
Abstract
Stem cell-based therapy represents a novel and more advantageous modality of treatment for tooth defect or loss. However, this strategy is challenged in the circumstances where tooth-derived stem cells are not readily accessible. In present study we sought to explore the possibility of utilizing dermal multipotent cells (DMCs) easily available from skin tissue for odontogenic induction. Using the limiting dilution technique, colony-forming cell population was isolated and characterized by proliferative activity and multilineage differentiation potential. By exposure to conditioned medium of embryonic and neonatal tooth germ cells in culture, the proliferation and mineralization activity of DMCs was elevated, while the embryonic tooth germ cell-conditioned medium (ETGC-CM) produced more significant effects. Meanwhile, ETGC-CM-treated DMCs phenocopied the odontoblasts in vitro as indicated by specific lineage markers. Following in vivo transplantation as cell pellet, ETGC-CM-treated DMCs were capable of producing blocks of mineralized tissues, which resembled those of dental pulp stem cell (DPSC) explants in the same subcutaneous pockets environment. These observations suggest that although more sufficient and continuous inductive microenvironment may be needed for undifferentiated DMCs to perform as odontoblasts, ETGC-CM-treated DMCs indeed acquire properties as those of DPSCs. Our work highlights the potential utility of DMCs as an alternative candidate cell source in hopes of developing more practical strategy of tooth regeneration research and offering promising opportunities for therapeutic approach.
Introduction
T
More recently, accumulating evidence has pointed to the critical role of stem cell niche in the regulation of stem cell functions [18 –20]. Signals provided by the specialized niche microenvironment are the key to homeostatic regulation of stem cell maintenance versus tissue regeneration [18]. Tooth development is characterized by the involvement of complex scenario of signaling pathways and growth factors [21,22]. Since tooth develops via the complex and dynamic interactions between the ectoderm and underlying ectomesenchyme, where the spectrum and concentration of molecules change continuously, application of a few growth factors is not likely to provide the complete repertoire of the molecules needed for odontogenic differentiation of undifferentiated cells. Recently, several studies showed that regulatory effects from exogenous microenvironment could be achieved by the application of conditioned medium of cultured cells [23–26]. These effects were supposed to be mediated by paracrine factors secreted into the medium and be able to dictate cell behavior influenced by them. We have previously found that the cocktail of soluble factors released from tooth germ cells could create the potent odontogenic microenvironment that promoted DPSCs to differentiate into functional odontoblasts and form the regular-shaped dentin-pulp complex in vivo [23]. Also, tooth germ cells provided a developmental odontogenic microenvironment that was strong enough to divert the differentiation of nonodontogenic cells as vibrissae follicle dermal papilla mesenchymal cells to go along the odontoblast lineage [27]. These results, together with the multipotency of dermal mesenchymal cells, prompted us to investigate the ability of dermal multipotent cells (DMCs) to response to primitive odontogenic environment and contribute to tooth regeneration.
To this goal, we have isolated and characterized DMCs as a population of adult stem cells from the skin dermal layer and demonstrated that embryonic tooth germ cell-conditioned medium (ETGC-CM), constituting the odontogenic niche, was capable of elevating the proliferation and mineralization of DMCs and stimulating their expression of odontogenic and mineralization markers in vitro. Thus we could postulate that DMCs have the odontogenic potential that could be stimulated by ETGC-CM, which provides a prospect avenue for acquiring candidate cells for tooth regeneration research.
Materials and Methods
Cell culture and analysis
All experiment procedures including the use of animals were performed in accordance with the Guidelines of the Animal Care Committee of Fourth Military Medical University. Rodent dermal fibroblasts were isolated from the foot palm skin of 1-day neonatal Sprauge-Dawley (SD; FMMU Medical Laboratory Animal Center, Xi’an, China) rats. After washed in sterile phosphate-buffered saline (PBS; Gibco-BRL, Grand Island, NY, USA), the subcutaneous tissues of specimens were carefully removed. The palm skins were then cut into small pieces and incubated in 0.1% Dispase II (Sigma, St. Louis, MO, USA) in PBS at 4°C overnight. Then, the underlying dermis was gently removed from the epidermis and minced into small pieces. Further digestion was performed with type I collagenase (0.3 mg/mL; Sigma) at 37°C for 1 h. Cells were gently dissociated by trituration. The solution was then filtered through a 70-µm cell strainer (BD Biosciences, Mississauga, ON, Canada), centrifuged, and resuspended in Dulbecco’s modified Eagle medium (DMEM; Gibco-BRL, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Gibco-BRL), 0.292 mg/mL glutamine (Invitrogen, Carlsbad, CA, USA), 25 mg/mL bovine pituitary extract (BPE; Gibco-BRL), 100 U/mL penicillin G, and 100 µg/mL streptomycin, and incubated at 37°C in 5% carbon dioxide.
