
Abstract
Postnatal mesenchymal stem/stromal-like cells (MSCs) including periodontal ligament stem cells (PDLSCs), dental pulp stem cells (DPSCs), and bone marrow stromal cells (BMSCs) are capable of self-renewal and differentiation into multiple mesenchymal cell lineages. Despite their similar expression of MSC-associated and osteoblastic markers, MSCs retain the capacity to generate structures resembling the microenvironments from which they are derived in vivo and represent a promising therapy for the regeneration of complex tissues in the clinical setting. With this in mind, systematic approaches are required to identify the differential protein expression patterns responsible for lineage commitment and mediating the formation of these complex structures. This is the first study to compare the differential proteomic expression profiles of ex vivo-expanded ovine PDLSCs, DPSCs, and BMSCs derived from an individual donor. The two-dimensional electrophoresis was performed and regulated proteins were identified by liquid chromatography—electrospray-ionization tandem mass spectrometry (MS and MS/MS), database searching, and de novo sequencing. In total, 58 proteins were differentially expressed between at least 2 MSC populations in both sheep, 12 of which were up-regulated in one MSC population relative to the other two. In addition, the regulation of selected proteins was also conserved between equivalent human MSC populations. We anticipate that differential protein expression profiling will provide a basis for elucidating the protein expression patterns and molecular cues that are crucial in specifying the characteristic growth and developmental capacity of dental and non-dental tissue-derived MSC populations. These expression patterns can serve as important tools for the regeneration of particular tissues in future stem cell-based tissue engineering studies using animal models.
Introduction
M
Gene microarrays have provided valuable information on the global gene expression pattern of several MSC types and dental cells based on mRNA expression. Studies have reported that the gene expression profiles of PDL fibroblasts and their physiologically neighboring gingival fibroblasts are significantly regulated reflecting their functional differences in vivo [16]. A comparative microarray analysis between BMSCs and DPSCs demonstrated similarities in gene expression profiles particularly for some collagens, non-collagenous extracellular matrix proteins, and several cell-adhesion molecules and various growth factors associated with initiation of osteogenesis and formation of mineralized bone matrix [17]. In addition, several differentially expressed genes were also identified although their potential role in mediating the distinct ontogeny and developmental potential of these MSCs requires further investigation. Interestingly, microarray analyses comparing PDLSCs and other MSC populations have not been reported to date. Although several genes involved in osteoblast differentiation including some essential to craniofacial bone development have been identified [18,19], gene, or more specifically, mRNA expression patterns, frequently do not correlate with translated protein expression at the functional level [20,21]. Furthermore, post-translational protein modifications that serve as important mechanisms in regulating signal transduction pathways are not detected by microarray or other gene-based analyses. A more accurate and complete pattern of differential gene expression between MSC populations may be derived from a proteomic investigation.
Proteomics provides a powerful method toward investigating the entire protein complement of a cell, that is, the proteome. Sequencing of the human genome [22] and the advent of mass spectrometry-based proteomics [23] has led to the ability to identify thousands of proteins expressed by human cells in a single experiment [24]. Proteomic-based analyses can generate reference maps that serve as invaluable databases of protein expression in MSCs cells under various experimental conditions [25,26]. Indeed, studies have demonstrated proteomic profiling to be an extremely effective tool in characterizing and comparing stem cell phenotypes and the mechanisms that control their self-renewal and differentiation potential [27 –29].
MSC-based tissue engineering strategies have provided promising alternatives to conventional therapies for successful and predictable regeneration of periodontal tissues in the clinical setting. Increasingly, studies are investigating the capacity of MSCs to regenerate periodontal tissues using large-animal models with surgically created periodontal defects representing periodontal disease [30 –33]. Recently, we isolated and ex vivo-expanded populations of ovine PDLSCs, DPSCs, and BMSCs, and performed initial characterization of these cells [14,34,35]. Importantly, ovine MSCs display a similar phenotype and genotype to their human counterparts including the expression of MSC- and bone-related markers and in terms of their developmental potential in vivo [14, 34]. Thus, ovine MSCs represent a potentially useful cell population for periodontal regeneration studies utilizing an ovine model of periodontal disease resembling periodontitis in the clinical setting.
The aim of this study was to identify proteins that are differentially expressed between ovine PDLSCs, DPSCs, and BMSCs derived from an individual donor. We hypothesize that MSC populations derived from different tissues differentially express a subset of proteins attributing to their specific growth and capacity to generate structures resembling the unique microenvironments from which they are derived in vivo. We anticipate that differential protein expression patterns can serve as important tools for the regeneration of dental and non-dental structures in future stem cell-based tissue engineering studies using animal models.
Materials and Methods
Dental tissue collection, preparation, and cell culture
Normal fully erupted incisors were extracted from 2 mature non-related Merino ewes (donors 1 and 2) under anesthesia. PDL was scraped from the middle third of the root surface. The dental pulp cavity was then exposed and pulp tissue separated from the crown and root. These procedures were performed in accordance with the Institute of Medical and Veterinary Science (IMVS) Animal Ethics Committee (130/06). Similarly, PDL and dental pulp tissue was also collected from normal human premolars extracted for orthodontic purposes following informed patient consent under approved guidelines set by the University of Adelaide Human Research Ethics Committee (H-125-2005). Tissues were digested in collagenase Type 1 (3 mg/mL; Worthington Biochemical Corporation, Lakewood, NJ) and dispase II (neutral protease; 4 mg/mL; Roche Diagnostics, Indianapolis, IN) for 2 h at 37°C to obtain single-cell suspensions of PDL and dental pulp cells. Primary PDLSC and DPSC cultures were established in tissue culture flasks and maintained in minimal essential medium (α-MEM; JRH Biosciences, Inc., Lenexa, KS) supplemented with 10% fetal calf serum (FCS), 2 mM
Bone marrow collection, preparation, and cell culture
Bone marrow aspirates were obtained from the posterior iliac crest from both sheep in accordance with the IMVS Animal Ethics Committee (130/06). Similarly, human bone marrow was obtained from one donor following informed consent under the approved guidelines of the Human Ethics Committee of the Royal Adelaide Hospital, South Australia. Bone marrow mononuclear cells (BMMNCs) were prepared as described previously [13]. Primary BMSC cultures were established in tissue culture flasks and maintained in α-MEM (JRH Biosciences, Inc.) supplemented with 10% FCS and additives as described above in a humidified atmosphere (37°C, 5% CO2).
