
Abstract
Following fusion, embryonic stem cells (ESCs) are capable of reprogramming somatic cells in cell hybrids. It has also been shown that transcriptional changes can occur in a heterokaryon, without nuclear hybridization. However, it is unclear whether these changes can be sustained after the removal of the dominant nucleus. In this study, we analyze the changes in embryonic stem (ES)–somatic heterokaryons following the removal of the ESCs nucleus. We also show that after ES–somatic cell fusion using tetraploid ESCs, a heterokaryon can be reverted to an autologous diploid state by differential enucleation of the denser tetraploid ES nucleus. To recover somatic cells from ES–somatic heterokaryons, we fused tetraploid ESCs containing the thymidine kinase (TK) suicide gene with mesenchymal stem cells containing a green fluorescent protein (GFP) transgene under the control of the OCT4 promoter. Following post-fusion enucleation (PFE), negative selection against the tetraploid ES genome was achieved using ganciclovir. The resulting GFP-positive clones were analyzed and shown to have undergone changes in growth characteristics, alkaline phosphatase activity, and gene expression using RT-PCR and microarray analysis. These results demonstrate that a change in transcriptional expression can be detected in somatic cells after the removal of the ES nucleus from ES–somatic heterokaryons.
Introduction
B
Heterokaryon formation (fused cells with two nuclei and a common cytoplasm) offers a unique tool to study the reprogramming process between two cell types. Interspecies heterokaryons formed from different adult cell lineages have revealed that the re-expression of silent genes can be detected within 24 h of fusion [6,7]. These changes in gene expression can take place in the absence of cell division or DNA replication, especially when the nuclear ploidy ratio is in favor of the dominant cell type [8,9]. Furthermore, reprogramming in heterokaryons appears to be a temporal process that involves the acquisition of the dominant gene expression program, followed by gene silencing in the nondominant nucleus [9]. More recently, the interspecies heterokaryon reprogramming of human lymphocytes by mouse ESCs revealed similar temporal transcriptional changes with the expression of pluripotency markers as early as 1 day after fusion, and the extinguishing of somatic genes after 2 days of fusion [10]. Despite the initiation of reprogramming in ES-somatic heterokaryons without chromosome intermixing [10], it remains unclear whether the reprogrammed state of a somatic nucleus can be maintained in the absence of the ES nucleus after heterokaryon formation.
We have previously described an approach termed post-fusion enucleation (PFE) that selectively removes one genome from heterokaryons without compromising cell viability [11]. Heterokaryons were generated by fusing tetraploid and diploid ESCs cells with subsequent enucleation of the denser tetraploid nucleus by centrifugation. This process did not have any adverse effects on pluripotency or viability following differential enucleation of the ES–ES heterokaryons. In the present study, we used a modification of this approach to investigate whether transcriptional changes of somatic cells in heterokaryons could be observed following enucleation of the dominant, pluripotent ES nucleus. We generated ES–somatic heterokaryons by fusing tetraploid ESCs and diploid somatic cells, which were then maintained in culture for 48 h. We identified conditions for the differential removal of the ESC nucleus, allowing for the recovery of somatic cells. Analysis of diploid PFE clones generated through this process, as identified by the expression of the OCT4-green fluorescent protein (GFP) transgene, revealed changes to growth characteristics and morphology, alkaline phosphatase activity, and gene expression as shown by RT-PCR and microarray analysis. These results suggest that transcriptional changes in a somatic nucleus can occur without nuclear hybridization and is observed after the removal of the ES nucleus from heterokaryons.
Materials and Methods
Cell culture
Feeder-independent mouse ESD3, tetraploid, and sorted ESC clones were cultured in DMEM (Invitrogen) supplemented with 10% Hyclone™ fetal bovine serum, 1 mM
OG2 MSC isolation
Femurs and tibias from 6- to 8-week-old OG2 mice [12] were dissected and the ends were removed to flush out the marrow using a 21-gauge needle containing MSC medium with 1% penicillin/streptomycin. The bones were cut up finely using a scalpel blade and briefly crushed with a mortar and pestle in 3% collagenase Type 1 (Gibco) in PBS and then incubated in a shaking water bath at 37°C for 45 min before filtering through a 70-µm strainer and washing again in PBS. The marrow and bone-derived cells were pooled and stained with CD31- and CD45-coupled PE and Sca1-FITC antibodies on ice in PBS for 15 min at 25 × 106 cells/mL and then washed with PBS. CD31 and CD45 negative, and Sca1 positive cells were FACS sorted and plated in MSC media at 2 × 105/cm2.
