
Abstract
Mesenchymal stem cells (MSCs) are highly useful in a variety of cell therapies owing to their multipotential differentiation capability. MSCs derived from umbilical cord blood are generally isolated by their plastic adherence without using specific cell surface markers and examined for their osteogenic, adipogenic, and chondrogenic differentiation properties retrospectively. Here, we report 2 subpopulations of MSCs, separated based on aldehyde dehydrogenase (ALDH) activity. MSCs with a high ALDH activity (Alde-High) proliferated more than those with a low ALDH activity (Alde-Low). Alde-High MSCs had a greater ability to differentiate than Alde-Low MSCs in in vitro culture. Transplantation of Alde-High MSCs into fractured mouse femurs enabled early repair of tissues and rapid bone substitution. Alde-High MSCs were also more responsive to hypoxia than Alde-Low MSCs, with the upregulation of Flt-1, CXCR4, and Angiopoietin-2. Thus, MSCs with a high ALDH activity might serve as an effective therapeutic tool for healing fractures within a short period of time.
Introduction
M
MSCs are self-renewing cells with the ability to differentiate into osteoblasts, chondroblasts, and adipocytes under appropriate cell culture conditions. They can also differentiate into nonmesenchymal cells, such as neurons, endothelial cells, skin cells, and hepatocytes [6 –10]. Due to their stem cell properties, MSCs have become an attractive source for clinical cell therapy for the repair of damaged tissues.
Since MSC-specific cell surface markers are yet to be defined, MSCs are currently isolated from various tissues by their capacity to adhere to cell culture dishes [11]. After cultivating adherent cells for a long period, they are examined for their ability to differentiate into 3 particular cell lineages, namely, adipocytes, chondrocytes, and osteocytes. Typical MSCs are defined by their positive expression of CD105, CD73, human leukocyte antigen (HLA)-ABC, CD29, CD44, CD71, CD90, CD106, CD120a, and CD129 markers and by their negative expression of CD45, CD14, and CD34 markers [12,13]. As it takes more than 4 weeks to characterize the adherent cells, total BM cells containing MSCs are used alternatively for cell therapy [13]. However, only a small population of total BM cells possesses the property of MSCs, and the rest are fibroblast-like cells, endothelial cells, or macrophages.
Aldehyde dehydrogenase (ALDH) is an enzyme responsible for oxidizing intercellular aldehydes and plays an important role in the metabolism of ethanol, vitamin A [14], and cyclophosphamide [15]. Based on the intensity of Aldefluor, ALDH-positive cells can be easily isolated by flow cytometry without the need for antibodies [16]. Interestingly, ALDH activity has been used to isolate immature cells from various tissues. For instance, stem cells from BM and intestinal crypt express ALDH at high levels [15,17]. UCB-derived hematopoietic stem cells (HSCs) with high ALDH activity have a higher hematopoietic progenitor function and ability to repopulate than those with low ALDH levels [18,19]. In addition, malignant stem cells have high ALDH activity [20].
Recently, we isolated 2 populations of endothelial progenitor cells (EPCs) from UCB according to their ALDH activity, and referred to them as Alde-Low and Alde-High [21]. Unlike HSCs, EPCs with a low ALDH activity (Alde-Low) were more actively proliferating than those with a high ALDH activity (Alde-High). Functional analyses clearly revealed that Alde-Low EPCs were more responsive to hypoxia than Alde-High EPCs, with the induction of vascular endothelial growth factor (VEGF), CXCR4, and Glut-1 mRNA expressions. The introduction of Alde-Low EPCs significantly reduced ischemic tissue damage in a mouse flap model. These results indicate that ALDH might be a potential biomarker for segregating functional stem cells from whole populations, which may cause the unexpected proliferation of other cell lineages.
In the present study, we isolated 2 populations of MSCs from UCB according to their ALDH activity (Alde-Low and Alde-High) and examined their surface markers and functions. While both MSC populations were almost similar in their cell surface marker expression, they differed significantly in their ability to proliferate and respond to hypoxia in vitro. Unlike EPCs, Alde-High MSCs were more highly proliferating than Alde-Low MSCs, and had a greater potential to differentiate into the 3 mesenchymal cell types (osteocytes, adipocytes, and chondrocytes). Introduction of Alde-High MSCs repaired fractured bone more rapidly than did Alde-Low MSCs. Alde-High MSCs were also more responsive to hypoxia than Alde-Low MSCs in inducing the expressions of Flt-1, CXCR4, and Angiopoietin-2 mRNAs. These data demonstrate that MSC isolation using Aldefluor offers a potential technique for obtaining functionally active and highly proliferating MSCs.
Materials and Methods
Isolation of MSCs and cell culture
Human full-term UCB samples were collected from umbilical cord veins with permission from the local ethics authorities at the University of Tsukuba. The samples were treated with HetaSep solution (StemCell Technologies) and RosetteSep solution (Cat. No. 15168; StemCell Technologies) to deplete cells expressing CD3, CD14, CD19, CD38, CD66b, and Glycophorin A as described previously [21]. The cells were laid onto a density gradient buffer (Histopaque, 1,083 g/cm3; Sigma-Aldrich) and centrifuged at 1,500 rpm for 20 min at room temperature. The cell layer between the plasma and the buffer was collected. The cells were then plated at a density of 2 × 105–2 × 106 cells/mL in a 25-cm2 culture flask (Sumitomo Bakelite) and maintained at 37°C in a humidified atmosphere of 5% CO2. The culture medium consisted of Iscove's Modified Dulbecco's Medium (IMDM) (Invitrogen) supplemented with 10% FBS (Hyclone), 2 mg/mL L-glutamine (Invitrogen), 10 ng/mL human b-FGF (Peprotech), and 0.1% (v/v) penicillin-streptomycin (100 U/mL penicillin, 0.1 mg/mL streptomycin; Invitrogen). The medium was replaced with fresh medium once a week.
The cell clusters formed in the flask were analyzed based on the uptake of phycoerythrin (PE)-conjugated low density lipoprotein from human plasma acetylated DiI complex (1,1′-dioctadecyl–3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate [DiI-Ac-LDL]) (Molecular Probes–Invitrogen) and viewed with an Olympus IX71 microscope (Olympus) under a UPlan FI (x4) lens. The fractions negative for DiI-Ac-LDL uptake, CD31, and CD45 were sorted using FACSVantageSE (BD Biosciences) as described previously [21]. The colony forming frequency of cells negative for DiI-Ac-LDL uptake was 1 out of 12 UCB samples when cells were maintained under the culture conditions described above.
When the sorted cells reached subconfluence, they were harvested with 0.05% trypsin-ethylenediaminetetraacetic acid (Invitrogen) and replated at a ratio of 1:4 or 1:5. Frozen cell stocks were prepared using Cell Banker solution (ZENOAQ) and stored in liquid nitrogen for future experiments. All experiments were performed using at least 3 distinct sources of UCB and gave reproducible results.
Human BM samples were collected from sternums with permission from the local ethics authorities at the University of Tsukuba. BM-derived MSCs were cultured in the same way as the UCB-derived samples described above.