Cell proliferation assay
Ovine PDLSCs, DPSCs, and BMSCs that had been expanded ex vivo were seeded at passage 3 (1,250 cells/well) in triplicate using a 96-well flat-bottom plate and maintained in 100 μL standard explant culture medium as described above for 5 days. After the incubation period, 10 μL of colorimetric Cell Proliferation Reagent WST-1 (Roche Molecular Biochemicals, Basel, Switzerland) was added per well, and samples were incubated a further 2 h. The absorbance of each well was then measured at a wavelength of 450 nm using a microtiter plate reader.
Preparation of whole-cell lysates for proteomic analysis
Ex vivo-expanded cultures of PDLSCs, DPSCs, and BMSCs obtained from both sheep were trypsinized (at passage 2–3), washed in Hank's balanced salt solution (HBSS; JRH Biosciences, Inc.) supplemented with 5% FCS, washed twice in ice-cold phosphate-buffered saline (PBS), and resuspended in 2 mL ice-cold 40 mM Tris buffer pH 7.4. Cells were lysed by four 15 s bursts of sonication (Soniprobe, Type 1130A; Dawe Instruments Ltd., London, UK) on ice with 30 s intervals to keep the samples cold. Endogenous protease activity was neutralized by the addition of 50 μL Protease Inhibitor Cocktail (for mammalian cell extracts; Sigma-Aldrich Inc.) during sonication and nucleic acids were then degraded on ice for 30 min following the addition of deoxyribnuclease II (2,000 U; Sigma-Aldrich Inc.), ribonuclease A (1.000 U; Sigma-Aldrich Inc.) and 50 mM MgCl2. Proteins within the whole-cell lysate were purified by precipitation following the addition of 4 volumes of ice-cold acetone with 50 mM DTT (Sigma-Aldrich Inc.) and incubated for 24 h at −20°C. Precipitated proteins were collected by centrifugation (10,000g, 15 min at 4°C) and the pellet then resuspended by vortexing in 2 mL ice-cold acetone with 50 mM DTT and washed by centrifugation (10,000g, 15 min at 4°C). The supernatants were then aspirated and residual acetone removed by evaporation. Purified samples were finally dissolved in rehydration extraction buffer #3 (Bio-Rad, Hercules, CA) containing 2 mM tri-n-butyl phosphine (TBP; Bio-Rad) and after 1 h at room temperature clarified by centrifugation (15,000g, 15 min at 18°C). The supernatants were then collected and stored at −80°C. Protein quantification was performed using an RC DC protein assay kit (Bio-Rad) according to the manufacturer's instructions.
Two-dimensional electrophoresis
Isoelectric focusing (IEF) was performed on 17 cm precast immobilized pH-gradient (IPG) strips with a pH range of 5–8 using a Protean IEF cell (all equipment and reagents from Bio-Rad unless stated otherwise). In brief, IPG strips were passively rehydrated in 330 μL rehydration/extraction buffer #3, containing 0.2% (w/v) pH 3–10 and 0.2% (w/v) pH 7–10 ampholytes, and 1.2% (v/v) De-Streak Reagent (GE Healthcare, Buckinghamshire, UK) for 24 h. For separation of proteins with a molecular weight (MW) range of 10–35 kDa or 25–110 kDa, samples containing 125 μg or 150 μg whole-cell lysate protein, respectively, were cup-loaded before IEF. In brief, the voltage was gradually increased to 10,000 V over 11 h as follows; increased to 150 V over 1 h (linear ramping unless otherwise stated), to 300 V over 5 h, to 600 V over 1.5 h, to 1,200 V over 1.5 h, followed by rapid ramping to 8,000 V over 1.5 h, and to 10,000 V over 30 min. Focusing occurred for 50,000 V hours (V.h) with a 50 μA/strip current limit and the temperature was maintained at 20°C. After 50,000 V.h had been achieved, IPG strips were maintained at 500 V until removed from the IEF unit and placed at −20°C. IEF was performed on each sample in triplicate (experimental replicates). Following IEF, each IPG strip was subjected to a 2-step equilibration process as previously described [36]. Large format (18 × 18 cm) polyacrylamide gels 8% T, 3.3% C (to resolve 25–110 kDa proteins) or 16% T, 3.3% C (to resolve 10–35 kDa proteins), containing Tris–HCl buffer pH 8.8, 0.1% SDS were cast without stacking gels in batches of 6 using the Protean II XL casting chamber. The second dimension, polyacrylamide gel electrophoresis (PAGE), was then performed at 5 mA per gel at 15°C using a Protean II XL Multi cell (6 gels per run) until the dye front reached the bottom of the gel.
Gel staining
Gels were fixed in 40% ethanol (v/v)/10% acetic acid (v/v) in Milli-Q (Millipore, Billerica, MA) water and stained with Flamingo Fluorescent Stain (Bio-Rad) according to manufacturer's instructions. Gels were then de-stained in 0.1% (v/v) Tween 20 in Milli-Q water for 10 min and rinsed several times in Milli-Q water prior to imaging.