BrdU labeling and detection
To track the nuclei by BrdU incorporation, 10 µM BrdU (Sigma) was added to growth phase cultures immediately after plating. For MSCs, BrdU was added for 72 h with a medium change at 48 h. For embryonic stem cells (ESCs), BrdU was added for 12 h. Cells were then washed three times in medium over 1 h before cell fusion or fixation and antibody detection. Fixation was performed with 4% paraformaldehyde (Sigma) and 0.625% Tween 20 (Sigma) for 30 min at 37°C and then washed three times in PBS (Invitrogen). DNA denaturation was performed with 2N HCl for 30 min at 37°C, followed by washing and incubation in a borate buffer (0.5 M, pH 8.5) for 10 min at room temperature. Cells were then washed three times in PBS. Fixed cells were incubated with a mouse anti-BrdU FITC-conjugated monoclonal antibody (Roche) diluted 1:25 in PBS + 0.1% BSA (Invitrogen) for 1 h at 37°C, washed three times with PBS and counterstained with DAPI (Vector Laboratories Inc.).
Cell fusions and heterokaryon enrichment
Generation of tetraploid ESCs. Tetraploid DsRed-expressing ESCs were generated by cell fusion between two clonal mouse ES D3 cell lines transfected with Ef1α-DsRed-neomycin and Ef1α-DsRed-hygromycin constructs (Clontech). respectively; whereas TK-sensitive 4N ESCs were generated by cell fusion between two clonal mouse ES D3 cell lines transfected with pSicoR-Hygro-Tk or pSicoR-Puro-Tk vectors, respectively. Cell fusion was performed in 4-well tissue culture dishes (Nunc) by plating the DsRed-neomycin D3 clone at 1.5 × 105 cells/cm2 overnight. A total of 1 × 106 DsRed-hygromycin ESCs were then spun on to the dense monolayer at 670g for 10 min. The medium was completely removed and fusion performed by gently adding 200 µL of 50% PEG6000/HEPES (150 mM, pH 7.5) and incubated at room temperature for 2 min. The PEG was aspirated and the cells were washed four times in calcium-and-magnesium-free PBS and allowed to recover in ESC medium in the incubator for at least 4 h. The cells were then trypsinized and transferred to a 10-cm dish and double antibiotic resistant clones selected over 10 days using either 300 µg/mL neomycin and 1.5 µg/mL puromycin or 150 µg/mL hygromycin and 1.5 µg/mL puromycin).
ES–somatic cell fusion and heterokaryon enrichment. ES–somatic cell heterokaryons were produced by cell fusion performed in a 6-well Falcon® Tissue Culture Plate. Tetraploid DsRed-expressing ESCs were plated at 2.5 × 106 per well and cultured overnight. A total of 3 × 106 constitutively GFP-expressing MSCs or OG2 MSCs prestained with SNARF-1 15 µM for 20 min in PBS and washed with media three times were spun on to the dense monolayer at 670g for 10 min. The culture medium was removed and fusion performed by adding 1 mL of 50% PEG6000 and incubating at room temperature for 2 min. The PEG was removed and the cells were washed four times in calcium-and-magnesium-free PBS, and the cells were allowed to recover in ES medium in the incubator for at least 4 h. The cells were harvested and resuspended in 1 mL medium for flow cytometry and/or stained with 10 µg/mL H33342 (Sigma) for 45 min. FACS analysis and sorting was performed using a MoFlo® DakoCytomation cell sorter, using the two parent cell lines and a mock-fusion control to set sorting gates. Fusion efficiency was determined by subtracting the control value from the fusion value. Heterokaryons were enriched for by sorting double-positive red and green cells or SNARF-1 positive cells with high H33342 staining. Sorted cells were then plated in cellular fibronectin-coated (Sigma) wells at 5 × 105 per 1-cm well. Once cells had plated down, heterokaryons containing multiple nuclei could be visualized by adding 5 µg/mL H33342 for 10 min and using phase contrast or epifluorescence on an inverted OlympusIX71. Images were captured by using an Olympus DP70 camera and DP Controller software.
