
Abstract
Mesenchymal stem cells (MSCs) are studied as a cellular source for the treatment of various diseases. In this work, we isolated and cultivated murine bone marrow-derived MSCs. After a first observation of a solid tumor in a mouse injected with these cells, we systematically explored their chromosomal stability. We observed in all the cytogenetically analyzed cases gross chromosomal alterations every time the MSCs went through the senescence crisis while the lymphocytes from the same animals showed a normal chromosome count. This observation was confirmed in different mouse strains, with different culture protocols, and even in short-term cultures after a hematopoietic cell negative immunodepletion performed in order to accelerate the isolation procedure. Therefore, we conclude that murine MSCs display high chromosomal instability and can generate tumors, and that care must be taken before using them for the evaluation of MSC therapeutic potential.
Introduction
M
However, the safety issue remains open. As MSCs have the ability to expand, one might wonder whether they may also be the seed for tumors. A few reports indicated spontaneous ex vivo transformation of these cells in mice [12,13]. In contrast and quite fortunately, in humans, if this phenomenon exists, it seems to be an exceptional event occurring only after long-term ex vivo culture [14 –16].
Here we report that bone marrow-derived mouse MSCs invariably undergo chromosomal alterations during the ex vivo culture, even before complete purification. Such a genetic instability thus prevents the use of murine MSC in any animal model of cell therapy.
Design and Methods
Mouse strains
BTBR-Pahenu2 /J mice were obtained from the Jackson Laboratory (Bar Harbor, ME), BTBR T+ tf//J and C57BL/6J from Charles River Laboratories (Bruxelles, Belgium). All animal protocols were approved by the local ethical review board.
Genotyping
Genetic characterization of BTBR-Pahenu2 /J animals was performed on DNA prepared from tail or tumor tissue using the High Pure PCR Template Preparation Kit (Roche Diagnostics, Vilvoorde, Belgium). The enu2 mutation introduces a BsmA1 restriction site in the exon 7 of the Pah gene. This mutation was detected after PCR amplification (sense primer: 5′-TGCTTTCCAGCTTGTACTGGTTTCC-3′; anti-sense primer: 5′-GATCCATGCCTAATGTACTGTGTGC-3′; 30 cycles: 30 s at 94°C, 30 s at 60°C, 30 s at 72°C) and digestion with BsmA1 restriction enzyme, 2 h at 55°C (New England Biolabs, Westburg, Leusden, The Netherlands). The digestion products were loaded on a 20% acrylamide gel and visualized by UV illumination after ethidium bromide staining.
Morphological evaluation of the tumor
Five-micron frozen sections of the tumor specimen were stained with hematoxylin eosin safran (HES).
MSC isolation and culture
If not otherwise stated, culture media components came from Lonza (Verviers, Belgium). All media were supplemented with 100 U/mL penicillin/streptomycin and 2 mM
The other media components used were: Horse serum and Perbio Hyclone Fetal Bovine serum (Belgium), Mouse Serum (Abcam, Cambridge, CB4, UK), hepatocyte growth factor (HGF; Peprotec, London, UK), leukemia inhibitory factor (LIF) (mouse; Sigma-Aldrich, Bornem, Belgium), EGF (epithelial growth factor) (human; Peprotec, London, UK). MSC purification was realized by adherence and passages until they were free from hematopoietic cells as assessed by flow cytometric analysis, or by immunodepletion as described below.
Bone marrow was collected by flushing mouse femurs and tibiae as described [17]. We tested different cell culture densities: in most cases, cells were maintained between 2,000 and 15,000 cells/cm2.
Hematopoietic cell immunodepletion from MSC cultures
At passage 2, MSCs were incubated with biotinylated anti-mouse CD34 (Serotec, Oxford, UK), CD45, and CD11b antibodies (BD Pharmingen, Erembodegem, Belgium). Afterward, streptavidin-coated microbeads were added before loading on a MS separation column from Miltenyi Biotech (Utrecht, The Netherlands).
MSC injection in mice
The 2 × 106 MSCs were injected into the spleen of BTBR-Pahenu2 /J mice, which had previously undergone a hepatic lesion induced by 2 intraperitoneal administration of 45 mg/kg allyl alcohol (Sigma-Aldrich, Bornem, Belgium).