The colony-forming unit assay was performed as previously described [1,28]. Single-cell suspensions of dermal fibroblasts (1 × 103 cells or 1 × 104 cells) within DMEM containing 15% FBS were seeded into culture plates (Corning, Lowell, MA, USA) and then incubated at 37°C in 5% carbon dioxide. After 14 days of cultivation, cultures were fixed with 4% formalin, and then stained with 0.1% Crystal Violet (Jiayuan Co., Hangzhou, China). Aggregates containing 50 or more cells were counted as colonies under the microscope. The colony-forming efficiency was defined by the percentage of cells plated that form a colony.
To obtain homogeneous populations of DMCs, subconfluent primary cultures of skin dermal fibroblasts were obtained using the limiting dilution technique as previously described [29,30]. In brief, cells were diluted into DMEM containing 15% FBS in 96-well culture plates (Corning) at a concentration of 10 cells/mL (100 µL/well). Each individual chamber was then assessed for the presence or absence of a single cell under the inverted microscope (Olympus, Tokyo, Japan). Wells containing only one cell were marked for further analysis. After 2–3 weeks original culture, the single cell-derived clones were then harvested and further expanded in the growth medium.
For population doubling assay, DMCs at each passage were seeded at 1 × 103 cells/well in six-well plates (Corning), then trypsinized and counted on day 3. Cell doubling was calculated at every passage as described [4]: number of divisions = log2 (number of cells at subculture/number of cells seeded).
As parallel control for in vivo transplantation, DPSCs were isolated and cultured according to our previous method [23,31]. In brief, the dental pulps were extracted from the lower incisors of 4-week-old SD rats, and were physically separated from the enamel organs and apical buds to obtain primary DPSCs. DPSCs were subsequently cultured in DMEM/F12 (Gibco-BRL) containing 10% FBS, 0.292 mg/mL glutamine, 100 units/mL penicillin G, 100 µg/mL streptomycin, 2.5 µg/mL ascorbic acid, and 25 mg/L bovine pituitary extract (Gibco-BRL). DPSCs at fourth to sixth passage were prepared for in vivo transplantation in parallel with DMCs.
Preparation of conditioned medium
Dental cells were prepared from first lower molars of 14-day embryonic (E14) and 1-day postnatal (1 dpn) SD rats, respectively. The first molar germs were carefully isolated using dental explorer under the stereomicroscope (Leica Microsystems, Germany) and were minced into small pieces in PBS. Tooth germ tissues were then digested with type I collagenase (0.3 mg/mL; Sigma) for 50 min at 37°C. Dissociated tooth cells and tissues were collected by centrifugation, and washed twice in DMEM containing 10% FBS. Single-cell suspensions were generated by filtration through 70-µm cell strainer, then placed into 75-cm2 culture flasks (Corning) at 1 × 105 cells/mL and incubated at 37°C in 5% carbon dioxide. The culture medium of primary tooth germ cells (TGCs) containing both epithelial and mesenchymal cells at 70% confluence was changed every day until full confluence for collecting the supernatant, which would be filtered through a 0.22-µm Millipore strainer (Carrigtwohill Co., Cork, Ireland) [23]. The supernatant was mixed with equal volume of fresh DMEM supplemented with 10% FBS and stored at −80°C to be used as ETGC-CM or neonatal tooth germ cell-conditioned medium (NTGC-CM).
Flow cytometry analysis
To identify the MSC phenotype, cells at third passage were trypsinized and centrifuged. Approximately 5 × 105 cells were incubated with anti-CD34 (1:100; R&D Systems, Inc., Minneapolis, MN, USA), anti-CD44 (1:100; AbD Serotec, Oxford, UK), fluorescein isothiocyanate (FITC)-conjugated anti-CD90 (1:100; BioLegend, San Diego, CA, USA), anti-CD45 (1:100; BioLegend), anti-CD73 (BD Biosciences Pharmingen, San Diego, CA, USA), and anti-CD105 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) according to the manufacturer’s protocol. FITC-conjugated isotype-matching immunoglobulins were used to determine nonspecific staining. The secondary reagents included goat anti-mouse and goat anti-rat IgG-FITC (Santa Cruz Biotechnology). Cells were analyzed on a FACS Caliber (Becton-Dickinson, San Jose, CA, USA), and the data were analyzed with CellQuest software.