Image acquisition and analysis
Stained gels were scanned using a Typhoon Trio Variable Mode Imager (Molecular Dynamics Inc., Sunnyvale, CA) using a Green Laser (532 nm) excitation source and 610 ± 30 nm band pass emission filter. Image analysis was performed using PD-Quest v7.2 software whereby protein spots were automatically detected and matched initially, then manually edited and normalized for total density of all detected spots in the gel. For each sheep, triplicate 2-DE gels of PDLSCs, DPSCs, and BMSCs were arranged as match-sets and proteins with quantitative differences in mean density between replicate groups were analyzed for statistical significance using a Student's t-test (P < 0.05). Proteins that were consistently significantly regulated between the same MSC populations within both sheep were flagged for identification.
Automated spot excision
Representative gels containing well-resolved significantly regulated protein spots for identification were placed onto a low-fluorescence glass plate containing 2 reference marker disks and scanned using an Ettan DIGE Imager (GE Healthcare, Rydalmere, Australia) with the filter settings corresponding to the measurement of Cy2 (excitation = 480/30 nm, emission = 530/40 nm), a 0.4s acquisition time and pixel size of 100 μm. The images were imported into DeCyder software (version 6.5, GE Healthcare) and spots detected using the automated method. Spots of interest identified from the previous image analysis were selected by inspection and the resulting pick-list was exported from DeCyder and imported into Spot Picker software (version 1.2, GE Healthcare) and spots were excised using an Ettan Spot Picker (GE Healthcare) according to the manufacturer's instructions. Gel plugs were washed with 0.1 M ammonium bicarbonate buffer followed by Milli-Q water, then dehydrated in acetonitrile (ACN) and dried.
Tryptic digestion of excised proteins
Each gel plug was digested with 10 μL of solution containing 100 ng trypsin in 5 mM ammonium bicarbonate containing 10% ACN for 16 h at 37°C. Peptides were extracted sequentially with 1% formic acid, 50% ACN/0.1% formic acid, and ACN and the combined extracts were concentrated by centrifugal evaporation and diluted in 6 μL 3% ACN/0.1% formic acid.
Liquid chromatography—electrospray-ionization tandem mass spectrometry
Peptides were separated using an Agilent Protein ID Chip column assembly (40 nL trap column with 0.075 × 43 mm C-18 analytical column) housed in an HPLC-Chip Cube Interface (Agilent, Forest Hill, Australia) connected to an a HCT ultra 3-D-Ion-Trap mass spectrometer (Bruker Daltonik GmbH, Germany). The column was equilibrated with 4% ACN/0.1% formic acid at 0.5 μL/min and the samples eluted with an ACN gradient (4%–31% in 32 min). Eluting peptides were fragmented by collision-induced dissociation (CID). Liquid chromatography—electrospray-ionization tandem mass spectrometry (MS and MS/MS) spectra were subjected to peak detection using DataAnalysis (version 3.4, Bruker Daltonik GmbH) and imported into BioTools (version 3.1, Bruker Daltonik GmbH) for database searching and annotation.
Database searching and de novo sequencing
Peptide spectra data were searched against databases using Mascot (version 2.2, Matrix Science:
Western blot analysis
Ovine and human PDLSCs, DPSCs, and BMSCs were grown in 10-cm dishes to 70–80% confluence and whole-cell protein lysates were obtained as previously described [38]. Equivalent amounts of protein lysates (15–50 μg) were separated on 12%T 3.3%C polyacrylamide gels by sodium dodecyl sulfate (SDS)-PAGE. Proteins were then electro-transferred to a Hybond™-P PVDF transfer membrane (GE Healthcare, Buckinghamshire, UK). Transfer membranes were blocked in 25 μg/mL membrane blocking agent (RPN2125V; GE Healthcare) and 1% (w/v) bovine serum albumin (BSA; Sigma-Aldrich Inc.) in 1% (w/v) Tris-buffered saline (TBS)/0.1% (v/v) Tween 20 and then probed overnight with primary antibody; anti-heat shock protein 27 (Hsp-27; mouse anti-human; 1 μg/mL; Rockland Inc., Gilbertsville PA), anti-Annexin A4 (AnxA4; rabbit anti-human; 2 μg/mL; Abcam, Sapphire Bioscience Pty. Ltd., NSW, Australia), anti-collapsin response-mediated protein 4 (CRMP-4; goat anti-human; 1 μg/mL; Santa Cruz Biotechnology Inc., Santa Cruz, CA), and anti-α-tubulin (rat anti-human; 0.25 μg/mL; Abcam) in 1% BSA 1% (w/v) TBS/0.1% (v/v) Tween 20 at 4°C. After washing in 1% TBS 0.1% Tween 20, membranes were incubated with the appropriate secondary antibody; anti-mouse, anti-rabbit, and anti-rat IgG AP-conjugated (Chemicon Australia Pty. Ltd., Melbourne, Australia), and anti-goat IgG AP-conjugated (Chemicon International, Temecula, CA) at 1 μg/mL. Proteins were detected using 1 mL ECF substrate (RPN5785; GE Healthcare) and scanned using a Typhoon 9410 variable mode imager (Molecular Dynamics Inc., Sunnyvale, CA). Expression of α-tubulin was used as an internal control of protein load.