Differential enucleation of cells
A total of 5 × 105 cells were plated in 1-cm tissue culture wells cut from NUNC™ 4-well coated with bovine cellular fibronectin at 2 µg/cm2, and grown in ESC medium for 48 h. Before enucleation, cells were treated with cytochalasin B (Sigma) (10 µg/mL) for 90 min at 37°C. The tissue culture wells were then placed upside down in a centrifuge tube and submerged in ES media containing 10 µg/mL cytochalasin B and 0.1 µg/mL aphidicolin D (Sigma) and spun for 10 min at 19,770g at 25°C. Cytochalasin B and aphidicolin D were washed off and replaced with fresh medium containing 5 µg/mL Hoechst 33342 for 10 min, and the cells were visualized as above. Wells containing heterokaryons that had been subjected to PFE and control wells that had not undergone centrifugation, were grown in medium containing 2 µM ganciclovir for negative selection against 4N hygro/puro-TK ESCs. The selection media was refreshed every 48 h.
Microsatellite PCR analysis
Genomic DNA was extracted from cells using DNeasy Blood and Tissue kit (Qiagen) and amplified using microsatellite primers. The PCR cycle parameters included an initial denaturation at 94°C for 5 min followed by 38 cycles of denaturation at 94°C for 1 min, annealing at 52°C for 35 s, and extension at 72°C for 1 min, followed by final extension at 72°C for 5 min. PCR products were run on a 3% agarose gel at 100 V for 5–6 h. The following microsatellite markers primers sequences, D1Mit206, D2Mit493, D3Mit259, D4Mit178, D5Mit246, D6Mit102, D7Mit31, D8Mit242, D9Mit311, D12Mit270, D13Mit3, D14Mit109, D16Mit57, D19Mit10, and DX170, were obtained from the Broad Institute (http://www.broad.mit.edu).
RT-PCR
Total RNA was extracted from cells using the RNeasy kit (Qiagen) according to the manufacturer’s instructions. To remove contaminating genomic DNA, the resulting total RNA was subjected to DNase I treatment using the DNA-free kit (Ambion). RNA (2 µg) was used to synthesize cDNA using the SuperScript III reverse transcriptase kit (Invitrogen). cDNA samples were subjected to PCR amplification with the following primer pairs:
Oct4: F, GTTCAGCCAGACCACCATCT; R, CCTGGGAAAG GTGTCCTGTAG
Rex1: F, AAGCAAGACGAGGCAAGGCCAGTCCAG; R, AG GACACTCCAGCATCGATAAGACACC
Nanog: F, AAAGGATGAAGTGCAAGCGGTGG; R, CTGG CTTTGCCCTGACTTTAAGC
Sox2: F, AACGGCAGCTACAGCATGA; R, GCAGTCCAGC CCTCACAT
SPARC: F, AATTTGAGGACGGTGCAGAGG; R, GGTTGTT GCCCTCATCTCTCT
Fibronectin: F, TGCTGTTAATGGCAGAGAGG; R, GGGAG TGGTGGTCACTCTGT
Cdkn2a: F, GCTGCAGACAGACTGGCCA; R, GTCCTCGC AGTTCGAATCTG
Aldha1: F, ggaagctgcagggaaaagcaatc; R, tccgggatgctgcgacaca
Bmp1: F, CGTTTGTGATTGGAGGGAAT; R, TGGCTGTAT GTTCTCGCGTA
Ctbp2: F, ACTGCTGGATGGCAGAGACT; R, GATTCGCAC GATCACTCTCA
Ccnd1: F, GAGAAATGTACTCTGCTTTGCTGAA; R, GGGCT GTAGGCACTGAGCAA
Igf1: F, GGACCAGAGACCCTTTGCGGG; R, GGCTG CTTTTGTAGGCTTCAGTGG
β-actin: F, GGAATCCTGTGGCATCCATGAAAC; R, AAAA CGCAGCTCAGTAACAGTCCG
The PCR cycle parameters included an initial denaturation at 94°C for 5 min followed by 30 cycles of denaturation at 94°C for 1 min, annealing at 52°C–58°C for 45 s, and extension at 72°C for 1 min, followed by final extension at 72°C for 5 min. PCR products were run on a 1% agarose gel at 100 V for 1 h.