Cell cycle analysis was performed among ETGC-CM- and NTGC-CM-treated DMCs. DMCs cultured in regular medium were used as controls. Following 5 days cultivation, single-cell suspensions of DMCs were harvested. After fixing in ice-cold 75% ethanol at 4°C, cell suspensions were subjected to Elite ESP flow cytometry (Beckman Coulter Inc., Fullerton, CA, USA) for cell cycle analysis.
MTT assay
DMCs were plated in a 96-well culture plate at a density of 4,000 cells per well in DMEM. Six hours later, wells were rinsed and two different TGC-CM or normal culture medium (DMEM) was added. At the end of the test time points (1, 3, 5, 7 days), cell proliferation of DMCs was assessed using methyl tetrazolium methods (MTT; Sigma) according to the manufacturer’s instructions. The optical density (OD) values were determined with a multi-plate reader (BIO-TEK, USA) at a wavelength of 570 nm. The experiment repeated at least three times.
Analysis of differentiation ability of DMCs
Single-cell-derived DMCs were analyzed for their capacity to differentiate toward adipogenic or osteogenic lineages [14]. Cells were seeded in six-well culture plates at a density of 1 × 105 cells/well. Adipogenic differentiation medium was DMEM supplemented with 5% FBS, 0.5 mM isobutyl methylxanthine (IBMX; Sigma), 2 µM insulin (Sigma), and 10 nM dexamethasone (Sigma). DMCs were cultured in adipogenic medium or in regular medium for 4 weeks. Osteogenic medium was DMEM supplemented with 5% FBS, 100 nM dexamethasone, 5 mM β-glycerophosphate (Sigma), and 50 µg/mL
Odontogenic differentiation of DMCs
Odontogenic differentiation environment was mimicked by indirect co-culture. The indirect co-culture using TGC-CM was performed as we previously described [23,27,32]. In brief, single-cell-derived DMCs at a density of 1 × 104 cells/mL were plated in 24-well culture plates (Corning). Controls were cultured in DMEM and experiments were carried out in ETGC-CM or NTGC-CM for 5 days. Both the TGC-CM as well as the control DMEM were renewed every 24 h.
Immunofluorescence staining
Immunofluorescence staining was performed on cells plated on 24-well culture plates. Cells were fixed in 4% paraformaldehyde for 20 min at room temperature, then were rinsed with PBS before being permeabilized with 0.1% Triton X-100 for 20 min at room temperature. Following permeabilization, the cells were rinsed with PBS and were blocked in 4% normal goat serum for 30 min at 37°C. Cells were then incubated with primary antibodies including rabbit anti-Ki67 (1:100; Abcam, Cambridge, UK), rabbit anti-DSP (1:100; Santa Cruz Biotechnology), goat anti-DMP-1 (1:50; Santa Cruz Biotechnology), and rabbit anti-ALP (1:100; Abcam) at 4°C overnight. After rinsed with PBS, cells were incubated with rabbit anti-goat or goat anti-rabbit secondary antibodies that were conjugated to FITC (1:200; Santa Cruz Biotechnology) or Rhodamine (1:200; Chemicon, Billerica, MA, USA) for 45 min at 37°C. Cell nuclei were counterstained in culture medium containing Hoechst 33342 (5 µg/mL; Sigma) for 15 min at 37°C. All antibodies were diluted in PBS. The optimal dilution of primary antibodies was determined by a gradient (dilution range 1:50–1:500). PBS instead of the primary antibodies was used for negative controls. The image collection and superimposition were processed by DP controller (Olympus) and DP manager. For quantification of the percentage of cells expressing the marker protein, at least three fields were randomly captured in each experiment per marker. Then, the number of positive cells were counted and calculated relative to the total number of Hoechst-labeled nuclei.