MSC differentiation into osteoblast, adipocytes, and chondrocytes
Differentiation Media Bullekit Osteogenic and Adipogenic from Lonza were used according to the manufacturer's instructions. Analysis was performed by measuring the Ca2+ deposition using the Calcium Stanbio Total Liquicolor kit (Medigal, Acoz-Lausprelle, Belgium), or by staining the lipids with Oil Red O (Sigma-Aldrich, Bornem, Belgium). Chondrocyte differentiation was realized by micromass culture of 5 × 105 MSCs in polypropylene tube for 21 days in the presence of chondrogenic differentiation medium composed of DMEM and 100 μg/mL pyruvate (GIBCO; Thermo Fisher Scientific, Erembodegem, Belgium), 500 ng/mL BMP-6 and 10 ng/mL TGF-b3 (Peprotec, London, UK), dexamethasone 10−7 M and 50 μ/mL ascorbate 2-phosphate and 40 μ/mL proline and ITS + 1 premix (Sigma-Aldrich, Bornem, Belgium). Negative differentiation control were realized by micromass culture by of 5 × 105 MSCs in polypropylene tube for 21 days in the presence of DMEM and 10% FBS (GIBCO; Thermo Fisher Scientific, Erembodegem, Belgium). MSC clumps were fixed with 4% paraformaldehyde, paraffin-embedded, and toluidine blue stained.
Flow cytometry
The 4 × 105 MSCs were incubated with primary antibodies against mouse CD45, CD106, CD34, CD11b, Ly6A/E (Sca-1), or CD90.2 (Thy1.2). The anti-CD11b and CD45 antibodies were PE-labeled. The CD90.2 antibody was FITC-labeled. Detection of CD106, CD34, and Sca-1 staining was performed with an APC-labeled secondary antibody. As negative controls, a PE-labeled immunoglobulin G isotype-matched control or FITC-labeled G isotype-matched control were used or the primary antibody was omitted. The antibodies were all from BD Pharmingen except for the CD90.2 antibody from eBioSciences (ImmunoSource, Halle-Zoersel, Belgium).
Analysis was performed on a BD FACSCalibur (BD Biosciences).
Metaphase spread
All media components were purchased from Gibco (Invitrogen, Thermo Fisher Scientific, Erembodegem, Belgium). MSCs were seeded on glass slides and incubated for 4 h with 100 ng/mL colcemid. Cells were incubated in hypotonic solution (0.9‰ NaCl) and fixed in methanol:acetic acid (3:1). The 0.5 mL heparinized blood was placed in culture for 72 h with 5 mL culture medium composed of RPMI, FBS 20%, PHA 3%, 100 U/mL penicillin/streptomycin. Blood cells were incubated for 1.5 h with 100 ng/mL colcemid. Cells were then incubated in hypotonic solution (0.075 M KCl) and fixed in methanol:acetic acid (3:1).
Results and Discussion
In the context of a cell therapy research project for phenylketonuria, we transfused bone marrow-derived MSCs, isolated from BTBR-Pahenu2 /J, in the spleen of syngeneic immunocompetent mice. Unexpectedly, 5 months later, 1 of the 6 transfused mice developed a solid tumor at the injection site. Because of the different status for the phenyl-alanine hydroxylase gene (Pah) of the MSC donor animal (Pah+/+) and the host mice (Pah−/−), we could genotype this tumor and confirm its donor origin.
HES-stained sections showed a tumor formed by malignant undifferentiated spindle cells focally organized in fascicles with elongated or pleomorphic nuclei and acidophilic poorly delineated cytoplasm (Fig. 1). The degree of nuclear atypia was highly variable, as well as the mitotic activity and the degenerative changes. The histology was thus compatible with a sarcoma and there was not any evidence of mineralized bone, osteoid, cartilage formation, or muscular differentiation within the tumor.

Morphological evaluation of the tumor. (
As it has been postulated that genome instability is critical for tumor formation [18], we performed metaphase spreading of the MSCs that have led to the tumor formation. All mitosis presented abnormal chromosomal count as depicted in Figure 2.