Immunohistochemistry
Ovine and human MSCs were plated at a density of 8,000 cells/well in 96-well plates and cultured in α-MEM supplemented with 10% FCS and additives as described earlier in a humidified atmosphere (37°C, 5°% CO2). At 70% confluence, cultures were fixed in 4% paraformaldehyde (PFA) in PBS for 20 min, washed in 0.1% Tween 20 PBS and endogenous peroxidase activity blocked with 0.5% H2O2 in methanol for 30 min. Cells were then incubated in 3% normal goat serum (NGS) or normal rabbit serum (NRS) in 0.1% Tween 20 PBS for 3 h to block the binding of non-specific immunoglobulins. Cells were then probed overnight at 4°C with primary antibody; anti- Hsp-27 (10 μg/mL), anti-CRMP-4 (10 μg/mL) and anti-AnxA4 (2 μg/mL), or isotype control immunoglobulin (Ig); normal mouse IgG (10 μg/mL; in-house antibody), normal goat IgG (10 μg/mL; Caltag Laboratories, Burlingame, CA), and normal rabbit IgG (2 μg/mL; Caltag Laboratories) in 0.1% Tween 20 PBS. After washing, cells were incubated with biotinylated secondary antibody; goat anti-mouse (10 μg/mL; Caltag Laboratories), rabbit anti-goat (10 μg/mL; Vector Laboratories Inc., Burlingame, CA), or goat anti-rabbit (10 μg/mL; Vector Laboratories Inc.) in 0.1% Tween 20 PBS for 1 h and then incubated with Vectastain® ABC Reagent for 45 min according to manufacturer's instructions (Vector Laboratories Inc.). Staining was visualized by the addition of AEC peroxidase substrate kit for 10 min according to manufacturer's instructions (Vector Laboratories Inc.). Cells were counterstained with Mayer's hematoxylin for 15 s and rinsed with PBS until clear.
Results
Relative proliferation rates of ovine MSCs
Ovine PDLSCs, DPSCs, and BMSCs demonstrated rapid proliferation in primary explant cultures (data not shown). However, the rate of cellular proliferation was significantly lower in ex vivo-expanded BMSCs compared to both PDLSCs and DPSCs at passage 3, while no significant difference was observed between PDLSCs and DPSCs (Fig. 1).

Rate of proliferation measured by mitochondrial dehydrogenase activity using Cell Proliferation Reagent WST-1 in cultures of ovine periodontal ligament stem cells (PDLSCs), dental pulp stem cells (DPSCs), and bone marrow stromal cells (BMSCs) plated at passage 3 (n = 3). The proliferation rate of BMSCs was significantly lower compared to both PDLSCs and DPSCs (* Student's t-test, P < 0.05). The proliferation rates of PDLSCs and DPSCs showed no significant difference. Values are shown as mean ± standard deviation (SD).
Differential protein spot expression analysis of ex vivo-expanded ovine PDLSCs, DPSCs, and BMSCs
The present study is the first to compare the proteomic expression profiles of ovine PDLSCs, DPSCs and BMSCs derived from the same donor that have been expanded ex vivo. Whole-cell lysate proteins derived from ovine PDLSCs, DPSCs, and BMSCs were separated by 2-dimensional electrophoresis (2-DE). Proteins within the isoelectric point range of pH 5–8 and a MW range of 10–110 kDa were resolved using 16% T, 3.3% C (16% PAGE) and 8% T, 3.3% C (8% PAGE) for resolution of proteins within a MW range of 10–35 kDa and 25–110 kDa, respectively. Proteins were detected with Flamingo fluorescent stain and analyzed using PD-Quest v7.2 software. Representative raw 2-DE gels and software-generated Gaussian images for 16% PAGE and 8% PAGE are shown in Figure 2A–2D. The total numbers of well-resolved protein spots used for analysis of each MSC population from both sheep were 157 and 467 (PDLSCs; 16% PAGE and 8% PAGE), 135 and 444 (DPSCs), and 151 and 366 (BMSCs). Gels were arranged into 2 match-sets per sheep each containing 3 experimental replicate gels of each MSC population (Match-set 1; sheep 1 using 16% PAGE, Match-set 2; sheep 1 using 8% PAGE, Match-set 3; sheep 2 using 16% PAGE, Match-set 4; sheep 2 using 8% PAGE). Within each match-set, 3 analysis sets were created (each comparing replicate gels of 2 MSC types) to identify significantly differentially expressed proteins (P < 0.05). In total, 58 detected protein spots showed significant differential expression between at least 2 MSC populations where the regulation was conserved in both sheep donors. The location of regulated protein spots on the Gaussian images are also shown in Figure 2B and 2D. The number of protein spots detected to be significantly up-regulated within each MSC population relative to 1 or 2 other MSC populations is listed in Table 1.

Representative raw 2-DE gels and Gaussian images of ovine mesenchymal stromal-like cell (MSC) whole-cell protein lysates. The 16% and 8% polyacrylamide gel electrophoresis (PAGE) was performed for resolution of proteins within a molecular weight (MW) range of 10–35 kDa (
PDLSCs, periodontal ligament stem cells; DPSCs, dental pulp stem cells; BMSCs, bone marrow stromal cells.
Identification of regulated protein spots
Differentially expressed protein spots identified by tandem mass spectrometry (MS/MS) and database searching are summarized in Table 2. Of the 58 regulated spots, 42 met the criteria for significant protein identification by MS and MS/MS requiring at least 2 peptides to be matched to the SwissProt or NCBInr databases with a Mascot score above the cut-off (threshold) score. Another 7 regulated protein spots were weakly matched by MS and MS/MS with fewer than 2 peptides giving ion-scores greater than the Mascot threshold, however, were considered to be significantly identified following de novo sequence interpretation (Supplementary Fig. 1; Supplementary materials are available online at
The criteria for a significant protein identification required a least 2 peptides to be matched to the SwissProt or NCBInr databases with a Mascot score above the cut-off (threshold) score. Some protein spots matched weakly with fewer than 2 peptides giving ion-scores greater than the Mascot threshold, however, were identified following de novo sequence interpretation. The table also lists the species to which the regulated protein spot was matched, the SwissProt database accession number, percentage of sequence coverage based on MS/MS spectra, number of unique peptides matched, and fold up-regulation of protein expression between mesenchymal stromal cells (MSCs) populations within each donor.
Denotes likely protein identity. More than one protein identified within spot (see Supplementary Table 2; Supplementary materials are available online at
Denotes protein identity uncertain. Multiple proteins identified within spot (see Supplementary Table 2).
Denotes protein identity confirmed by de novo sequencing (see Supplementary Fig. 1).