Reverse transcription-polymerase chain reaction and real-time PCR
Total cellular RNA was extracted using TRIZOL® Reagent (Invitrogen) and cDNA was prepared using a Superscript II first-strand cDNA synthesis kit (Invitrogen Life Technologies) according to manufacturer’s instructions. Reverse transcription-polymerase chain reaction (RT-PCR) was carried out including the following primers: peroxisome proliferator-activated receptor-γ2 (PPAR-γ; GenBank Accession No. NM 013124); Leptin (GenBank Accession No. NM 013076); alkaline phosphatase (ALP; GenBank Accession No. NM 013059); osteocalcin (OCN; GenBank Accession No. NM 013414). Primer sequences were listed as follows: PPAR-γ-sense, 5′-GAA CGT GAA GCC CAT CGA GGA C-3′, and PPAR-γ-antisense, 5′-TGG AGC ACC TTG GCG AAC AGC-3′; leptin-sense, 5′-GAA CGT GAA GCC CAT CGA GGA C-3′, and leptin-antisense, 5′-TGG AGC ACC TTG GCG AAC AGC-3′; ALP-sense, 5′-TTT GCT ACC TGC CTC ACT TCC G-3′, and ALP-antisense, 5′-GGC TGT GAC TAT GGG ACC CAG-3′; OCN-sense, 5′-AGA CTC CGG CGC TAC CTC AAC AAT-3′, and OCN-antisense, 5′-CAG CTG TGC CGT CCA TAC T-3′; β-actin-sense, 5′-TGG AAT CCT GTG GCA TCC ATG AAA C-3′; β-actin-antisense, 5′-TAA AAC GCA GCT CAG TAA CAG TCC G-3′. In some experiments, real-time PCR was performed with a 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA) by using SYBR® Green PCR Master Mix (Applied Biosystems) according to the manufacturer’s instructions. The following primers were incorporated: dentin matrix protein 1 (DMP-1; GenBank Accession No. NM 203493), dentin sialophosphoprotein (DSPP; GenBank Accession No. NM 012790), ALP, bone sialoprotein (BSP; GenBank Accession No. NM 012587), and OCN. Primer sequences were listed as follows: DMP-1-sense, 5′-TCG CTG TCA CCT TGC TCC TC-3′, and DMP-1-antisense, 5′-GAC AAG ACC CCA GCA GTG AG-3′; DSPP-sense, 5′-GCG ACA GCA GTG ACA GTA GC-3′, and DSPP-antisense, 5′-GCT GTC GCT ACT GTC ACT GC-3′; ALP-sense, 5′-AAC GTG GCC AAG AAC ATC ATC A-3′, and ALP-antisense, 5′-TGT CCA TCT CCA GCC GTG TC-3′; BSP-sense, 5′-TGT GGA ATG GTG CTA CGG TCT C-3′, and BSP-antisense, 5′-GAT CAA CAG CCC TGA TTT ACG ATG-3′; OCN-sense 5′-GGT GCA GAC CTA GCA GAC ACC A-3′, and OCN-antisense 5′-AGG TAG CGC CGG AGT CTA TTC-3′; glyceraldehyde-3-phosphate (GAPDH)-sense, 5′-GAC AAC TTT GGC ATC GTG GA-3′, and GAPDH-antisense, 5′-ATG CAG GGA TGA TGT TCT GG-3′.
In vivo transplantation
To further investigate the in vivo differentiation capacity of DMSCs co-cultured with TGC-CM, an in vivo transplantation assay was performed. Following 5 days pretreated with ETGC-CM or NTGC-CM, aggregates of ∼2.0 × 106 DMCs were implanted with fibrin gel as a carrier into subcutaneous pockets of 6-week-old nude mice (BALB/c-nu; FMMU Medical Laboratory Animal Center). Meanwhile, DMCs cultured in regular medium at 2.0 × 106 with fibrin gel carrier were transplanted into the opposite site of the same host as the control. DPSCs pellets were prepared in the same way for in vivo transplantation in parallel with DMCs transplants. Each group consisted of 15 cell pellets. All procedures were performed under institutionally approved guidelines for the use of animals in research. After 4 weeks ectopic development, the transplants were harvested for histology and immunochemistry studies.
Histology and immunochemistry analysis
Normal developing mandible samples from E14 and 1 dpn SD rats and the transplant samples were harvested for further histological and immunochemistry study. Following decalcification with 10% EDTA (pH 8.0), the samples were embedded in paraffin and 5-µm sections were prepared. For histological analysis, sections were stained with hematoxylin and eosin (H&E) and Masson’s Trichrome (Baso Diagnostic Inc, Zhuhai, Guangdong, China), respectively. For immunohistochemical studies, sections were incubated with primary antibodies of rabbit anti-BSP (1:200; Abcam) and rabbit anti-DSP (1:200; Santa Cruz Biotechnology) and detected using the Dako REAL EnVision detection system (Dako, Carpinteria, CA, USA). Then sections were counterstained with hematoxylin. PBS instead of primary antibodies was used as negative control.
Statistical analysis
All data were expressed as mean ± SD and analyzed by two-tailed unpaired Student’s t-test using SPSS software (version 12.0, SPSS, Chicago, IL). P-values <0.05 were considered statistically significant.
