis play a major positive role in attenuating ongoing inflammation and fibrosis. Of note, through plasmapheresis, the goal is not to systemically inhibit or remove blood factors, a challenge that has been raised in most targeted therapy. Instead, the aim is to reduce concentrations of these factors to reach their optimal levels, which productively activate the physiological repair mechanisms (Fig. 3). To date, no studies have investigated calibration of circulating factors after MI via TPE.
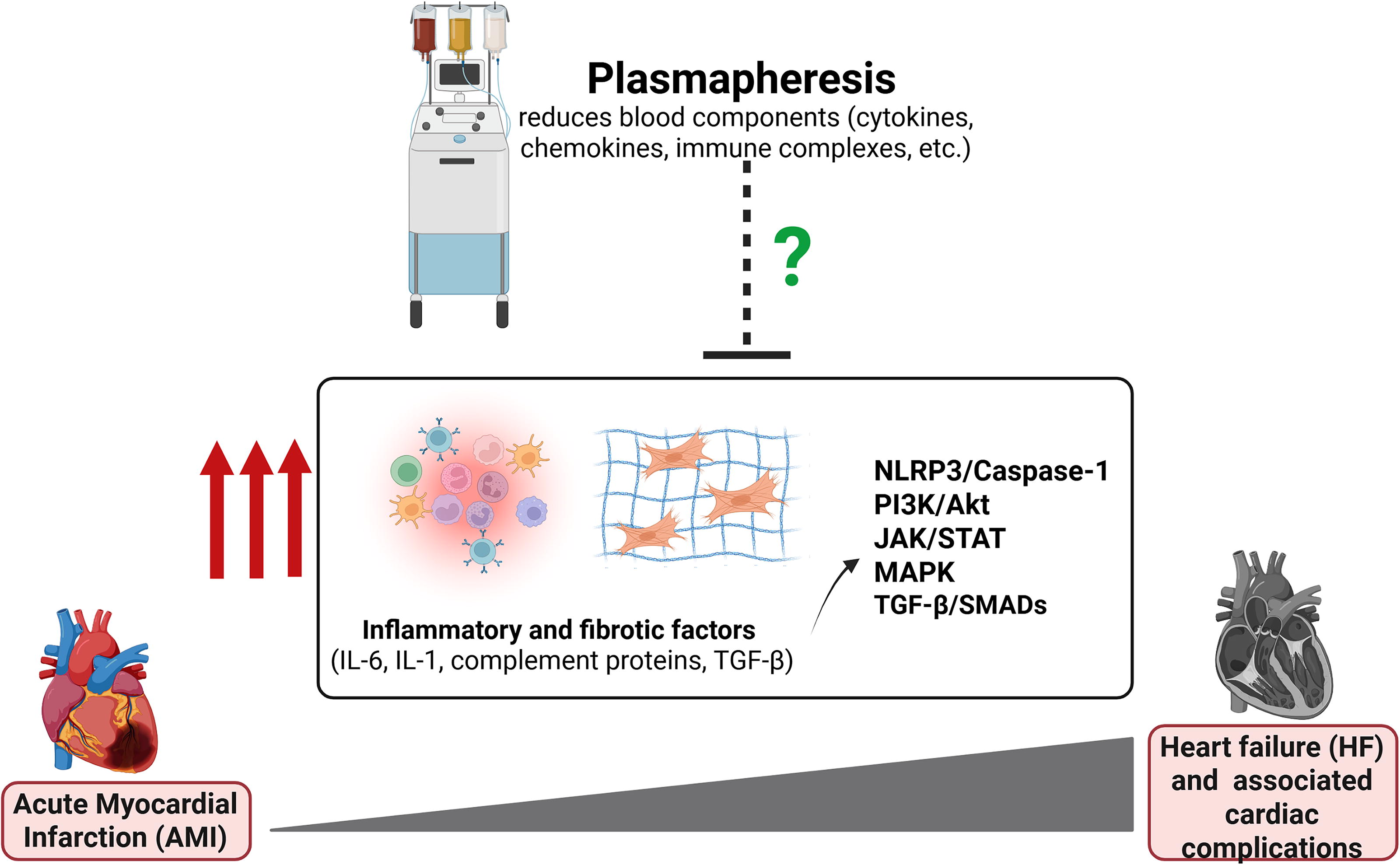
Therapeutic potential of plasmapheresis for acute myocardial infarction (AMI). Heart failure (HF) and associated cardiac complications following AMI are brought by excessive inflammatory and fibrotic factors which play a role in activating networks of signaling pathways, such as NLRP3/Caspase-1, PI3K/Akt, JAK/STAT, MAPK, and TGF-β. In this review, we highlight the potential of plasmapheresis, known to reduce cytokines, chemokines, immune complexes through blood dilution, to address these post-MI complications.