Subcellular localization of regulated proteins in cultured ovine PDLSCs, DPSCs and BMSCs
Differentially expressed proteins were classified according to their subcellular location (Fig. 3A). The majority of identified proteins were cytoplasmic in origin (42%) while others were localized in the nucleus (4%) mitochondria (8%), endoplasmic reticulum (4%), and cell membrane (4%). Another 13% are found in both the cytoplasm and nucleus, 13% in the cytoplasm and cell membrane, and 10% are cytoskeleton-related. The subcellular location of one protein (SSP 6207; NmrA-like family domain-containing protein 1) was unknown. The subcellular localization of each regulated protein is shown in Supplementary Table 1.
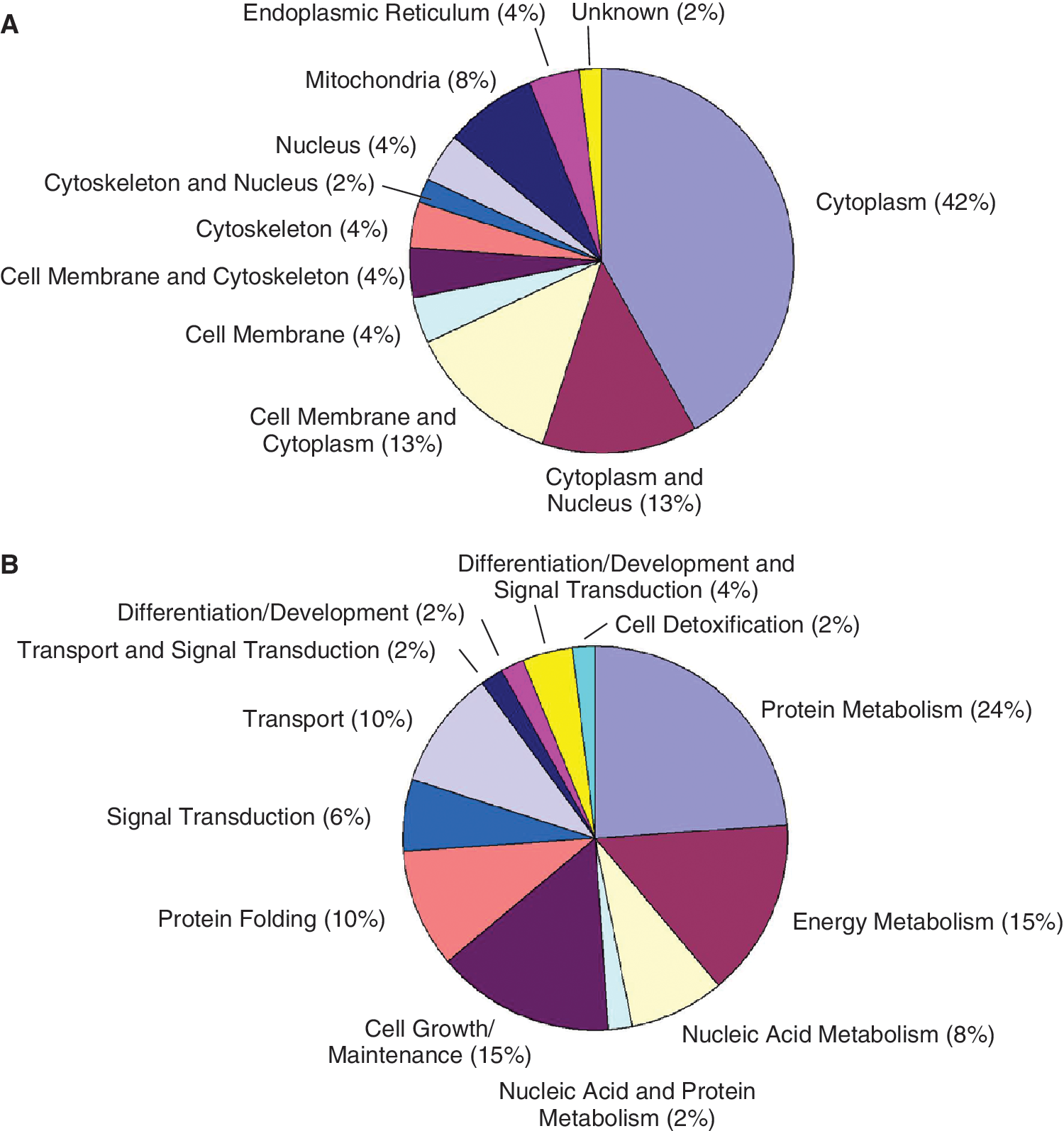
Classification of identified differentially expressed proteins between ovine periodontal ligament stem cells (PDLSCs), dental pulp stem cells (DPSCs), and bone marrow stromal cells (BMSCs). The charts illustrate the distribution of regulated proteins according to their subcellular localization (
Biological function of regulated proteins in cultured ovine PDLSCs, DPSCs, and BMSCs
Identified regulated proteins were also grouped according to their biological function (Fig. 3B). Many of these proteins were related to protein (24%), energy (15%), and nucleic acid (8%) metabolism. Another 15% are involved in cell growth and maintenance particularly in actin organization, cell motility, and cell adhesion. Other proteins are involved in transport (10%), protein folding (10%), and signal transduction (6%), and 6% are related to differentiation and development. The biological function of each differentially expressed protein is shown in Supplementary Table 1.