Results
Isolation and characterization of skin dermal-derived multipotent fibroblasts
Recent studies have highlighted the existence of skin dermal-derived multipotent cells, and several findings have lend support to dermis-derived cells having some of the differentiation potential as MSCs [17,33 –35]. Using the limiting dilution technique, we isolated a population from skin dermal-derived fibroblasts (Fig. 1A–D), termed DMCs. Resemble to MSCs, the isolated dermis-derived cells were capable of forming adherent colonies. The colony-forming efficiency (20–30 colonies/103 cells plated; 40–50 colonies/104 cells plated) indicated the presence of a clonogenic cell population in dermal fibroblasts (Fig. 1E). While at early passages, DMCs exhibited prolonged doubling time, and an increasing growth rate was observed with passage accumulation (Fig. 1F).
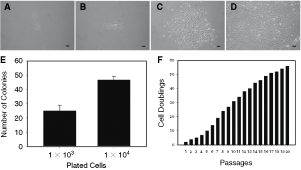
Identification of dermal multipotent cells (DMCs) from skin tissues. (
As described by previous studies [5,10,36,37], MSCs could be characterized by some nonspecific surface antigens. To further characterize the stem cell phenotypic markers of single-colony-derived DMCs, the expressions of CD44, CD90, CD73, CD105, CD34, and CD45 were analyzed by flow cytometry. We observed that a high percentage of DMCs expressed CD90 (Fig. 2A), CD44 (Fig. 2B), CD105 (Fig. 2E), and CD73 (Fig. 2F), respectively. In contrast, only 1.2% (Fig. 2C) DMCs expressed CD34 and 0.2% (Fig. 2D) DMCs expressed CD45.
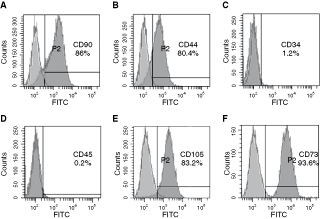
Flow cytometric analysis of ex vivo expanded dermal multipotent cells (DMCs). Representative diagrams are given for: CD90 expression (
To further test the differentiation potential of DMCs, the functional capacity of DMCs was assessed by multipotent differentiation assay. DMCs cultured in regular medium showed no staining of Oil Red O (Fig. 3A) or Alizarin Red S (Fig. 3D). After exposure to adipogenic medium for 21 days, Oil Red O staining demonstrated that DMCs exhibited the adipogenic phenotype (Fig. 3B). Similarly, DMCs were proved to be capable of osteogenic differentiation under the osteogenic induction (Fig. 3E). Further confirmation was provided by mRNA expression of PPARγ2 and leptin for adipocyte phenotype (Fig. 3C), as well as ALP and OCN for osteogenic phenotype (Fig. 3F). These results were consistent with previous stem cell properties elucidated in skin-derived MSCs or SKPs [14,35,38], indicating that DMCs possessed the characteristics of MSCs.

Adipogenic and osteogenic differentiation of dermal multipotent cells (DMCs). (
Cell proliferation of TGC-CM-induced DMCs
Since completely dissociated dental epithelial and mesenchymal cells of embryonic murine could reproduce the interaction between epithelial and mesenchymal cells in early tooth organogenesis [39], and previous studies have proved that tooth germ cell-conditioned medium successfully mimicked the dentinogenic microenvironment from TGCs in vitro [23], we used the conditioned medium from the first molar tooth germs of embryonic and neonatal rats as inductive agent. The first molar tooth germ of E14 rats was in cap stage, while that of 1 dpn rats formed distinct pre-dentin and enamel representative of late bell stage (Supplementary Fig. 1; Supplementary materials are available online at http://www.liebertpub.com.) To determine whether TGCs secreted factors that could have effects on DMCs proliferation activity, DMCs were treated with both TGC-CM. After 72-h cultivation, the initially same seeding DMCs displayed more rapid proliferation ability in TGC-CM than in those cultured in regular medium by detection of microscope. The increase in cell number was significantly higher for cells cultured in ETGC-CM than for those cultured in NTGC-CM in the following culture time as proved by MTT analysis (Fig. 4A). Further support was obtained with cell cycle analysis (Fig. 4B–D). DMCs treated with ETGC-CM presented a higher percentage of cells in S (25.1%) and G2M phases (19.2%) compared with cells treated with NTGC-CM (S phase: 14.5%; G2M phase: 15.2%), suggesting that ETGC-CM may stimulate DMCs to shift from G0G1 phase to G2M or S phases. After 3-day co-culture with TGC-CM, proliferating cells in DMCs was validated by immunostaining analysis of proliferation marker Ki67 (Fig. 4E–H). More proportion of DMCs treated in ETGC-CM (45.7%) was positive, in comparison with cells treated in NTGC-CM (27.2%) or regular medium (12.5%). In general, during the entire co-culture period, DMCs cultured with ETGC-CM underwent a more dramatic change than NTGC-CM-treated DMCs in proliferation activity. Our data suggested that ETGC-CM played a crucial role in regulation of DMCs proliferation activity.