Furthermore, plasmapheresis has been shown to improve rejuvenation of three germ layers (liver, brain, and muscles) in murine models by resetting the old systemic milieu to a youthful state. 12 Hence, it would be interesting to investigate the effects of this procedure on the health of cardiac tissue.
Amid this promising potential of plasmapheresis as a treatment for MI, several factors are yet to be investigated. Compared to the diseases that use plasmapheresis, inflammation, and fibrosis after MI occur acutely, mainly during the initial steps of cardiac repair, ranging from onset of injury until 7 to 14 days. 46 Some reviews in this topic suggest that the initial reparative period after infarction could be as early as 24 hours. 18 Hence, the timing of plasmapheresis should be considered and coordinated with the event of MI. Moreover, the number and length of exchanges to reach the optimal levels of blood factors should also be determined.
In addition, vascular access is also an important factor to consider for doing plasmapheresis. It is necessary to have a steady flow of blood while doing the procedure, as interrupted flow can disrupt the procedure and alter its effectiveness. Most apheresis devices require blood flow rates of 50–120 mL/min. 108 Although vascular patency and angiogenesis are highly affected in MI cases, a therapeutic opportunity was demonstrated in a study on rats, which showed that the vasculature in infarcted areas, remains patent until the gradual progression of fibrosis contributes to flow impedance. 109 Moreover, reperfusion therapies have significantly improved over the decades and were proven to restore blood flow in the ischemic area. 110 Thus, we posit that the benefits of plasmapheresis can be maximized if it is performed during the early stages of MI after a reperfusion therapy was applied, to ensure stable blood flow during the TPE.
Conclusion
MI management has been improving over the past decade, yet many challenges still exist. Although several therapies have shown promising outcomes in animals, their translation to MI patients remains without success. Here, we propose the use of plasmapheresis to address the excessive inflammation and fibrosis following MI, through diluting plasma, thereby systematically reducing the MI-elicited excessive inflammatory, profibrotic factors, and immune cells. Although more research is needed to explore its application for MI and other cardiovascular diseases, plasmapheresis has long been used to treat diseases with the same pathogenic features (excessive inflammation, immune cells, toxins, etc.). Moreover, as more studies highlight the involvement of systemic responses after MI, TPE seems to be perfectly suited to ameliorate the MI pathology. Lastly, more work is needed to understand MI-related changes in plasma proteomes, which would aid in the design of future therapies.
Footnotes
Authors’ Contributions
J.M.C.C. and I.M.C. cowrote the article. J.M.C.C., J.L.G., A.R.M., A.J.A.M., J.V.S.P., and J.B.N. did the literature search. J.M.C.C., A.J.A.M., and J.V.S.P. contributed the figures. All the authors listed have made a substantial contribution to the work. All the authors have read and approved the article.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
This work was supported by Department of Science and Technology-Philippine Council for Health Research and Development (DOST-PCHRD) Research Enrichment Program (REP) grant (J.M.C.C.), Commission on Higher Education (CHED) Philippine-California Advanced Research Institutes-Institute for Health Innovation and Translational Medicine (PCARI IHITM) 2018-033 grant (J.M.C.C. and I.M.C.), National Institutes of Health-National Heart, Lung and Blood Institute (NIH-NHLB) R01 139605 (I.M.C.), National Institutes of Aging (NIA) R01AG071787 (I.M.C.), Open Philanthropy award (I.M.C.).