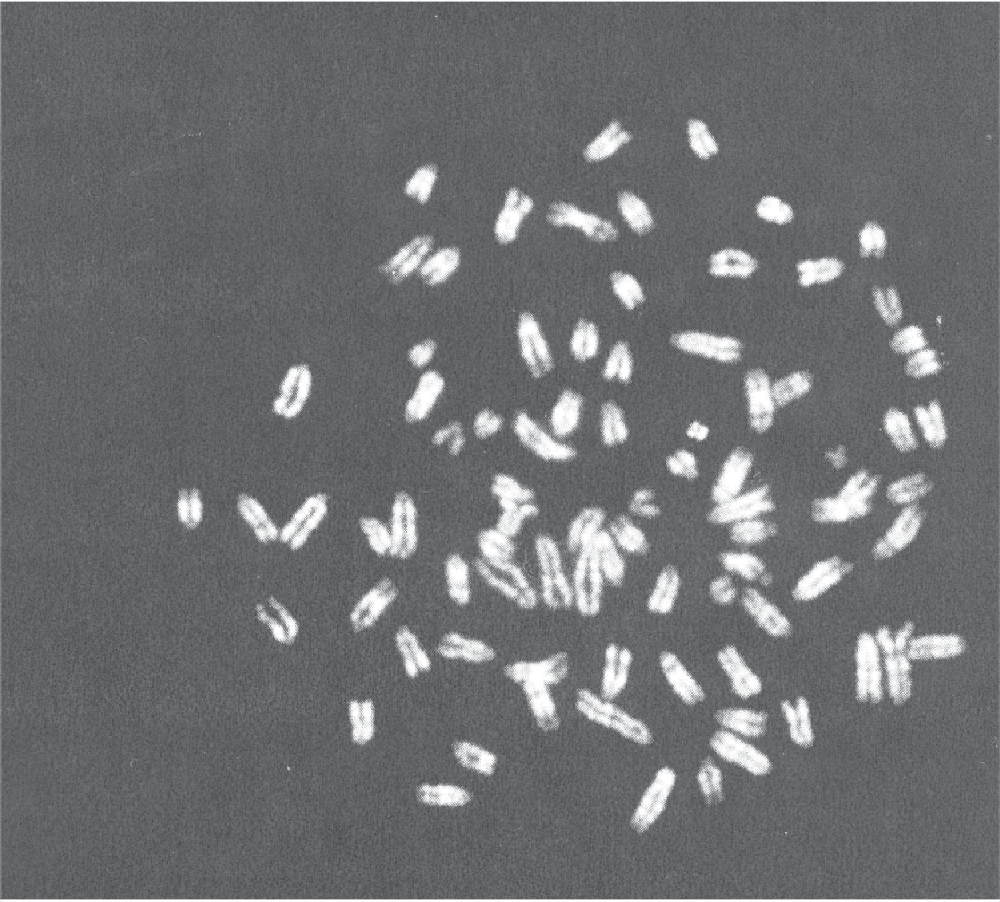
Metaphase spread of the tumor-inducing murine bone marrow-derived mesenchymal stem cells. Here, 84 chromosomes are present. Images were acquired with Olympus BX51 microscope (Olympus, Aartselaar, Belgium) and the Genetix Applied Imaging acquisition software (Applied Imaging, France).
In order to obtain MSCs with normal karyotype, we compared different culture protocols. We tested several media such as RPMI, DMEM, DMEM/F12, or Murine Mesencult. We also used serum from different species (horse or mouse) in combination with fetal bovine serum (10% FBS, 20% FBS, 10% FBS + 10% horse serum, 10% FBS + 10% mouse serum). We added different growth factors such as HGF [19], LIF, and EGF [20] (each at a concentration of 20 ng/mL). Moreover, we reduced the duration of the ex vivo culture by immunodepletion of the hematopoietic cells as recommended by Schrepfer and colleagues [19]. This operation was realized at passage 2 or 3. We also tested different cell densities. Most of the MSC cultures (around 70%) did not pass through the senescence and the crisis phases (∼2–3 passages). But when the cells survived after the senescence and the crisis phases, we observed systematically that independently of the culture medium used and of the realization of an hematopoietic cell immunodepletion, they always presented an abnormal chromosome number. We already observed this phenomenon at passages 2 and 3, just after the senescence phase, 2 months after isolation. As shown in Table 1, meta-phase spreads of 21 different MSCs cultures were realized and all of them showed abnormal chromosomal count, ranging from 30 to 158 chromosomes, with most of the mitosis presenting around 70 chromosomes. We never observed a normal count in MSCs while lymphocytes from the same mouse strain presented a regular 40 chromosomes karyotype. Moreover, we obtained exactly the same results when MSCs were isolated from other mouse strains: BTBR T+ tf/J or C57BL/6J.
MSC cultures were realized from bone marrow of different mouse strains BTBR-Pah enu2 /J, BTBR T+ tf/J or C57BL/6. MSC purification was performed by adherence and passages until they were free from hematopoietic cells as assessed by flow cytometry, or by immunodepletion.