Validation of regulated protein expression
To confirm the regulated expression of selected proteins including Hsp-27, AnxA4, and CRMP-4, additional studies were performed to investigate expression levels in ovine PDLSCs, DPSCs, and BMSCs derived from an individual sheep or human equivalent MSCs derived from different donors. Using western blot analysis, expression of AnxA4 was found to be higher in both ovine and human PDLSCs compared to both DPSCs and BMSCs (Fig. 4A I–II). Hsp-27 expression was also up-regulated in human PDLSCs compared to both DPSCs and BMSCs by western blot analysis (Fig. 4A II); however, no reactivity was observed with the anti-Hsp-27 antibody to ovine MSC whole-cell protein lysates (data not shown). In addition, no reactivity was observed with the anti-Hsp-27 or anti-AnxA4 antibodies to ovine or human MSCs by immunocytochemistry (data not shown). CRMP-4 expression levels were confirmed to be higher in both ovine and human DPSCs relative to BMSCs by western blot analysis and immunocytochemistry, respectively (Fig. 4B I–V). No reactivity was observed with the anti-CRMP-4 antibody to ovine MSCs by immunocytochemistry (data not shown).
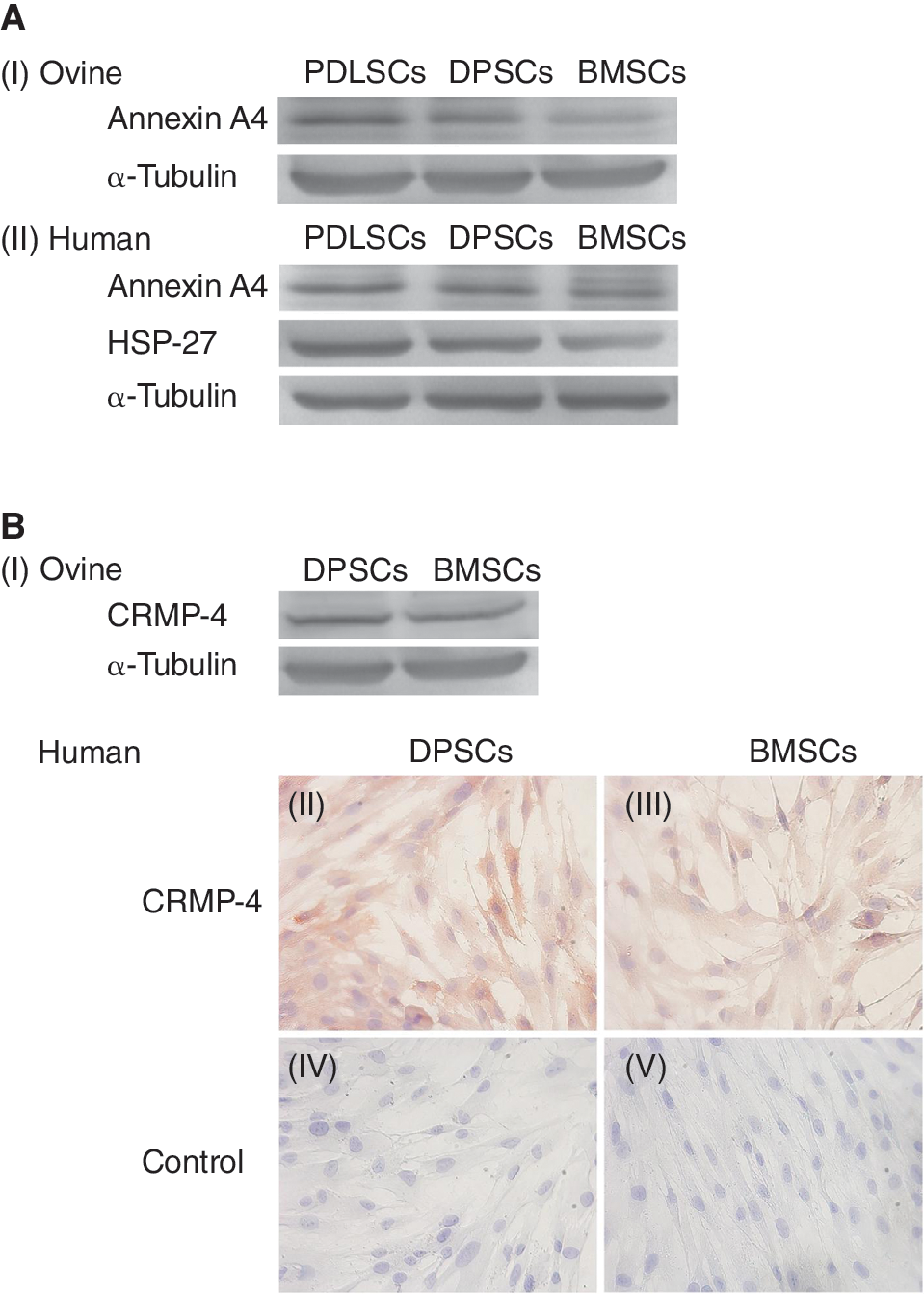
Validation of differential expression of selected proteins between mesenchymal stromal-like cell (MSC) populations. (
Discussion
MSCs derived from ovine periodontal ligament, dental pulp and bone marrow exhibit a similar surface marker phenotype and osteogenic differentiation potential in vitro and in vivo, which is consistent with human-derived MSCs [14,34,35]. However despite these similarities, MSC populations retain a capacity to regenerate the unique microenvi-ronments from which they are derived highlighting their functional differences in vivo [9,10,13,15]. The purpose of this study was to compare the protein expression profiles of dental and non-dental ovine MSC populations from the same donor and investigate the potential significance of differentially expressed proteins in relation to the ontogeny, function, and developmental capacity attributed to tissue-specific MSCs. In total, 49 of 58 differentially expressed proteins between ex vivo cultured PDLSCs, DPSCs, and BMSCs were significantly identified, most of which were categorized as cytoplasmic proteins. Differentially expressed proteins were also classified according to their biological function, the majority relating to cellular metabolism and cell growth and maintenance. Of the 58 regulated proteins, only 12 were found to be up-regulated in 1 MSC population relative to the other 2 MSC populations. Interestingly, the regulation of selected proteins including Hsp-27, AnxA4, and CRMP-4 between ovine MSCs was similarly observed between equivalent human MSC populations suggesting proteins identified in this study may also have important implications in the context of human MSC biology. These proteins may provide valuable insight into the molecular mechanisms attributing to the unique regenerative capacity of tissue-specific human MSCs, such as the formation of alveolar bone, cementum, and PDL by PDLSCs. Performing similar comparative studies with human MSCs derived from individual donors would be difficult given the likely technical issues in obtaining multiple human MSC types from one individual donor in order to eliminate inter-donor variation.
