
Abstract
The cerebellum has critical roles in motor and sensory learning and motor coordination. Many cerebellum-related disorders indicate cell therapy as a possible treatment of neural loss. Here we show that application of inductive signals involved in early patterning of the cerebellar region followed by application of different factors directs human embryonic stem cell differentiation into cerebellar-like cells such as granule neurons, Purkinje cells, interneuron, and glial cells. Neurons derived using our protocol showed a T-shaped polarity phenotype and express similar markers to the developed human cerebellum. Electrophysiological measurements confirmed functional electrical properties compatible with these cells. In vivo implantation of differentiated human embryonic stem cells transfected with MATH1-GFP construct into neonatal mice resulted in cell migration across the molecular and the Purkinje cell layers and settlement in the internal molecular layers. Our findings demonstrate that the universal mechanisms involved in the development of cerebellum can be efficiently recapitulated in vitro, which enables the design of new strategies for cell replacement therapy, to study early human development and pathogenesis of neurodegenerative diseases.
Introduction
T
Methods
Cell culture
Primary hESC colonies (H1 or H9 lines; WiCell) were mechanically dispersed into several small clumps, which were cultured on fresh commercially available human foreskin fibroblasts (American Type Culture Collection) inactivated by mitomycin C in hECS medium containing knockout-Dulbecco's modified Eagle's medium (DMEM; Invitrogen), 100 μM β-mercaptoethanol (Sigma), 1 mM
Differentiation toward cerebellar neural cells
To initiate differentiation and to form embryoid bodies (EBs), undifferentiated hESC colonies (Stage I) were detached and transferred to low-attachment plates (Costar). The EBs were formed after 1 day and maintained in hESC medium for 4 days (Stage II). The medium was changed every day. After this period the EBs were transferred to neural induction medium consisting of DMEM/F12 with glutamax (Gibco), N2 supplement (Gibco), and heparin (2 μg/mL). Human FGF8 (100 ng/mL) and retinoic acid (RA, 10 μM) (Sigma) were added and the EBs were cultured for an additional 7 days (Stage III). To induce further differentiation toward cerebellar neurons, the EBs were transferred to plates previously coated with laminin (2 μg/mL) and fibronectin (5 μg/mL) (Sigma) and further cultured in basal medium eagle (BME) medium (Invitrogen) supplemented with insulin, transferrin, selenite (ITS) (Gibco), human FGF8 (100 ng/mL), human FGF4 (100 ng/mL), and human bFGF (20 ng/mL) (Invitrogen) for 4 days. Then, the cells were cultured for an additional 5 days in DMEM/F12 medium supplemented with human FGF8 (100 ng/mL), human wingless-related MMTV integration site 1 (WNT1) (50 ng/mL), and WNT3A (50 ng/mL) (Stage IV). To promote further differentiation, we cultured the cells in BME medium supplemented with N2, B27 (Invitrogen), human BMP4 (50 ng/mL), human BMP6 (20 ng/mL), human BMP7 (100 ng/mL), and human growth differentiation factor 7 (GDF7) (100 ng/mL) for additional 8 days (Stage V). To induce axon growth and proliferation (Stage VI), the cells were then disaggregated, replated, and cultured for another 8 days in BME medium supplemented with N2 and B27, to which the following factors were added: human BMP4 (50 ng/mL), human BMP6 (20 ng/mL), human BMP7 (100 ng/mL), human GDF7 (100 ng/mL), human Sonic hedgehog (100 ng/mL), human neurotrophin 3 (NT3) (100 ng/mL), jagged 1 (JAG1) (20 ng/mL), human brain derived neurotrophic factor (BDNF) (100 ng/mL). The human growth factors FGF8, BDNF, BMP7, and WNT1 were purchased from Peprotech, and factors BMP4, BMP6, SHH, Wnt3a, JAG1, NT3, and FGF4 from R&D Systems. Figure 1 summarizes the differentiation/experimental protocol. Eight independent experiments were performed for each described method.