Culture duration until the metaphase spread study is indicated, as well as the culture passage numbers. When indicated, an hematopoitic cell immunodepletion was realised using anti-CD34, anti-CD45 and anti-CDllb antibodies. The culture medium used is specified for each culture (FBS : foetal bovine serum, HS : horse serum, HGF : hepatocyte growth factor). Some MSC cultures were characterized by flow cytometry for the murine mesenchymal stem cell markers Sca-1, CD106 and CD90.2, and to assess their purity, for the hematopoietic cell markers CD34, CD45 and CD11b. MSC differentiation potential into osteoblasts, adipocytes and chondrocytes was checked after cell culture in specific differentiation media. Calcium deposition measurement was realised in the osteoblasts, lipid vacuoles were stained with Oil Red O for adipocytes, and toluidin blue staining was realized on micromass culture for chondrocytes. The total number of analysed mitosis is indicated, and the chromosome count is divided into 3 subcategories : nearly normal (around 40 chromosomes), nearly tetraploïd (around 70 chromosomes), and hyperploïd (around 120 chromosomes). ND: not determined; MSCs, mesenchymal stem cells.
We carefully checked if the cells we isolated presented MSC properties: they were able to form single colony clusters (CFU-F). By flow cytometry analysis, we verified that these cells present murine MSC markers (Fig. 3): they harbored Sca-1 and CD106 cell markers, and half of these MSC cultures presented the CD90.2. The differentiation capacities of these MSCs were also explored (Fig. 4 and Table 1).They always preserved their osteogenic potential. Their adipogenic potential was lost in half of the cases. Their chondrogenic potential was tested by micromass culture in presence or absence of chondrogenic differentiation medium. Although, we observed a week toluidine blue staining of the micromass even in the absence of chondrogenic differentiation medium, 3 of the 6 tested MSC cultures presented a stronger staining after incubation with the chondrogenic medium.

A representative flow cytometric analysis of purified bone marrow-derived murine mesenchymal stem cells (MSCs). (

Osteoblastic, adipocytic, and chondrogenic differentiation. Cells were cultured in osteoinductive medium for 2–3 weeks or left untreated and Ca2+ deposition was measured. (
Genomic instability of the murine MSCs has previously been reported by 2 other research groups who have also shown occasional sarcoma formation in mice injected with bone marrow-derived MSCs [12,13,21]. Miura et al. showed that karyotypic abnormalities occurred frequently and very early in the purification process. They characterized in details the genomic aberrations observed by spectral karyotyping, FISH analysis, and telomerase activity assays. In accordance with our results, the group of Tolar et al. confirmed that this phenomenon is strain-independent.
In opposition to these previous studies, the transformation was systematic and, critically, could not be avoided. In our hands, all the culture protocols and different mouse strains led finally to genetically abnormal cells. We cannot explain this discrepancy between our work and the other reports as we used similar isolation and culture protocols. Miura et al. observed the transformation of 5/100 single colony-derived BM-MSC clones; so it is likely that they would have observed a systematic transformation if cultures had been derived from whole bone marrow isolates like we did.
Chromosomal instability is increasingly recognized as a key component of tumorigenesis [22]. Moreover, it is likely that the properties of MSCs as engraftment, homing, rooming, immunosuppressive, or therapeutic potential could be affected by the genetic modifications observed.
A single report showed human MSC transformation after long-term ex vivo culture [14]. This emphasizes the differences between murine and human cells. Human cells require more genetic changes before going into neoplastic transformation than their murine counterparts [23,24]. Moreover, telomere biology, which is often associated with chromosomal instability in cancer, differs between the 2 species [25].
Maslov and Vijg described that an optimal balance between maintaining a pool of stem cells for tissue regeneration and eliminating severely damaged stem cells ensures maximal longevity, free of cancer, and degenerative tissue decline [26]. Greater genomic instability is observed in mouse MSCs compared to human. As a consequence, mice are expected to develop more tumors than humans, or to present a shorter lifespan. Indeed, in case of mutations following genomic instability for example, MSCs undergo apoptosis leading to the rapid exhaustion of the MSC pool destinated for tissue regeneration. This difference in genomic stability could in part explain the much shorter lifespan of the mice.
In conclusion, we observed systematic murine bone marrow-derived MSC transformations during isolation and purification. Other groups have reported similar results and this event appeared to be species-specific. These data warn the research community about the scientific relevance of murine MSCs for transplantation models. As these cells present drastically damaged genome even after short ex vivo culture, their biological properties are likely to be modified. Therefore, they might not be suitable to predict the outcome of any clinical protocol with human MSCs.
Footnotes
Acknowledgments
The authors would like to thank Sandra Ormenese (GIGA-Imaging platform—Liege University, Belgium) for the FACS analysis and Yasmina Amkrane for her excellent technical assistance. We thank P. Roncarati and Dr. A. Thiry for technical and scientific support. This work was supported by a Waleo grant from the Region Wallonne.
Author Disclosure Statement
No competing financial interests exist.