Microarray
Microarray analysis was performed using the Oligo GEArray® Mouse Stem Cell Microarray from SuperArray Biosciences Corporation according to the manufacturer’s instructions. In brief, 3 µg of purified total RNA (kit) was used to amplify Biotinylated-UTP labeled cRNA overnight using the TrueLabeling-AMP™ kit and purified using the ArrayGrade™ cRNA Cleanup Kit. Labeled cRNA (4 µg) was hybridized to HybTube format membranes at 60°C overnight. Chemiluminescent detection was performed using the CDP-Star® Chemiluminescent kit, and membrane images were captured using Kodak X-ray film and scanned into digital format. The GEArray Expression Analysis Suite was used to analyze the expression profiles of 263 genes related to the identification, growth, and differentiation of stem cells. Two biological replicates for each cell line were analyzed following background correction and normalization to Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Peptidylprolyl isomerase A (cyclophilin A) (PPIA). A clustergram was generated using the software, while a Venn diagram was produced using present/absent calls from the three groups of cells whereby a minimum of two out of three present/absent calls was required for the PFE group.
Alkaline phosphatase
Alkaline phosphatase activity was detected with an AP kit (Chemicon) according to the manufacturer’s instructions.
Results
Identification of differential enucleation conditions for ES–somatic heterokaryons
First, to determine differential enucleation conditions for ES–somatic heterokaryons, constitutively DsRed-expressing tetraploid ESCs were fused to constitutively GFP-expressing MSCs to allow for the identification of fused cells. DsRed-expressing tetraploid ESCs were generated by fusing two clonal ES lines that had been transfected with plasmids containing a DsRed gene and either a neomycin or hygromycin resistance gene. Double antibiotic resistant clones were selected over 10 days and cultured as described previously [13]. DsRed ESCs (4N) were fused to constitutively GFP-expressing mouse MSCs and heterokaryons could be enriched by FACS (fusion efficiency of 2.28% ± 0.89 SD, n = 7), sorting for DsRed and GFP fluorescence (Fig. 1A).
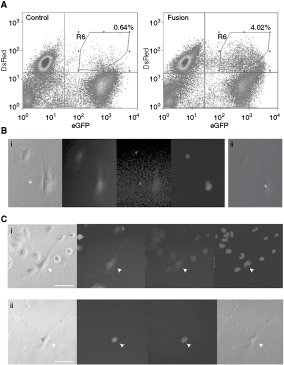
Generation of embryonic stem (ES)–somatic heterokaryons and differential enucleation of 4N ES nucleus. (
To demonstrate that heterokaryons contain nuclei from the two parental cells, nuclei were prelabeled for identification. Before fusion, cells were treated with BrdU, a base analog of thymidine, which is irreversibly incorporated into the DNA and allows for tracking of labeled nuclei. GFP-MSCs were labeled for a period of 72 h, giving rise to >90% labeled cells (Supplementary Fig. 1; Supplementary materials are available online at http://www.liebertpub.com). They were fused to unlabeled 4N DsRed ESCs and heterokaryons were identified with only one BrdU-labeled nucleus following fixation (Fig. 1B). These results demonstrate that GFP/DsRed-positive heterokaryons contain intact nuclei from the two parental cells and that BrdU labeling can be used to track the nuclei from the donor and recipient cells.
Owing to the adherent properties of ESCs and MSCs, enucleation can be performed by centrifugation. In brief, cells were grown on tissue culture wells, treated with the actin depolymerization agent, cytochalasin B, and inverted and submerged into centrifugation tubes. Centrifugation was then used to expel the nuclei, leaving behind enucleated cells (cytoplasts). Conditions were established whereby the nuclei from 4N DsRed ESCs could be expelled, while under the same conditions the diploid nuclei of MSCs were retained (Supplementary Fig. 2). The differential enucleation was applied to GFP/DsRed-positive heterokaryons, which had been maintained in culture for 48 h resulting in ∼30% elimination of one nucleus. To conclusively demonstrate PFE of the tetraploid ESC nucleus and retention of the somatic nucleus in heterokaryons, MSCs were prelabeled with BrdU before fusion. Following PFE of heterokaryons, the remaining nucleus was identified to be BrdU positive (Fig. 1C). These results demonstrate that both the remaining nuclei are that of the somatic cell, and that nuclear hybridization with an ES nucleus had not occurred (supported by the microsatellite analysis of recovered diploid clones described later).