Antibodies
The antibodies used in this study were fluorescein isothiocyanate (FITC)-labeled HLA-ABC (W6/32), PE-labeled anti-CD31 (WM59), allophycocyanine-labeled anti-CD45 (HI30) (BioLegend), FITC-labeled anti-CD105 (SN6) (Serotec), PE-labeled anti-CD166 (3A6), anti-CD73 (AD2), anti-CD14 (M5E2), allophycocyanine-labeled anti-CD34 (581), and anti-mouse IgG (BD Biosciences). The cells were stained with the fluorochrome-conjugated antibodies and sorted using FACSVantageSE (BD Biosciences) as described previously [22].
In vitro differentiation assay of MSCs
To induce osteogenic differentiation, 5 × 104 cells were treated with osteogenic differentiation medium in 4-well plates (Nalge Nunc) for 4 weeks. The medium consisted of IMDM supplemented with 1% FBS, 0.1 mM dexamethasone (Sigma-Aldrich), 10 mM β-glycerol-2-phosphate (Sigma-Aldrich), 0.2 mM ascorbic acid (Sigma-Aldrich), and 50 ng/mL human epidermal growth factor (Wako) [23]. The culture medium was replaced with fresh medium once or twice a week. Alkaline phosphatase activity was examined histologically using a Leukocyte Alkaline Phosphatase Kit (Sigma-Aldrich) according to the manufacturer's instructions.
To evaluate the formation of mineralized matrix, the cultured cells in osteogenic differentiation medium were fixed with 10% formaldehyde (Wako) and stained for 5 min with 1% Alizarin red S solution (Kodak) made up with water. The cells were dissolved in 0.2 M HCl (Wako) and 5% sodium dodecyl sulfate, and the absorbance was measured at 480 nm.
Adipogenic differentiation was induced in 4-well plates for 4 weeks using adipogenic differentiation medium consisting of IMDM supplemented with 10% FBS, 0.1 mM dexamethasone (Sigma-Aldrich), 0.5 mM 3-isobutyl-1-methylxanthine (IBMX; Sigma-Aldrich), 2 mg/mL insulin (Wako), and 0.1 mM indomethacine (Sigma-Aldrich). The culture medium was replaced with fresh medium once or twice a week. The cultured cells in adipogenic differentiation medium were fixed with 10% formaldehyde (Wako) and stained with Oil-Red O solution (Muto Pure Chemicals) for 30 min at 42°C. After staining, the cells were dissolved with 4% IGEPAL CA630 (Sigma-Aldrich) in isopropanol and the absorbance was measured at 480 nm.
To promote chondrogenic differentiation, cells were treated with chondrogenic differentiation medium in 96-well spheroid plates for 4 weeks. The medium consisted of IMDM supplemented with 1% FBS, 0.1 mM dexamethasone (Sigma-Aldrich), 1 mM sodium pyruvate (Invitrogen), 0.25 nM ascorbic acid, 50 mg/mL ITS premix (BD Biosciences), 40 mg/mL proline (Sigma-Aldrich), 10 ng/mL TGF-β1 (Wako), and 10 ng/mL BMP-2 (Wako). The culture medium was replaced with fresh medium once or twice a week. To evaluate chondrocyte differentiation, spheroids were fixed with 4% paraformaldehyde and stained with Toluidine blue solution (Muto Pure Chemicals). Spheroids were also stained with anti-human type II collagen antibody (N-19; Santa Cruz). After washing with PBS, spheroids were incubated with biotinylated goat anti-rabbit antibody (Vector Laboratories), followed by streptavidin-conjugated ABC complex (Vector Laboratories). Specific staining was viewed by incubation in 3,3′-diaminobenzidine, as described before [22]. Counterstaining was performed using Nuclear Fast Red.
Analysis of ALDH activity in MSCs
ALDH activity in MSCs was analyzed with Aldefluor reagent (StemCell Technologies) according to the manufacturer's specifications and a previous report [21]. Aldefluor substrate (0.625 μg/mL) was added to 1 × 106 to 2 × 106 MSCs in Aldefluor assay buffer and incubated for 30 min at 37°C to induce the conversion of Aldefluor substrate to its fluorescent product. For each experiment, an aliquot of Aldefluor-stained cells was immediately quenched with 5 μL of 1.5 mM diethylaminobenzaldehyde (DEAB), a specific ALDH inhibitor. After treatment with Aldefluor reagent, cells were stained with fluorochrome-conjugated antibodies and sorted using FACSVantageSE.10.
Growth curve
Alde-Low and Alde-High MSCs were plated at a density of 4 × 104 cells per 35 mm tissue culture dish (Sumitomo Bakelite) and cultured under normoxic (20% O2) and hypoxic (5% O2) conditions. Cell culture medium was changed once a week. The number of live cells in triplicate dishes was scored using a hemocytometer at 24-h intervals for 11 days. Dead cells were excluded using trypan-blue staining solution (Invitrogen).
Colony assay
Alde-Low and Alde-High MSCs were plated at a density of 2 × 103 or 4 × 103 cells per 35 mm tissue culture dish (Sumitomo Bakelite) and cultured with Amniomax medium (Invitrogen) as described previously [24]. The medium was changed once a week. The number of colonies in triplicate dishes was scored under a microscope after 10 days of plating.
Analysis of MSCs in a bone fracture mouse model
A bone fracture mouse model was generated according to the modified method reported by Taguchi et al. [25]. Adult C57/BL6 mice were anesthetized, and closed transverse fractures were produced in the midsections of femurs. After reconnecting the fractured femurs with pins, incisions were made at the new joints using a 27-gauge needle (2 mm in diameter).
MSCs (5 × 105) were plated on 2 mm × 2 mm Gelforms (Pfizer) and incubated at 37°C for 2 h before transplanting into the mice at the fractured sites. Two days before assaying, immunosuppression was started by the intraperitoneal injection of cyclosporin-A (Wako) at 20 mg/kg of body weight and continued daily for the entire period of the assay. Adult mice with BALB/c nu/nu genetic background were also used in this study without cyclosporin-A treatment. Mice were examined by X-ray 28 days posttransplantation, and the density of the bone-gap area was measured with NIH imaging software.
Thighbones were histologically stained as follows. Thighbones were fixed with 4% paraformaldehyde (Wako) for 7 days and decalcified with Plank-Rychlo solution (Muto Pure Chemicals) for 30 days. Paraffin-embedded samples were sectioned at 7 μm and stained with hematoxylin-eosin solution (Muto Pure Chemicals). Frozen sections were obtained 7 days posttransplantation and fixed with 4% paraformaldehyde (Wako) [26]. Immunohistochemistry using anti-mouse CD31 antibody (MEC13.3; BD Biosciences) was performed as described previously [22]. The number of CD31-positive cells was scored in distinct sections derived from different mice.