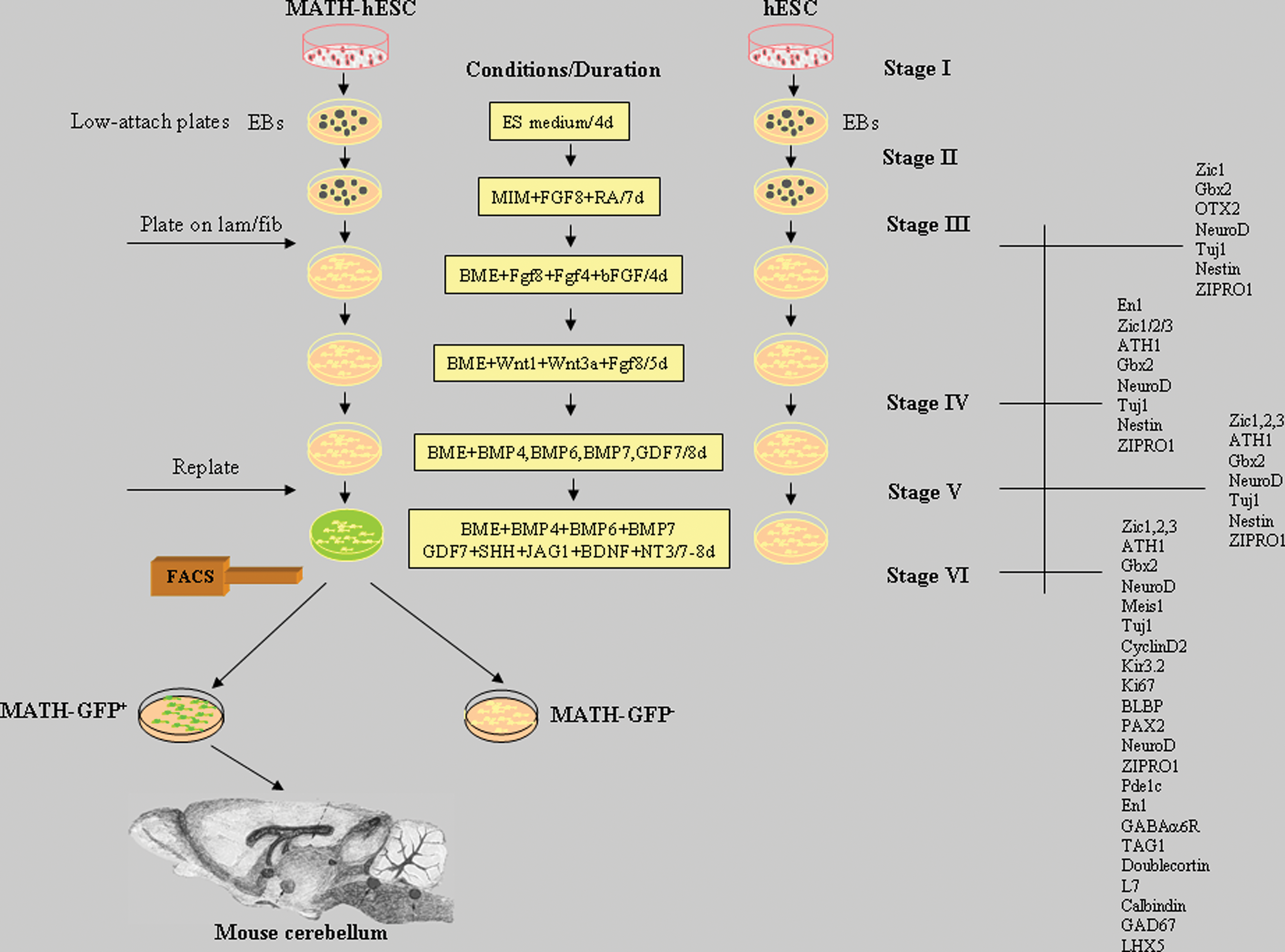
Overview of the experimental schedule and the protocol of differentiation of hESCs toward cerebellar cells. hESCs or MATH1-hESCs were cultured in medium supplemented with soluble factors as described in the Methods section. The cellular marker genes expressed by differentiated hESCs detected by immunofluorescence and real time (RT)–polymerase chain reaction are indicated for each stage on the right side of the panel. After the Stage VI, cell sorting (FACS) was applied using differentiated MATH1-hESC line. The sorted cells were analyzed and transplanted to the developing mouse cerebellum. hESC, human embryonic stem cell; EBs, embryonic bodies. Color images available online at
RNA extraction and reverse transcription–polymerase chain reaction analysis
Total RNA was extracted using High Pure RNA Isolation Kit according to manufacturer's instructions (Roche Diagnostics). cDNA was synthesized using High Capacity cDNA Archive Kit (Applied Biosystems). Amplification was performed on the cDNA using Taq polymerase (Invitrogen). Polymerase chain reaction (PCR) conditions included a first step of 3 min at 94°C, a second step of 35 cycles of 15 s at 94°C, a 30 s annealing step at 56°C, 45 s at 72°C, and a final step of 8 min at 72°C. Glyceraldehyde-3-phosphate dehydrogenase was used as a control gene to evaluate and compare the quality and quantity of different cDNA transcripts. Primer sequences will be provided upon request. As a positive control for developing brain and cerebellum, we used human total RNA Master Panel II (Clontech), from which we used human fetal brain total RNA pooled from 21 male/female Caucasian fetuses obtained from autopsy materials (ages 26–40 weeks), human whole brain total RNA pooled from 2 male Caucasians (ages 47–55 years), and human cerebellum total RNA (Clontech) pooled from 10 male/female Caucasians (ages 22–68 years).