Identification of cells following post-fusion enucleation
The differential enucleation conditions for ES–somatic heterokaryons were used on a second lot of fusions that utilized a reporter system to recover and analyze somatic cells that had undergone PFE. To maximize the identification of clones that had undergone changes following fusion and PFE, a two step strategy using both positive and negative selection was employed (Fig. 2A). First, to identify clones that had undergone transcriptional changes following PFE, MSCs were isolated from OG2 mice which contain a GFP transgene under the control of the OCT4 promoter (OCT4-GFP MSCs) [12]. Both FACS and microscopic analysis of OG2 MSCs before fusion confirmed that the cells were negative for GFP expression (Supplementary Fig. 3). Second, to remove the possibility of nuclear hybridization and contamination by ESCs in the analysis, tetraploid ESCs containing the thymidine kinase (TK) suicide gene were generated by fusing two clonal ES lines that had been transfected with pSicoR-Hygro-Tk or pSicoR-Puro-Tk vectors. These 4N hygro/puro-TK ESCs were shown to be sensitive to ganciclovir with complete cell death after 5 days of selection (Supplementary Fig. 4). Negative selection using ganciclovir would allow for selection against 4N ESCs and any hybrid cells that had undergone nuclear hybridization after the fusion and before the enucleation process. Coculture of somatic cells with ESCs that were not subjected to the fusion protocol did not result in GFP reactivation. In other parallel control experiments, ESCs and MSCs were fused and sorted but were not subjected to PFE, no GFP-positive colonies were observed after ganciclovir treatment.
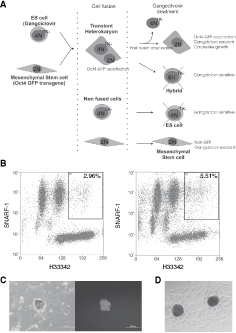
Generation and identification of autologous diploid cells following embryonic stem (ES)–somatic cell fusion. (
Oct4-GFP MSCs were stained with the intracellular dye SNARF-1 and fused to hygro/puro-TK 4N ESCs. Cells were then stained with Hoechst 33342 (H33342), and heterokaryons were enriched by sorting for red fluorescence and a high DNA content ≥6N by FACS (Fig. 2B). Sorted cells were plated on to cellular fibronectin-coated enucleation wells and treated with cytochalasin B and aphidicolin D for 48 h. PFE was then performed and the cells were returned to culture and exposed to ganciclovir for 7 days, after which GFP-positive colonies were identified (Fig. 2C). The efficiency of obtaining the PFE clones was calculated as 2.08 ± 0.05 SD (n = 3) PFE clones per 100,000 cells sorted. These cells were expanded on either gelatin or a MEF feeder layer by either trypsinization or manual passage techniques. However, the growth rate of the GFP colonies resulting from PFE was restricted compared with ESCs. The ability of ESCs to reprogram MSCs following fusion was confirmed by the generation of hybrids when no enucleation or ganciclovir treatment was performed and these cells were used in subsequent gene expression analysis.
Three clones, designated PFE A, B, and C, were expanded and shown to be diploid by chromosome counting, and autologous to the somatic cell donor (OG2) mice by microsatellite analysis (Supplementary Fig. 5). The colonies were positive for alkaline phosphatase activity (Fig. 2D). The cells could be freeze/thawed and could be maintained in culture for further 8–9 passages before reaching senescence. Any outgrowths from the colonies would frequently revert to a fibroblast-like morphology characteristic of MSCs (Fig. 2C).