Microscopy analysis
Cell samples were viewed with an Olympus IX71 microscope system (Olympus) using UPlanF objective lenses 4x/0.13PhL and 10x/0.30Ph1. Sample slides were viewed with an Olympus BX51 microscope system (Olympus) using UPlanSApo objective lenses 4x/0.16PH, 10x/0.40PH, and 20x/0.75PH (Olympus) and a mounting reagent (Muto Pure Chemicals). Data acquisition was achieved using a DP70 digital camera attached to the microscope and DP controller software (Olympus). Images were processed using Adobe Photoshop version 8.0 (Adobe System).
Reverse transcriptase (RT)-polymerase chain reaction and quantitative polymerase chain reaction
Total RNA (1 μg) was reverse transcribed using an reverse transcriptase (RT)-polymerase chain reaction (PCR) kit (BD Biosciences) as described previously [21]. The resulting cDNAs were amplified by a GeneAmp PCR System 9100 (Applied Biosystems) for 23–35 cycles of 95°C for 5 s followed by 68°C for 30 s. The reaction mixtures for quantitative PCR were prepared using POWER SYBR® Green PCR master mix (Applied Biosystems) and analyzed by a 7700 Sequence Detector (Applied Biosystems). Experiments were performed in triplicate, and data were calculated by the double delta cycle number of threshold (DDCt) method.
The sequences of the primer sets used for the PCR reactions are listed in Table 1.
PPARγ, peroxisome proliferator-activated receptor γ; LPL, lipoprotein lipase; VEGF, vascular endothelial growth factor.
Western blot analysis
Cells were cultured under 1% O2 for 6 h, and nuclear extracts were prepared as described previously. Nuclear extracts (30 μg) underwent electrophoresis in 8.5% sodium dodecyl sulphate–polyacrylamide gel electrophoresis gels. Rabbit anti-hypoxia inducible factor (HIF)-1α antibody (Novus), anti-HIF-2α antibody [27], and anti-HIF-3α antibody (Santa Cruz) were used for immunoblotting. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Invitrogen) or goat anti-mouse IgG (Invitrogen) was used as secondary antibody and detected by enhanced chemiluminescence (GE Healthcare Bio-Science). Goat anti-Lamin B antibody (Santa Cruz) was used to monitor protein loading and transfer.
Statistical analysis
Data were statistically evaluated using the Student's t-test for per-comparison analysis. Data are presented as means ± SD.
Results
Isolation of UCB-derived MSCs
To separate MSCs from UCB, we developed a novel isolation procedure and culture conditions by modifying the previously reported methods [21,23]. After depleting hematopoietic cells by density gradient centrifugation, mononuclear cells were cultured in IMDM/10% FBS with b-FGF in a flask without a specific surface coating. Two distinct types of adherent cells were observed and colonized in a flask after 10 days of cultivation (Fig. 1A). A DiI-Ac-LDL incorporation analysis clearly demonstrated that cells positive for this complex were EPCs as previously reported [21]. There were significantly less DiI-Ac-LDL-negative colonies than positive ones (3.1 + 2.7 and 16.6 + 8.9, respectively, per 25 cm2 flask, N = 6).

Isolation of MSCs from human UCB. (
Cells were trypsinized and CD45/DiI-Ac-LDL-double negative cells were sorted and cultured in IMDM/10% FBS supplemented with b-FGF (Fig. 1B). These cells were more fibroblastic in nature (Fig. 1C) compared with the UCB-derived EPCs observed in our previous study and did not form tube-like structures in Matrigel (Supplementary Fig. S1, available online at
Characterization of CD45/DiI-Ac-LDL-double negative cells
To ascertain whether the adherent cells derived from UCB contained MSCs, we examined their ability to differentiate into osteogenic, adipogenic, and chondrogenic cells (Fig. 2A). CD45/DiI-Ac-LDL-double negative cells were cultured in appropriate differentiation culture medium. These cells differentiated into an osteoblast lineage, as shown by alkaline phosphatase staining (Fig. 2Ad) and Alizarin red staining (Fig. 2Ae), and also into an adipogenic lineage, as shown by oil red-O staining (Fig. 2Af). After growth in serum-free chondrogenic differentiation medium for 4 weeks, the cells were harvested and their chondrogenic potential evaluated. We isolated mRNA from the cell pellet and determined by RT-PCR that type II collagen was expressed (data not shown). The pellet was also positive for Toluidine blue staining (Fig. 2Ah). Taken together, these results indicate that a subset of the CD45/DiI-Ac-LDL-double negative cells derived from UCB exhibits properties of MSCs.