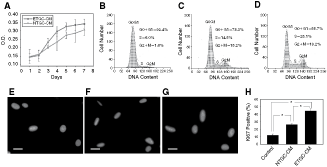
Effects of tooth germ cell-conditioned medium (TGC-CM) on proliferation of dermal multipotent cells (DMCs). (
Mineralization ability of TGC-CM-induced DMCs
We have demonstrated that TGC-CM, especially ETGC-CM, could modify the growth of DMCs. In an attempt to further identify whether TGC-CM-secreted factors were effective to cell differentiation, we examined the cells grown in CM for indications of mineralization ability. The osteogenic potential of DMCs was determined after 14 days and 28 days in vitro culture in ETGC-CM and NTGC-CM, respectively. At day 14, the calcified nodules were observed in ETGC-CM-induced DMCs as determined by Alizarin Red S staining. In contrast, DMCs grown in NTGC-CM did not show obvious staining. Following 28-day cultivation, quantification of the Alizarin Red S staining showed calcified nodule per microscopical field was 40.0% in ETGC-CM, 20.1% in NTGC-CM, and 0.6% in regular medium-treated cells (Fig. 5A–C), indicating a moderate increase of mineralization ability in ETGC-CM-induced DMCs compared with NTGC-CM-induced DMCs (Fig. 5D). We next examined mRNA relative intensities compared to GAPDH expressions of corresponding genes. After 3-day co-cultured with TGC-CM, DMCs expressed ALP, BSP, and OCN mRNA. Relative quantity showed 1.4- to 1.6-fold higher expression of ETGC-CM-treated DMCs compared to NTGC-CM-treated DMCs (Fig. 5E). Meanwhile, TGC-CM-treated DMCs presented an odontogenic expression profile, as evidenced by increased expression level of DMP-1 and DSPP mRNA (Fig. 5E). The expression levels were higher in ETGC-CM-treated DMCs than in NTGC-CM-treated DMCs. These results indicated that the in vitro mineralization of DMCs induced by TGC-CM was characterized by tooth-specific phenotype.
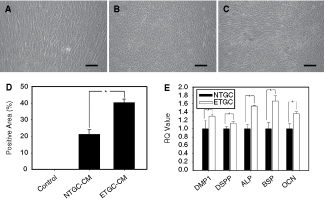
Effects of tooth germ cell-conditioned medium (TGC-CM) on mineralization potential of dermal multipotent cells (DMCs). (
Characterization of the phenotype of TGC-CM-induced DMCs
Soluble factors released from dental epithelial and mesenchymal cells in developing tooth germ cells played a paramount role in the differentiation and dentinogenesis process mediated by DPSCs [23], together with the ability of TGC-CM to regulate the proliferation and mineralization of DMCs, suggesting an odontogenic environment provided by TGC-CM. To further verify the results of mRNA expression of DMP-1 and DSPP, we explored whether DMCs could be directed toward odontogenic-like cells by phenotype transformation. DMCs were exposed to ETGC-CM or NTGC-CM, which was hypothesized to provide a growth condition with multiple molecular signals or growth factors necessary for tooth organogenesis. Protein expression of odontogenic cell markers was determined by immunofluorescent staining. DMCs cultured in regular medium negatively expressed the marker protein (Fig. 6A–C). DMCs cultured with NTGC-CM for 72 h weakly expressed DSP, DMP-1, and ALP (Fig. 6D–F). In contrast, after 72 h of cultivation with ETGC-CM, DMCs expressed all of these odontogenic and mineralization proteins (Fig. 6G–I). Quantification of the staining in three markers showed positive cells per microscopical field were significantly higher in ETGC-CM-cultured DMCs, compared to those in NTGC-CM- and regular medium-cultured DMCs (Fig. 6J–L). These results suggested that medium conditioned by embryonic tooth germ cells was sufficient to activate odontogenic genes and efficient cell differentiation proceeded.