Gene expression analysis of PFE clones
To determine whether global transcriptional changes had occurred following cell fusion and enucleation, a mouse stem cell array was used to compare ESCs, MSCs, ES–MSC hybrids, and three PFE clones that had been expanded for 6–7 passages. A clustergram revealed that varying levels of global transcriptional changes had occurred across the PFE clones away from the parental MSCs; however, their expression profiles remained similar to MSCs than to ESCs (Fig. 3A). Using present/absent calls for the 263 genes on the array, a Venn diagram was constructed to identify genes that were either switched on or off in the PFE cells when compared to the parental ESCs and MSCs (Fig. 3B). Four genes, the cell cycle regulators Ccnd1 (cyclin D1) and Cdkn2a (cyclin-dependent kinase inhibitor 2A); Ctbp2 (C-terminal binding protein 2), involved in the Notch signaling pathway; and tfrc (transferrin receptor) involved in hematopoiesis, found to be expressed in ESCs, were identified to be expressed in PFE cells as well. Five genes expressed in MSCs, Actc1, Akp5, Col1a1, Igf1, and Pdgfb, were identified to be turned off in PFE cells. The metabolic marker Aldh1a1 (aldehyde dehydrogenase family 1, subfamily A1) and Bmp1 (Bone morphogenetic protein 1) were identified to be expressed in PFE cells alone.
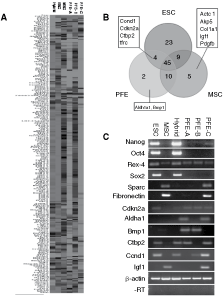
Gene expression analysis of post-fusion enucleation (PFE) clones. (
The expression of genes identified in the array along with some key pluripotency and MSC markers was further analyzed by RT-PCR (Fig. 3C). These results confirmed that in PFE cells, a number of genes expressed in MSC had been switched off including Sparc, fibronectin, and Igf1. Although a number of genes were activated in PFE cells, low expression of the ES marker Nanog in one clone and the expression of Ccnd1, Cdkn2a, and Ctbp2 identified by microarray analysis were confirmed by RT-PCR. The clones were analyzed after expansion from single colonies and despite the activation of the Oct4-GFP transgene no endogenous Oct4 expression was detected during the RT-PCR and microarray analysis.
Discussion
In the present study, we show by using tetraploid ESCs for cell fusion with MSCs, an ES–somatic heterokaryon can be reverted to a diploid state by exploiting this difference in ploidy. Following ES–somatic cell fusion, heterokaryons were maintained in culture for 48 h, a period where reprogramming of the somatic nucleus has previously been shown to take place [1,4,9,10,14], without nuclear hybridization. Fused cells were then reverted to a diploid autologous state using a combination of enucleation of the tetraploid ES nucleus, along with selection against the ES DNA using the TK suicide gene. Following PFE, the cells were analyzed and shown to have undergone a number of morphological and gene expression changes following exposure to the ES nucleus in a transient heterokaryon.
Transient heterokaryons formed from somatic cells of different lineages [7,15,16], and more recently in ES–somatic heterokaryons [10], have been reported to undergo gene expression changes. This study, however, is the first to examine gene expression changes after the removal of the dominant nucleus from heterokaryons. Indicative of the reprogramming process, which requires extinguishing of the somatic-specific transcriptional program and activation of the ES transcriptional program [9,10], we showed that a number of genes expressed in MSCs such as Actc1, Akp5, Col1a1, Igf1, and Pdgfb are switched off, while genes expressed in ESCs involved in cell cycle regulation (Ccnd1 and Cdkn2a) and involved in the Notch signaling pathway (Ctbp2) are switched on in PFE clones after expansion. Furthermore, these results are consistent with the findings that nuclear hybridization is not required for changes to the transcriptional program to occur following interspecies ES–somatic cell fusion [10].
Although the complete restoration of pluripotency was not observed following enucleation of heterokaryons, these results demonstrate that gene expression changes can be maintained in the absence of the dominant nucleus. We did observe changes in morphology resulting in colony-like growth along with alkaline phosphatase activity, and these initial changes are consistent with the stepwise process of iPS cell reprogramming [17,18]. Interestingly, the expression of the endogenous Oct4 gene was not detected in PFE clones despite the activation of the Oct4-GFP transgene. This would suggest that factors from the 4N ES nucleus were able to reactivate the Oct4 promoter-driven GFP transgene in the OG2 MSCs while the nuclei were together in the heterokaryon; however, this was not enough to activate or maintain the expression of endogenous Oct4. The PFE clones were also varied in their gene expression profiles, with PFE clone C showing weak expression of nanog; however, this clone also had the least amount of inactivation of somatic cell markers suggesting that following enucleation from heterokaryons the clones were at various stages of transcriptional changes, which was not maintained after PFE. These results indicate that the exposure of a somatic nucleus to an ES nucleus in a heterokaryon was not enough to completely reprogram the transcriptional profile of the somatic cell to a pluripotent state, and the cells appear to be reverted to an intermediate state that mostly resembles the donor somatic cell. Furthermore, the reversion of a partial or incomplete reprogrammed state back to the somatic cell state has also been described when reprogramming with dox-inducible iPS factors. Halting the expression of the transgenes by the removal of dox during iPS induction resulted in the cells reverting to the somatic cell state, suggesting that reprogramming is a gradual and reversible process [17].