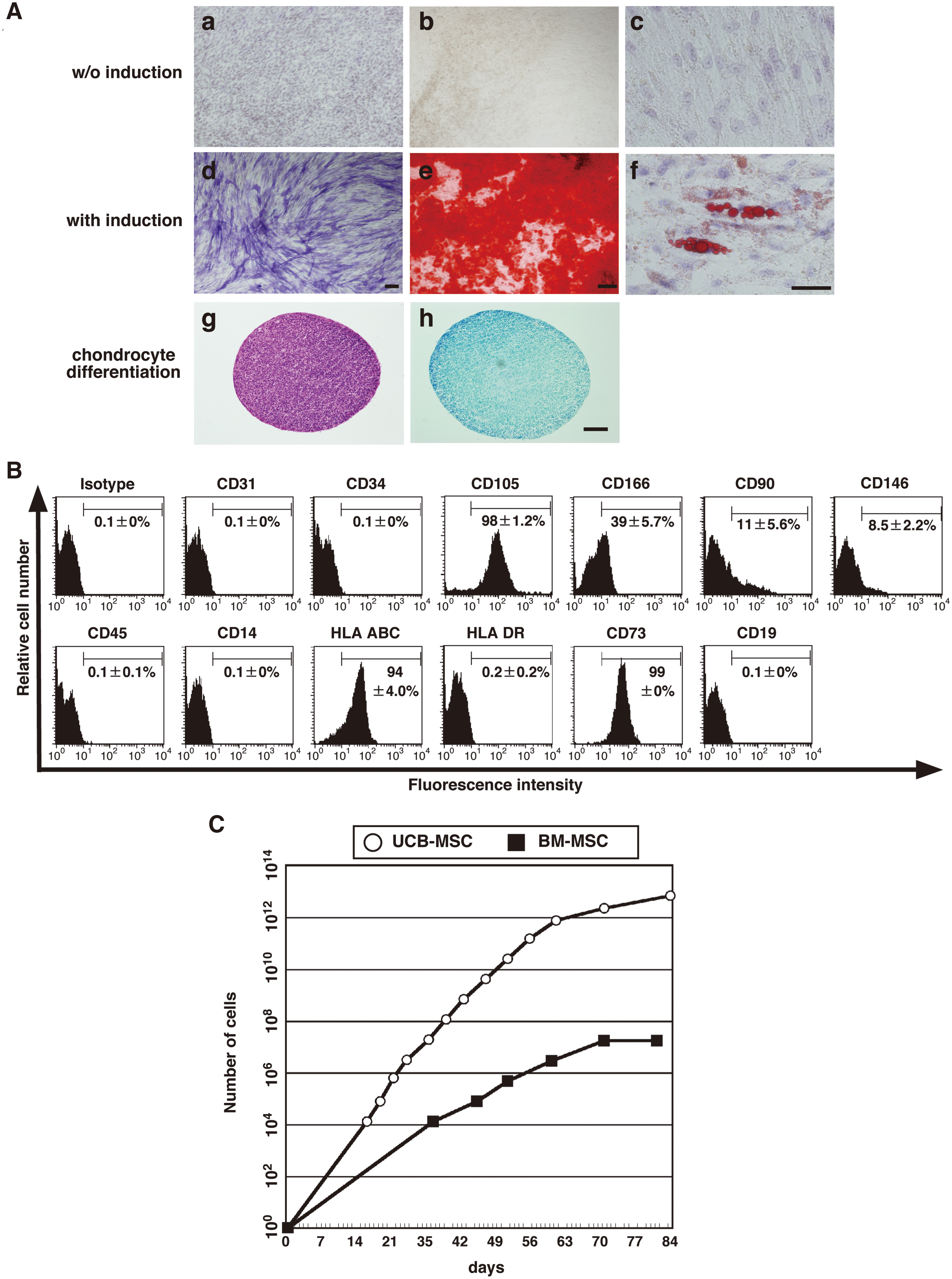
Examination of the differentiation and proliferation of CD45/DiI-Ac-LDL-double negative cells. (
We further examined cell surface markers of the CD45/DiI-Ac-LDL-double negative cells. The cells were positive for CD105 (SH-2), CD166, HLA-ABC, and CD73 (SH-3 and SH-4) and negative for EC-specific markers (CD31 and CD34), hematopoietic cell-specific markers (CD45, CD14, and CD19), and HLA-DR (Fig. 2B). The expression of these markers is consistent with previous reports for UCB-[23] and BM-[12] derived MSCs. Dominichi et al. reported that over 95% of human MSCs are positive for CD90 [13]. However, it is still controversial whether CD90 can be a specific marker for human MSCs [23,28]. In this study, only 11.0% ± 5.6% of the CD45/DiI-Ac-LDL-double negative cells expressed CD90, consistent with the finding for UCB-derived MSCs in the literature [23]. CD146 is also suggested as a specific marker to isolate MSCs from BM and umbilical cord tissue [29,30]. CD146 is expressed at a high level in MSCs originating from perivascular cell populations in multiple tissues [5]. Most of the UCB-derived cells in this study were negative for CD146 expression, suggesting a minor contribution of perivascular cells in the CD45/DiI-Ac-LDL-double negative cell population.
We compared the proliferation of UCB-derived MSCs with that of adult BM-derived MSCs through serial passage (Fig. 2C). UCB-derived MSCs continued to proliferate for more than 80 days, while the growth of adult BM-derived MSCs ceased by 70 days. At 84 days of culturing, the total number of UCB-derived MSCs was 106-fold greater than that of BM-derived MSCs. These results are consistent with previous data [31].
Collectively, CD45/DiI-Ac-LDL-double negative cells from UCB were characterized as MSCs by their multilineage differentiation and cell surface marker expression profile and found to be more highly proliferating than BM-derived MSCs.
Separation of MSCs according to their ALDH activity
Previously, we found that UCB-derived EPCs with a low ALDH activity possessed a greater ability to proliferate and migrate compared with those with a high ALDH activity [21]. We therefore assessed if the expression level of ALDH can be used as a marker for segregating MSCs with distinct functional properties.
We divided UCB-derived MSCs into Alde-negative (R1), Alde-Low (R2), and Alde-High (R3) fractions according to the expression level of ALDH (right panel, Fig. 3A). Alde-High and Alde-Low cells were found at frequencies of 1.8% + 1.7% and 12.8% + 7.9%, respectively (N = 4). As expected, ALDH activity was significantly blunted in the presence of DEAB, a specific ALDH inhibitor (middle panel, Fig. 3A). The Alde-Low and Alde-High fractions were found in the DEAB-treated cells after several passages, negating the assumption that variability in ALDH activity is observed only transiently in a limited culture period (Supplementary Fig. S2, available online at
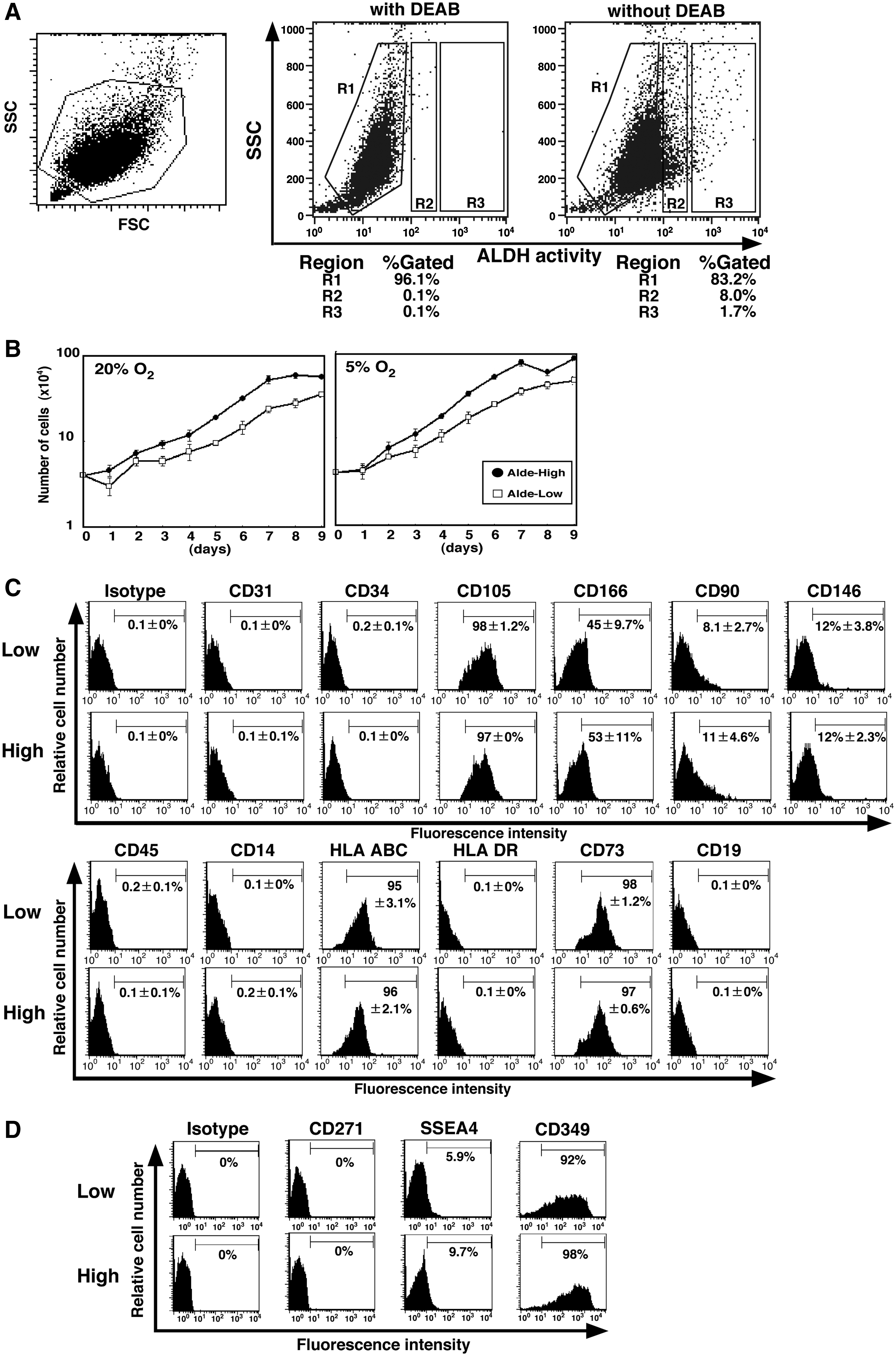
Isolation of Alde-High and Alde-Low MSCs. (
Unlike EPCs, Alde-High MSCs had a shorter doubling time than Alde-Low MSCs under normoxic conditions (38.5 + 1.3 vs. 47.3 + 0.9 h; N = 3, P < 0.01) (Fig. 3B). Both proliferated faster when exposed to hypoxia (Alde-High, 35.0 + 1.5 h; Alde-Low, 42.4 + 2.4 h; N = 3, P < 0.01). Despite their rapid proliferation in a liquid culture condition, neither Alde-Low nor Alde-High MSCs formed colonies (data not shown). Given that these cells showed clonogenic growth at the beginning of culture (Fig. 1A), we assume that their colony forming activity might be lost during several passages.