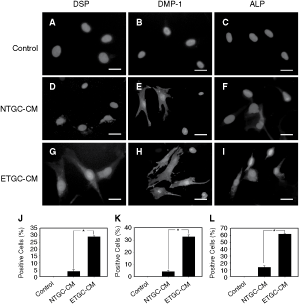
Effects of tooth germ cell-conditioned medium (TGC-CM) on differentiation potential of dermal multipotent cells (DMCs). After 72 h of cultivation, the immunostaining of odontogenic and osteogenic markers in DMCs. (
In vivo differentiation potential of TGC-CM-induced DMCs
To verify the changes observed in vitro, we performed in vivo transplantation experiment. Cultured in ETGC-CM or NTGC-CM or regular medium for 5 days, aggregates of DMCs were implanted with fibrin gel as carrier into subcutaneous pockets of 6-week-old nude mice for 4 weeks. While H&E staining and Masson’s trichrome staining showed DMCs cultured in regular medium formed mostly connective and collagenous tissues and NTGC-CM-treated DMCs only generated occasionally observed mineral deposits (Fig. 7A, B, D, and E), both of which were demonstrated to be BSP-negative (Fig. 7C and F), ETGC-CM-treated DMCs formed blocks of bone-like tissue with typical lacuna structures (66.7% of the transplanted cell pellets, Fig. 7G and H) that positively expressed BSP (Fig. 7I). These histological observations resembled those of DPSCs explants in the same subcutaneous pockets environment, except for the difference that in vivo incubation of DPSCs results in larger amount of bone-like tissues with strongly positive BSP staining (Fig. 7K–M). However, the mineralized tissues formed by both DPSCs and ETGC-CM-treated DMCs did not show positive staining against odontoblast-specific marker DSP (Fig. 7J and N) and this was in accordance with above histologically identified bone-like tissues. These results indicated that ETGC-CM-treated DMCs differed with NTGC-CM-treated and nontreated DMCs in that they acquired mineralization traits that were featured by bone-like structure, resembling those of DPSCs following subcutaneous implantation with fibrin gel as carrier.
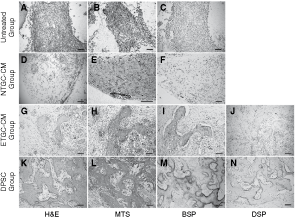
Effects of tooth germ cell-conditioned medium (TGC-CM) on in vivo differentiation potential of dermal multipotent cells (DMCs). (
Discussion
Dermal fibroblasts are the major cell type in dermis that has been identified as MSCs with multilineage differentiation potential [14,16,27,33]. In accordance with these facts, we found that single-colony-derived DMCs showed certain stem cell properties including clonogenicity, high number of population doublings, multipotent differentiation, and expression of MSC markers. Based on these properties, we hypothesized that DMCs possess plasticity in differentiation and might response to primitive odontogenic niche microenvironment created by developing tooth germs.
As far as present studies show, tooth development is a process that occurs between mesenchymal and epithelial cells via sequential and reciprocal complicated interactions [21,22,40,41]. The spectrum and concentration of a great number of substances change continuously in the local microenvironment of tooth development [21]. The combined effect of these molecules may be additive, synergistic, or antagonistic, and the timing of their use may be critical [42]. The dynamic process of inducing and regulating the odontogenic differentiation is not likely to be manipulated by several defined molecular factors. Indeed, completely dissociated single cells from epithelial and mesenchymal tissues of tooth germ at cap stage in E14.5 mice generated structurally correct tooth [39], together with that dissociated epithelial and mesenchymal cells of developing tooth could regenerate tooth structure [43,44], suggesting the re-organized odontogenic microenvironment could continuously favor the tooth formation. At this stage, tooth-forming potential is directed by a specific group of signaling molecules that shifts back and forth between epithelium and mesenchyme prior to terminal differentiation before eruption [22]. These studies suggested that developing tooth germs could provide suitable odontogenic environment that consisted of the critical components for tooth development. Thus, this encouraged us to use developing tooth germs as the microenvironment and to explore its function in the induction of odontogenic process of candidate cells.
To test this possibility, ETGC-CM and NTGC-CM was used to induce the DMCs. Both conditioned medium elevated the proliferation activity of DMCs, while the ETGC-CM produced more significant effects as analyzed by cell cycle and MTT results. According to studies focused on lineage reprogramming [45,46], stimulating cell proliferation may facilitate cell type conversions. Notably, while under induction of both CM the DMCs exhibited mineralization activity, we found that calcified nodules were formed earlier and in greater amounts in the ETGC-CM-treated DMCs than those of NTGC-CM group over the 28-day duration. This reflected the intrinsic difference of the two types of CM with respect to odontogenic microenvironment. Meanwhile, these results suggested that the paracrine effect mediated by relatively stable factors secreted by tooth germ cells into culture medium could enhance the proliferation and mineralization activity of DMCs, implying the possible odontogenic differentiation of DMCs by virtue of ETGC-CM-produced microenvironment.