The restoration of pluripotency to a differentiated mammalian nucleus without modification to the genome has been a challenge since the first demonstration of somatic cell reprogramming by somatic cell nuclear transfer [19]. The exposure of differentiated cells to cell extracts made from amphibian oocytes, or cell extracts made from mammalian embryonal carcinoma and ESCs have been shown to alter gene expression profiles toward an embryonic state [20 –23].
Cell fusion has also been proposed to be an alternative method for generating autologous pluripotent cells; however, this would require the removal of the ESC-derived DNA following reprogramming [2,3]. One method involves the removal of the specific chromosomes responsible for self-recognition. In this approach, a universal chromosome elimination cassette that induces cre-mediated sister chromatid recombination during cell division to delete ES chromosomes carrying the MHC molecules, resulting in a hybrid aneuploid cell that is immune matched to the somatic cell [24]. Alternatively, the successful reprogramming of a somatic cell in a heterokaryon could potentially lead to the generation of autologous pluripotent cells after the removal of the ESC-derived DNA following the enucleation process described in the present study.
Despite a number of gene expression changes occurring without nuclear hybridization, this technique would need to be further modified to yield a fully reprogrammed autologous somatic cell. We hypothesize that the use of a more potent ESC with upregulated reprogramming factors such as Nanog shown to increase reprogramming by cell fusion [14], or the upregulation of the four reprogramming factors used to generate iPS cells, may result in complete reprogramming of the somatic nucleus in a heterokaryon. Alternatively, the somatic genome could be made more amenable to reprogramming before cell fusion or in heterokaryons through the use of chromatin modifiers [25,26] or cell synchronization [27]. Furthermore, in the present study somatic cells containing a random insertion of the Oct4 promoter driven GFP transgene were used as a marker for reprogramming/transcriptional changes in PFE cells, the use of endogenous Oct4 or Nanog knock-in selectable markers [17] would allow for the identification of cells that had undergone transcriptional reprogramming at the endogenous loci. The technique could also be used to reprogram or dedifferentiate the somatic cell to a more primitive cell type that has limited differentiation potential rather than a pluripotent state.
More recently, the integration and constitutive expression of viral vectors containing the transcription factors oct4, sox2, c-myc, and klf-4 or lin28 has been shown to reprogram differentiated mouse and human cells to produce what are termed, iPS cells, which display most characteristics of ESCs [28 –33]. Furthermore, progress has been made to reduce the number of viral factors required to restore pluripotency [34 –36], and minimize viral integration [37,38].
The generation of nonmodified patient-specific pluripotent cells without the use of SCNT still remains a challenge. The ability to revert a fused ES–somatic cell to a diploid state, without modification to the somatic genome, suggests that a cell fusion based approach could potentially be used to generate autologous pluripotent cells. Furthermore, this approach may also help to elucidate the mechanism underlying fusion-mediated cell reprogramming through the identification and analysis of species-specific transcripts in interspecies ES–somatic heterokaryons and recovered somatic cells.
Footnotes
Acknowledgments
We would like to thank R. Jaenisch and O. Kirak for the gift of pSicoR-Hygro-Tk and pSicoR-Puro-Tk vectors and helpful discussions, P. Simmons and B. Short for the gift of GFP-positive MSCs and protocols, and P. Hutchinson for FACS assistance. This project was supported by funding from the Australian Stem Cell Centre. H.S. receives a NH&MRC Biomedical Training Fellowship supported by an Establishment Gift from the Clive and Vera Ramaciotti Foundation.