The expression profile of cell surface markers in Alde-Low and Alde-High MSCs (Fig. 3C) was similar to that of parental cells (Fig. 2B). There was no significant difference in the expression of these markers between the two MSC populations.
Recently, it was reported that SSEA-4, CD271, and CD349 (frizzled-9) are employed as specific MSC markers for BM and placenta [32 –34]. SSEA-4 and CD271 have been used to isolate MSCs from BM [32,33], and CD349 has been used to isolate MSCs from placenta [34]. We examined whether the expression of these markers correlate with ALDH activity in UCB-derived MSCs (Fig. 3D). Both Alde-Low and Alde-High MSCs were positive for CD349 but negative for SSEA-4 and CD271. Importantly, there was no significant difference between Alde-Low and Alde-High MSCs in the expression of these markers. Taken together, these results suggest that the expression of MSC markers might be partially different among BM, placenta, and UCB.
In vitro analysis of MSC differentiation
We examined the potential of Alde-Low and Alde-High MSCs to differentiate into the 3 mesenchymal cell lineages. TDifferentiation into an osteogenic lineage was determined by Alizarin red staining, and the efficiency of this differentiation was quantified using an absorbance meter (Fig. 4A). While both populations of MSCs were able to differentiate into an osteogenic lineage, Alde-High MSCs did so more efficiently (Alde-High: 0.68 + 0.03; Alde-Low: 0.49 + 0.02; N = 4, P < 0.01).
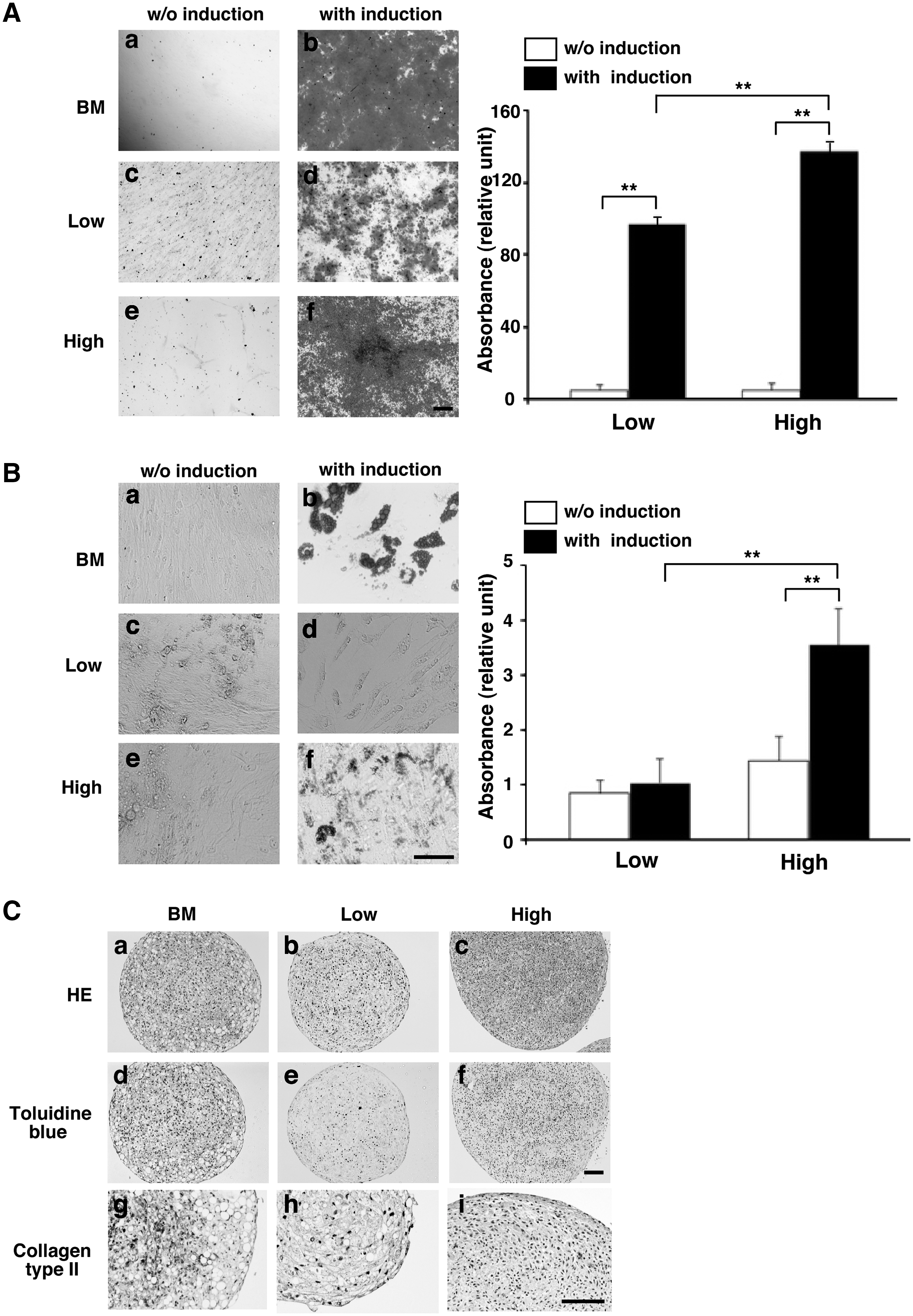
Analysis of Alde-High and Alde-Low MSC differentiation. (
The adipogenic potential of the 2 MSC populations was quantified by absorbance following oil red-O staining (Fig. 4B). As in osteogenic differentiation, Alde-High MSCs showed greater ability to differentiate into an adipogenic lineage (Alde-High: 0.07 + 0.01; Alde-Low: 0.02 + 0.01; N = 4, P < 0.01). Importantly, Alde-Low MSCs could differentiate into adipocytes to a similar extent as Alde-High MSCs after a long-term culture for adipogenic differentiation (38 days and 45 days of culture, Supplementary Fig. S3, available online at
The chondrogenic differentiation of the cells was assessed by Toluidine blue staining and anti-collagen type II immunostaining (Fig. 4C). Alde-Low MSC pellets were only faintly stained, while Alde-High MSCs were strongly stained. In summary, these data indicate that both Alde-High and Alde-Low MSCs can differentiate into the 3 mesenchymal cell lineages, although the differentiation potential of Alde-Low MSCs is not as significant as that of Alde-High MSCs in vitro.