Quantitative PCR analysis further supported aforementioned results by determining the intrinsic expression pattern of mineralization- and odontogenesis-related differentiation. The expression of BSP, ALP, and OCN mRNA well unraveled the biological fundamental basis of cell mineralization. Furthermore, the cell phenotype of ETGC-CM-induced DMCs presents several crucial characteristics of odontoblast as assessed by the qualitative immunostaining of DMP-1 and DSP and quantitative mRNA expression of DMP-1 and DSPP. While the immunostaining revealed higher positive rate of marker protein in ETGC-CM-treated DMCs than that in the qPCR assay, both assays suggested the dissimilarity of differentiation ability of ETGC-CM- and NTGC-CM-induced DMCs. DMP-1 is a key regulator of both early odontoblast differentiation and late mineralization [47]. DSPP is a compound gene encoding for two proteins namely dentin sialoprotein (DSP) and dentin phosphoprotein (DPP), which is produced by terminally differentiated odontoblasts and is known to modulate dentin mineralization [48]. The expression of these specific markers indicated that DMCs phenotypically adopted the odontoblast lineage especially after induction with ETGC-CM, which was hence supposed to provide more optimal microenvironment for this diversion process. Although it is unlikely to explicitly define the factors in ETGC-CM that contributed to this function, it is worth noting that the E14 tooth germ includes both early dental epithelium and mesenchyme according to histological evidence. In agreement with other reports [43,44], despite enduring with enzymatic dissociation and subsequent culturing, the tooth germ cells still maintain potent odontogenic capability and thus could produce a complex scenario of soluble factors and signaling molecules representative of odontogenic microenvironment.
We further showed that following in vivo transplantation as cell pellet only ETGC-CM-treated DMCs were capable of producing blocks of mineralized tissues that were morphologically and immunochemically identified as bone-like tissues. Although this demonstrated that ETGC-CM indeed had conductive effect for DMCs in stark contrast to NTGC-CM and regular medium, these in vivo results seemed a paradox with respect to the in vitro study that demonstrated the odontoblast lineage differentiation of ETGC-CM-induced DMCs. It was implied that the in vivo circumstances where the odontogenic microenvironment no longer existed abrogated the ability of DMCs to differentiate into odontoblasts and form typical dentin structure reminiscent of tooth development. Correspondingly, previous studies have demonstrated that in vivo-incubated DPSCs were able to alternatively producing dentin or bone tissues [1,23,49 –51], the adoption of which depended on specific extracellular microenvironmental cues. The present results using DPSCs as positive control well agree with previous findings that prove DPSCs exhibit bone-producing capacity [52] in contrast to those that under specific induction can form typical dentin-pulp complex when transplanted under the renal capsules representative of more superior in vivo incubation loci [23]. In addition, the species disparity maybe another explanation for the difference of in vivo results of DPSCs between previous studies [1,3] and the present results. The human-derived DPSCs, which were utilized in the experiments, might have relatively prior odontogenic ability to form regular dentin–pulp structure. However as shown here, the rodent-derived DPSCs usually formed osteodentin-like structure as proved by our experiments. Our results further suggest that the extrinsic niche where cells reside in, regardless of the cell origin, is continuously required to maintain odontogenic differentiation. More suitable and optimal odontogenic microenvironment need to be further exploited and will be of substantial utility in the context of choosing DMCs for tooth regeneration.
Taken together, our results have characterized the stem cell properties of single-colony-derived DMCs population. More importantly, we demonstrated that DMCs possessed the potential to be directed into odontoblast-like cells by exposure to secreted factors of ETGC-CM in culture. The in vivo results added weight to the notion that emphasizes the indispensable and critical role of microenvironment in diverting DMCs to functionally go along odontogenic differentiation. Our work provided an innovative strategy for the rational and practical design of tooth regeneration research by seeking available candidate cell sources. More challenges are to be addressed with respect to faithfully mimicking these microenvironmental cues conducive for nondental DMCs to function as dentin-producing cells.
Footnotes
Acknowledgment
This work was supported by a grant from the Nature Science Foundation of China (Project No. 30572042).