Several proteins were significantly up-regulated in PDLSCs relative to both DPSCs and BMSCs including Hsp-27. In addition to its ATP-independent molecular chaperone and cytoprotective properties during cellular stress [39 –41], increased Hsp-27 expression levels have been reported during cellular transition from proliferation to differentiation [42 –44]. Furthermore, immunoreactivity studies have demonstrated spatiotemporal expression of Hsp-27 during the maturation of cementoblasts and endothelial cells within PDL, and also odontoblasts and dental pulp fibroblasts [45,46]. Hsp-27 is also thought to be involved in maintaining and regulating actin filament stability and dynamics, and spatiotemporal expression of Hsp-27 has been observed to be almost identical to actin filaments in some dental cells [46 –48]. These findings suggest Hsp-27 may play an important role in cell shape alterations during differentiation processes. Significantly higher expression of Hsp-27 in PDLSCs may reflect their regenerative properties during the high turnover of periodontal tissues [49] relating to constant mechanical and biological stresses in comparison to bone that undergoes slow turnover and dentin that exhibits a limited reparative capacity.
Annexins A3 and A4, which belong to a family of calcium-dependent phospholipid membrane-binding proteins, were also found to be up-regulated in PDLSCs relative to both DPSCs and BMSCs. Although their precise biological function remains unknown, studies have proposed annexins play a variety of roles including signal transduction, regulation of phospholipase activity, organization of membrane fusion, exocytosis and endocytosis, and cytoskeleton-membrane interaction [50,51]. Some annexin family members are also thought to play an important role in the initiation of mineralization processes by mediating Ca2+ influx into cells and matrix vesicles [52,53], increasing AP activity [54] and transducing mechanical signals into cellular responses in osteoblasts [55]. Annexin A3 is considered to possess inositol 1,2-cyclic phosphate 2-phosphohydrolase activity, which metabolizes and regulates cellular concentrations of inositol cyclic phosphates, components of the phosphatidylinositol signaling pathway. The high turnover of membrane-bound phosphatidylinositol (PtdIns) and their phosphorylated products, phosphoinositides, occurs following stimulation by various extracellular agonists such as platelet-derived growth factor and epidermal growth factor [56 –58]. These phosphoinositide–protein interactions lead to downstream signaling events participating in cell growth and proliferation, apoptosis, actin remodeling, cell motility, and vesicle trafficking [59]. Transduction of these extracellular signals is mediated by fluctuations in Ptdlns, in particular phosphatidylinositol 4,5-bisphosphate (PIP2), or by various metabolites generated by phospholipases including cyclic and noncyclic inositol phosphates acting as secondary messengers [60,61]. Given a capacity to regulate cellular concentrations of inositol cyclic phosphates, Annexin A3 may therefore play a role in PtdIns-mediated signaling. Interestingly, a recent study by Roubelakis and colleagues also demonstrated expression of Annexin A3 in amniotic fluid (AF)-derived MSCs but not in BMSCs. In contrast, both AF MSCs and BMSCs expressed AnxA4; however, their relative levels of expression were not shown [29]. Significant up-regulation of AnxA4 protein levels has also been demonstrated in PDLSCs and BMSCs during osteogenic differentiation compared with undifferentiated MSCs [62,63]. These findings suggest a potential role of AnxA4 in contributing to the capacity of MSCs to form mineralized tissue and in maintaining tissue homeostasis. Similar to Hsp-27, higher expression of AnxA4 in PDLSCs compared to other MSCs may again reflect the high turnover of periodontal tissues and constant requirement to regenerate mineralized tissues.
Other regulated proteins expressed higher in PDLSCs compared to DPSCs and BMSCs included DNase I and ubiquitin-activating enzyme E1. DNase I plays an active role in cell apoptosis by hydrolyzing DNA [64] and is known to have tight-binding affinity for actin thereby decreasing the pool of free monomers for actin filament polymerization [65]. Actin–DNase I complexes appear to increase cofilin-binding affinity suggesting that elevated expression of DNase I in PDLSCs may reflect the high activity of cells undergoing cytoskeletal remodeling and differentiation in periodontal tissues. Similarly, the 2-fold up-regulation of ubiquitin-activating enzyme E1 in PDLSCs relative to other MSCs may indicate greater levels of ubiquitination-related processes occurring in PDLSCs including proteolysis and other non-proteolytic events such as membrane–protein endocytosis, intracellular trafficking, transcription regulation, DNA repair, and assembly of signaling complexes [66,67].
Ubiquitin C-terminal hydrolase L1 (UCH-L1) is strongly implicated in maintaining neuronal structure, function, and health. We detected UCH-L1 levels to be significantly higher in DPSCs compared to both PDLSCs and BMSCs. UCH-L1 is a neuronal de-ubiquitinating enzyme that facilitates ubiquitin/proteasome-mediated protein degradation by recycling free ubiquitin following cleavage from degraded ubiquitinated peptides [68,69]. UCH-L1 is a major target of oxidative damage in neurodegenerative brains, which may cause irreversible alteration in the conformation and/or enzymatic activity of UCH-L1 leading to deleterious effects on neuronal function and survival [70 –72]. In addition, UCH-L1 is also reported to have an unusual ubiquitin-ligase activity, which may suggest a critical role in neuronal proteasomal protein degradation [73]. The up-regulation of UCH-L1 in DPSCs relative to BMSCs possibly reflects the neural crest origin of DPSCs during development and is consistent with the demonstrated ability of DPSCs to differentiate into functionally active neurons under the appropriate inductive conditions [74 –76]. Furthermore, it suggests that BMSCs may possess a lower neuronal differentiation capacity consistent with their non-neurectodermal origin. Higher expression of UCH-L1 in DPSCs compared to PDLSCs suggests that DPSCs may also have an increased tendency to undergo neuronal differentiation compared to PDLSCs despite their common migrating neural crest origin [77]. Interestingly, several other proteins up-regulated in DPSCs relative to PDLSCs or BMSCs are also implicated in neuronal development and maintaining neuronal structure, function, and health including collapsin response mediator proteins-2 and −4 [78 –81], triosephosphate isomerase, [82 –85], and fascin-1 [86 –88].