Analysis of mRNA expression during MSC differentiation
We examined the expression of cell-type specific genes in MSCs induced to undergo osteogenic or adipogenic differentiation (Supplementary Fig. S4, available online at
After 7 or 14 days of osteogenic induction, mRNA was isolated and osteocyte-specific gene expression was tested by RT-PCR (Supplemental Fig. S4A). While the expression levels of osteopontin and osteocalcin increased following osteogenic induction, their expressions seemed slightly greater in Alde-High MSCs than in Alde-Low MSCs. The expression of Runx2 was undetectable prior to osteogenic induction (day 0), but could be detected after 7 and 14 days of induction. The expression level of collagen type I was stable during differentiation.
After adipogenic induction, the mRNA expression of Apolipoprotein C-II (Apo C2), a component of very low density lipoprotein [35], was high in Alde-High MSCs but undetectable in Alde-Low MSCs (Supplemental Fig. S4B). The mRNA expression of peroxisome proliferator-activated receptor γ (PPARγ) was greater in Alde-High compared with Alde-Low MSCs on day 14 after induction. However, the mRNA expression of lipoprotein lipase (LPL) did not differ significantly between the 2 cell populations.
We next examined by real time PCR the molecular markers for undifferentiated cells that are expressed in embryonic stem cells [36 –38]. The immature stem cell markers Oct-4, NANOG, SOX2, and Rex1 were expressed to similar extents in Alde-Low and Alde-High MSCs (Supplemental Fig. S4C).
Analysis of the ability of MSCs to repair bone fractures
Our cell culture experiments showed that Alde-High MSCs have a greater ability to proliferate and differentiate than Alde-Low MSCs. We tested whether transplanting MSCs into leg fractures would facilitate new bone formation (Fig. 5).
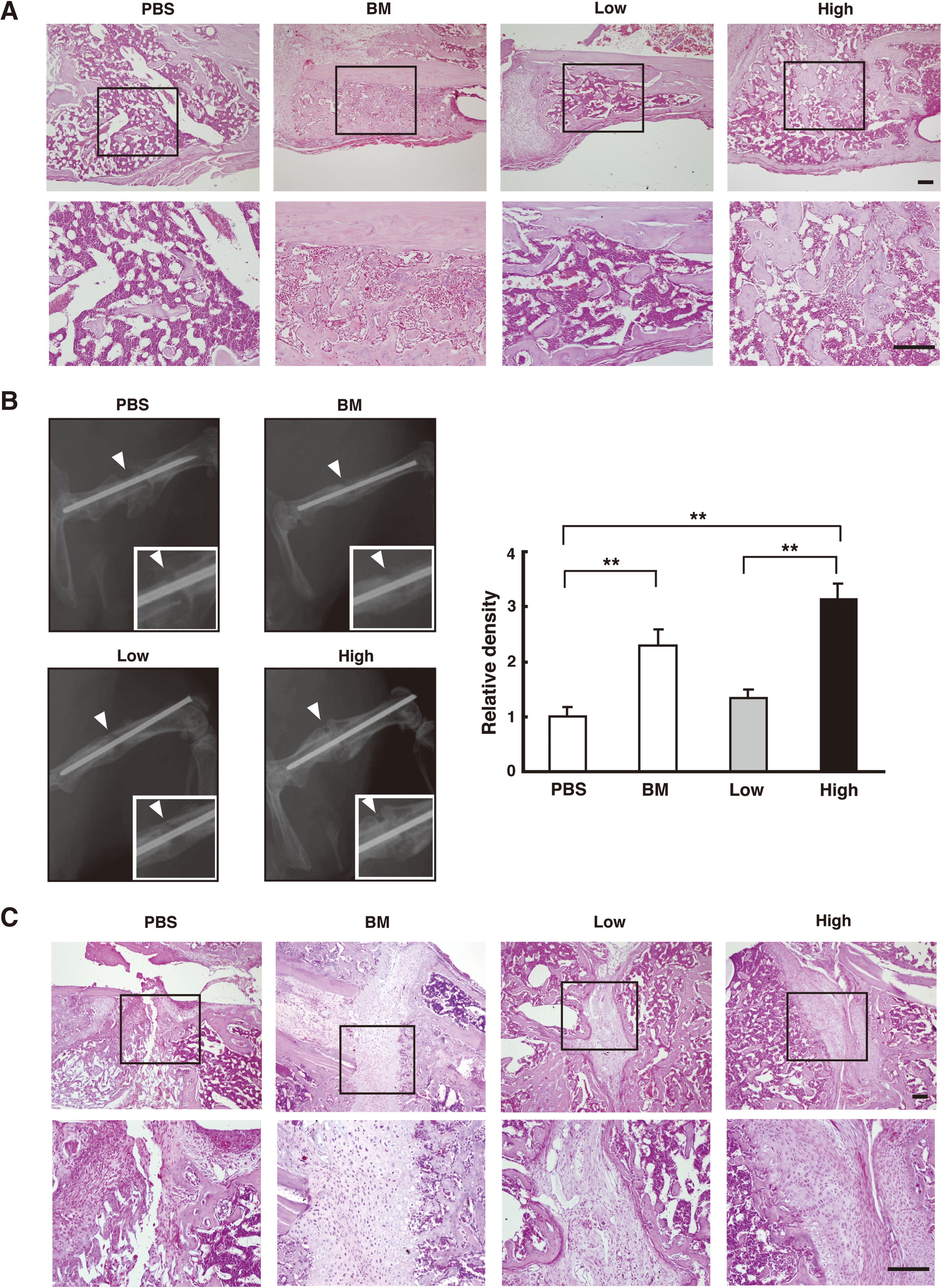
Repair of bone fractures by MSC transplantation. Alde-Low MSCs (Low), Alde-High MSCs (High), BM-derived MSCs (BM), or no cells (PBS) were transplanted into mice at the bone fracture sites using Gelform (collagen-based gelatin sponge) as a carrier vehicle. The morphological structure (HE staining;
The transplanted regions at the bone fracture sites were covered by mononuclear cells including inflammatory blood cells and osteocytes, which were easily distinguishable by their morphologies (hematoxylin-eosin staining; Fig. 5A). Osteogenesis was significantly improved at the sites transplanted with whole BM cells or ALDH-High MSCs, whereas inflammatory blood cells were largely observed at the sites with no cell (PBS) transplant or an Alde-Low MSC transplant. These experiments were performed using immunocompetent C57/BL6 mice treated with cyclosporin-A. To exclude the effects of cyclosporin-A on the functions of MSCs, the same experiments were done using BALB/c nu/nu mice not treated with cyclosporin-A (Supplementary Fig. S5, available online at
The degree of osteogenesis was also evaluated by X-ray images of bone calcification following cell transplantation at the fractured sites (Fig. 5B). Alde-High MSCs were found to induce osteocalcification more rapidly than Alde-Low MSCs.
Further, histological analysis revealed that lamellar formation was prominent where BM cells or Alde-High MSCs had been transplanted into the joints (Fig. 5C). Lamellar formation on preexisting hyaline cartilage represents bone substitution during the repair process. In contrast, woven bone with fibroblastic cells was observed at the joints injected with PBS or Alde-Low MSCs. Although more fibroblastic cells filled the joints after the introduction of Alde-Low MSC transplants than with PBS alone, osteogenic differentiation was obviously delayed compared with that in the BM or Alde-High MSC transplants. These data demonstrate that transplantation of Alde-High MSCs facilitates bone substitution and repair in our mouse model.
Immunohistochemistry with anti-human HLA-ABC antibody was used to examine the distribution of implanted Alde-High MSCs in our mouse model. A small number of HLA-ABC-positive cells was detected on the surfaces of osteoid cells. However, most of the newly formed osteoid cells at the joints were negative for the HLA-ABC antigen (data not shown).
Analysis of the response of MSCs to hypoxia
We previously demonstrated that UCB-derived Alde-Low EPCs are more sensitive to hypoxic stimuli than Alde-High EPCs [21]. Under physiological conditions, MSCs in the body exist at a lower oxygen tension than in ex vivo culture (20% O2) [39,40], and their differentiation is markedly impaired under hypoxic conditions [41,42].
Western blot analysis demonstrated that the protein level of HIF-1α was greater in Alde-High MSCs than in Alde-Low MSCs under hypoxic conditions (Fig. 6A). HIF-2α was barely detectable in either cell fraction, whereas HIF-3α levels were comparable in both. These results indicate that HIF-1α plays a major role in the response to hypoxia in UCB-derived MSCs. Since its expression level was greater in Alde-High MSCs, genes targeting HIF-1α may contribute to the greater ability of Alde-High MSCs to differentiate and proliferate.
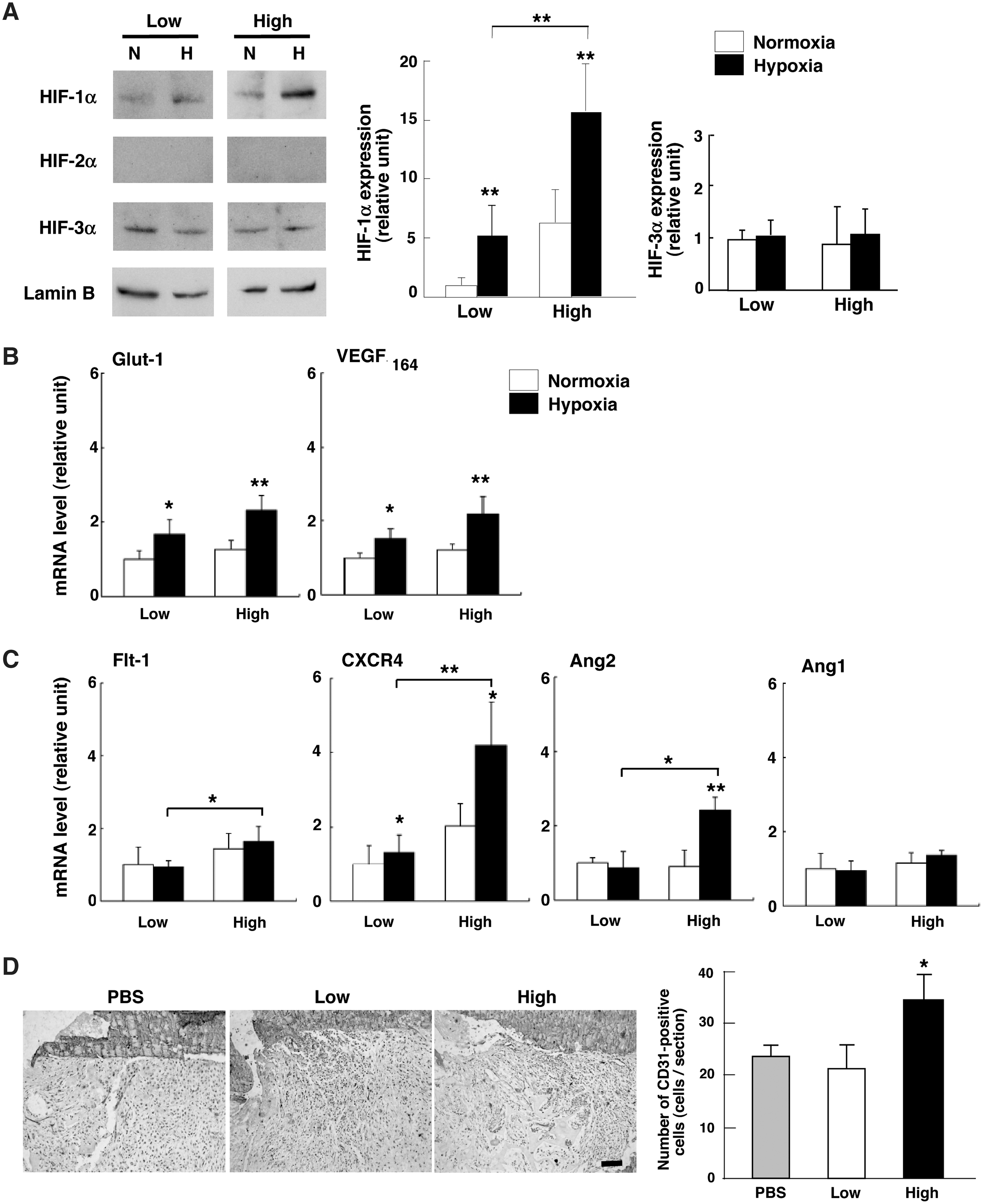
Analysis of the response of Alde-High and Alde-Low MSCs to hypoxia. (
We therefore evaluated the expressions of hypoxia-responsive genes in MSCs cultured under hypoxic conditions. The expressions of Glut-1 and VEGF mRNAs, which are known target genes of HIF-1α (Fig. 6B), were upregulated in both Alde-Low and Alde-High MSCs. Despite the expression level of HIF-1α being higher in Alde-High MSCs, Glut-1 and VEGF mRNA expressions were similar in both MSC populations.