Expression of the interferon-γ (IFN-γ)-induced enzyme tryptophanyl-tRNA synthetase (TTS) [89 –91] was also significantly up-regulated in DPSCs relative to both PDLSCs and BMSCs. TTS is required for protein synthesis and is an important enzyme for cell survival and proliferation [92,93]. IFN-γ also induces indoleamine 2,3-dioxygenase (IDO) expression in antigen-presenting cells that catalyzes the degradation of tryptophan within the microenvironment [94] leading to inhibition of T-cell proliferation and diminished T-cell-mediated immune responses [95 –97]. Regulation of cellular TTS expression may be a mechanism by which IDO-expressing cells protect themselves from tryptophan self-starvation [98,99]. Interestingly, recent studies have demonstrated that PDLSCs, DPSCs [100], and BMSCs [101 –103] exhibit immunosuppressive effects on T cells by IDO-mediated tryptophan degradation. The fact that TTS expression in DPSCs is ∼2-fold higher compared to both PDLSCs and BMSCs may suggest that DPSCs have a higher rate of protein synthesis or reflect their need to counteract an enhanced capacity of IDO-mediated tryptophan deprivation and, as such, may possess a greater immunosuppressive capacity. This may have important implications in tooth regeneration applications utilizing allogeneic DPSC donors.
Rho GDP-dissociation inhibitor 1 (Rho GDIα) was also up-regulated in DPSCs compared to both PDLSCs and BMSCs. Rho GDIα controls Rho protein cycling between the cytosol and membrane as well as interaction with effector targets [104]. Rho GDIα is ubiquitously expressed and binds with multiple Rho-signaling proteins including Rho, Rac, and Cdc42 thus regulating their activity [105 –107]. Rho proteins play an important role in signaling cascades by interacting with protein kinases, phospholipid kinases, and phospholipases leading to actin cytoskeleton rearrangements such as membrane ruffling, formation of focal adhesions, and stress fibers [108 –111]. In addition, Rho proteins have been implicated in the formation of podosomes [108,112], the linking of cytoskeletal remodeling in fibroblasts to cell cycle progression, and TGF-β-mediated gene transcription [111,113,114]. Furthermore, it has been suggested that differentially activated Rho proteins play an important role in regulating neuronal morphology [115]. Taken together, increased expression of Rho-regulating protein Rho GDIα may have important implications in cell functions involving massive cytoskeletal changes such as in proliferation, differentiation, migration, and adhesion. However, the significance of increased expression in DPSCs relative to PDLSCs and BMSCs requires further investigation. Interestingly, expression of 2 downstream signaling effectors of Rho GDIα, gelsolin, and fascin-1, known to play important roles in actin cytoskeleton rearrangements, are also up-regulated in DPSCs compared to PDLSCs or BMSCs. This observation suggests an entire pathway linking signal transduction and actin cytoskeleton dynamics may be more activated in DPSCs compared to other MSC populations.
The enzyme ribonucleoside diphosphate reductase (RNR) is involved in the biosynthesis of deoxyribonucleotides and maintains a highly regulated and balanced pool of DNA precursors available for DNA replication and repair. RNR activity is tightly adapted to the cell cycle and levels are at their highest during S phase in eukaryotes when requirement for dNTPs is greatest [116]. However, induction of RNR also occurs during cellular stress in order to control DNA replication [117,118]. We found the large subunit of RNR (R1), containing both the catalytic and allosteric regulatory sites of the protein [116], to be up-regulated in DPSCs relative to PDLSCs and BMSCs; however, the significance of this regulation is unknown.
In summary, this study is the first to compare the proteomic profiles of ex vivo-expanded ovine PDLSCs, DPSCs, and BMSCs derived from an individual donor. The observed differential expression of selected proteins between ovine MSCs was demonstrated to be conserved between equivalent human MSC populations suggesting all regulated proteins identified in this study have potential importance in the context of human MSC biology. Further investigations are required to determine the functional significance of these and other differentially expressed proteins (within pI ranges 3–5 and 8–10) in specifying the characteristic growth and developmental capacity of dental and non-dental MSC populations. We anticipate that differential protein expression profiling will provide a basis for elucidating the protein expression patterns and molecular cues that are crucial in mediating MSCs to regenerate the tissue from which they are derived in vivo. Furthermore, these expression patterns can serve as important tools in selecting an ideal MSC population for the regeneration of specific tissues in future stem cell-based tissue engineering studies using animal models. These studies are paramount for successful and predictable regeneration of damaged and diseased periodontal tissues and other complex structures.
Footnotes
Acknowledgments
This research was supported by grants from the National Health and Medical Research Council (NH&MRC) and the Australian Dental Research Foundation (ADRF). We greatly acknowledge Mr. Victor Marino for ovine incisor extractions and Dr. Peter Psaltis for collection of ovine bone marrow. Preliminary work was presented at the 14th Annual Proteomics Symposium, Australasian Proteomics Society, February 2009, Lorne, Victoria, Australia (poster presentation).
Author Disclosure Statement
For each author, no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.