In the process of tissue repair following damage from ischemia, both EPCs and MSCs are mobilized in peripheral blood and recruited to the ischemic region [43]. MSCs can also enhance angiogenesis by secreting angiogenic factors in a paracrine manner [44,45]. VEGFR1 (Flt-1) and CXCR4 are associated with migration in response to hypoxic stimuli, and their mRNA expression levels were greater in Alde-High MSCs than in Alde-Low MSCs under hypoxic conditions (Fig. 6C). Angiopoietin-2 (Ang2) mRNA expression was significantly upregulated by hypoxia only in Alde-High MSCs. Conversely, Angiopoietin-1 (Ang1) mRNA expression was not induced by hypoxia in either Alde-High or Alde-Low MSCs (Fig. 6C).
Angiogenesis is more likely supported by the more hypoxic responsive Alde-High MSCs, and the bone repair seen in their presence might have transpired through angiogenic factors. In fact, CD31-positive endothelial cells were more predominant at the fractured sites transplanted with Alde-High MSCs than with PBS or Alde-Low MSCs (Fig. 6D).
Discussion
In this study, we demonstrated that MSCs derived from UCB have the potential to differentiate into osteogenic, adipogenic, and chondrogenic cell lineages and can be divided into 2 populations based on ALDH activity. We showed using our mouse transplantation model that Alde-High MSCs, rather than Alde-Low MSCs, are capable of repairing bone fractures. Alde-High MSCs are also more sensitive to hypoxic stimuli than Alde-Low MSCs. These findings strongly suggest that MSCs enabling tissue repair are a superior therapy to effect fast recovery of patients or when unfractionated MSC transplantation is not effective.
Current studies have revealed that MSCs are multipotential in their differentiation into various lineages, which include not only mesenchymal but also other lineages, affirming MSCs as very useful adult-derived stem cells for the repair of damaged tissues [9]. However, there are no decisive cell surface markers for isolating MSCs specifically, and methods to determine functionally active MSCs have not been established. MSCs are recognized as adherent cells in culture, along with fibroblasts, endothelial cells, and macrophages, albeit at a very low frequency (approximately one-fifth that of UCB-derived EPCs). For these reasons, it takes more than a few weeks to obtain an established population of MSCs. Therefore, establishing a prospective purification method for MSCs would prove very meaningful to developing clinical cell therapy. We examined the expressions of markers in UCB-derived MSCs by FACS and found no significant differences between Alde-Low and Alde-High MSCs in this regard (Fig. 3D). Thus, while these markers are tissue-specific markers for the isolation of MSCs, they may not be suitable for the isolation of MSCs from UCB.
Recently, we isolated 2 populations of EPCs from UCB according to their ALDH activity (Alde-Low and Alde-High) and examined their functions [21]. Alde-Low and Alde-High EPCs differ significantly in their ability to proliferate and respond to hypoxia in vitro and in vivo. Further, Alde-Low EPCs have a higher rate of proliferation than Alde-High EPCs, unlike HSCs [18].
Although a previous study reported that MSCs can be isolated from human BM on the basis of ALDH activity simultaneously with the isolation of hematopoietic and endothelial progenitors [46], the characteristic difference between Alde-High and Alde-Low MSCs had not been fully determined. Recently, Capoccia et al. reported that human BM cells with high ALDH activity (Aldehigh cells) represent a progenitor-enriched population of several cell lineages including multipotent hematopoietic progenitors and mesencymal stromal cells [24]. They showed that Aldehigh cells possess greater activity in forming both hematopoietic and mesenchymal colonies and a higher differentiation activity toward osteocytes, adipocytes, and chondrocytes in vitro compared with cells with low ALDH activity. These data are consistent with our findings in the present study that Alde-High MSCs are superior to Alde-Low MSCs in both cell growth and differentiation (Figs. 3B and 4). Collectively, these data suggest that Alde-High MSCs might be in an immature state compared with Alde-Low MSCs both in BM and UCB. Whereas Aldehigh cells from BM showed high colony forming activity, both Alde-High and Alde-Low MSCs in our study proliferated without forming colonies even under the conditions for clonogenic assay described by Capoccia et al. [24]. Given that these cells showed clonogenic growth in the beginning of culture (Fig. 1A), we assume that the colony forming activity might be lost during several passages of MSCs. Further studies are required to determine the exact status of maturation in these MSC populations. BM is an attractive source of MSCs for autologous transplantation in clinical cell therapies. However, we found that BM-derived MSCs seemed to lose their growth activity after a couple of passages, whereas UCB-derived MSCs continued to grow rapidly (Fig. 2C). Thus, UCB-derived MSCs might have an advantage over BM-derived MSCs in providing a number of cells in a short period of culture for clinical application. Thus, the present study shows that separation of active progenitor cells based on ALDH activity would be a powerful tool not only for autologous transplantation but also for allogenic transplantation in clinical cell therapy.
The transplantation of MSCs is effective in the repair of injured tissue [47], which suggests that the migratory activity of MSCs is also important for tissue regeneration. Rosová et al. reported that preculturing MSCs in hypoxic conditions increased their potential to migrate and regenerate tissue [40]. Further, Okuyama et al. stated that VEGFR1 expression was upregulated by HIF-1α after culturing MSCs in hypoxic conditions [48]. While both Alde-Low and Alde-High MSCs were responsive to hypoxic culture conditions, the expression of genes such as Flt-1 and CXCR4 associated with cell migration was greater in Alde-High MSCs than in Alde-Low MSCs (Fig. 6). This might explain the earlier recovery of fractured bone when transplanted with Alde-High compared with Alde-Low MSC transplants (Fig. 4).
In addition, neovascularization is crucial for restoring injured tissue, and MSCs are known to be involved in the formation of new vessels [49]. Since transplanted MSCs were found in close proximity to newly formed vessels, MSCs may have a paracrine effect in forming new vessels. Under hypoxic conditions, VEGF mRNA expression was upregulated to similar extents in Alde-Low and Alde-High MSCs (Fig. 6B). In contrast, Ang2 was highly expressed in Alde-High MSCs exposed to hypoxia compared with Alde-Low MSCs. Ang2 stimulates the dissociation of pericytes from endothelial cells, thereby inducing the reorganization of vessel formations [50]. Ang2 also plays a role in endothelial cell migration, proliferation, and sprouting in the presence of VEGF [51]. Based on these, Alde-High MSCs may have a greater tendency to effect remodeling of preexisting vasculature at the injured site and support new vessel formation compared with Alde-Low MSCs (Fig. 6D).
In summary, our data demonstrate that the isolation of MSCs high in ALDH activity from UCB provides an efficient and reproducible method for obtaining MSCs with a high potential for proliferation. Cell therapy using these cells may be a promising means by which to accelerate the repair of damaged tissue. Further investigation is necessary to completely understand the functional properties of MSCs following transplantation.
Footnotes
Acknowledgments
The authors thank Ms. Naomi Kaneko for excellent work on immunohistochemistry. The authors also thank Tania O'Connor for critical reading of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.