Immunocytochemistry
At different stages of differentiation, the cells were washed in phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS for 10 min. Fixed cells were washed twice with PBS before staining. After permeabilization in 2% Triton and blocking in 1% bovine serum albumin for 30 min, primary antibodies were applied in blocking buffer for 2 h at room temperature. The cells were washed 3 times in blocking buffer before secondary antibody application. Secondary antibodies of goat anti-mouse Alexa conjugated, goat anti-rabbit Alexa conjugated (Molecular Probes) were diluted at 1:500 in blocking buffer and applied to cells for 1 h at room temperature. After 2 washes in PBS, mounting medium containing 4',6'-diamidino-2-phenylindole was applied for nuclear staining. Cells were observed under the fluorescence microscope 5 (Axiowert). For negative controls, primary antibodies were omitted and the same staining procedure was followed. Also, nonrelevant antibodies that have the same immunoglobulin isotope as specific antibodies were used and no positive reaction was found. The following primary antibodies and dilutions were used: rabbit anti-Nestin (1:250; Millipore), rabbit anti-ATH1 (1:200; Millipore), mouse anti-beta III tubulin-Tuj1 (1:2000; Abcam), rabbit anti-Zic1 (1:200; Abcam), rabbit anti-Zic2 (1:200; Millipore), rabbit anti-Zic3 (1:200; Millipore), rabbit anti-Gbx2 (1:200; Millipore), rabbit anti-pax6 (1:100; Millipore), rabbit anti-NeuroD (1:200; Millipore), rabbit anti-ZIPRO1 (RU49) (1:250; Applied Biosystem), anti-pcp2 (1:100; Abgent), rabbit anti-Kir3.2 [G protein-gated K+ current (GIRK)] (1:100; Sigma), mouse Calbindin28K (1:100; Sigma), rabbit anti-pax2 (1:100; Invitrogen), rabbit anti-En1 (1:100; Millipore), rabbit anti-otx2 (1:250; Millipore), rabbit anti-PDE1C (1:200; Abcam), rabbit anti-BLBP (1:100; Abcam), rabbit anti–γ-aminobutyric acid 6αR (anti-GABA6αR) (1:200; Chemicon, Millipore), mouse anti-GAD67 (1:200; Sigma), rabbit anti-LHX5 (1:200; Chemicon, Millipore), rabbit anti-Ki67 (1:200; Abcam), mouse anti-Meis1 (1:250; Upstate), rabbit anti-GFP (1:500; Abcam), mouse anti-human nuclei (1:100; Millipore), rabbit anti-doublecortin (1:100; Millipore). The cytochemistry experiments were done at least 5 times with each hESC line. For image analysis, 8 microscopic fields from each samples were taken randomly using × 20 objective. Two independent investigators counted the number of immunoreactive cells for each staining. The percentage of immunopositive cells was calculated dividing the number of specific cells by total cell number (4′,6-diamidino-2-phenylindole–stained cell nuclei).
Generation of MATH1-GFP hESCs
The MATH1-GFP transgenic hESCs were generated by introducing the Math1-enhancer–Venus-Neo/pBluescript SK+ plasmid [11] into hESCs using Fugene. The expression of Venus (GFP) is driven by human β-promotor and mouse Math1 enhancer (AF218258), which has 75% homology with the human sequence of Math1 enhancer (AF 218259). Briefly, the undifferentiated hESC colonies were maintained on Matrigel (Sigma) in human fibroblast-conditioned medium for 3 passages. The cell colonies were transfected with 10 μg of plasmid. After transfection, the cells carrying the plasmid were selected with 200 μg/mL of Gentamicin (Sigma). Strong Venus expression was detected upon differentiation (Fig. 5). Three independent MATH1-hESC lines were generated and differentiated and Math1 induction efficiency was similar in all 3 lines.
Electrophysiology
All reagents were from Sigma, unless otherwise stated. For current recordings, an EPC-10 amplifier was employed (HEKA GmbH). The holding potential (V h) was −70 mV in all cases. The bath solution consisted of the following components (in mM): 140 NaCl, 4 KCl, 2 CaCl2, 10 HEPES, 5 glucose, 20 mannitol (pH 7.4). The electrode solution contained the following components (in mM): 144 KCl, 2MgCl2, 10 HEPES, 5 EGTA (pH 7.2). The pipette resistance was 1.5–2 MΩ. Series resistance was compensated for >70% and leak subtraction was performed. Data were low pass-filtered at 3.3 kHz and sampled at 10 kHz. Applied pulse protocols and analysis were carried out by Pulse software (HEKA). Action potentials were evoked by applying a variable pulse of current (40–60 pA) to neurons in current-clamp configuration. In the voltage clamp configuration, squared pulse depolarization voltage steps were applied to −10 mV or to +60 mV from V h. Tetrodotoxin (TTX) (Tocris Laboratories) at 1 μM was used to block TTX-sensitive voltage-dependent Na+ channels. Replacement of K+ by Cs+ in the internal solution or external application of 2 mM 4-aminopyridine blocked outward currents. Receptor agonists: glutamate (Glu) at 100 μM, Glu at 100 μM plus glycine at 50 μM (Glu + Gly), GABA at 100 μM, or acetylcholine (Ach) at 100 μM were tested. The antagonists bicuculline (Tocris Laboratories), 6-cyano-7-nitroquinoxaline 6,2-amino-5-phosphonopentanoic acid (AP5), and α-tubocurarine were examined on neurotransmitter-mediated currents at the same concentration as the agonists. Data were averaged and represented as mean ± standard error of the mean.
In vivo transplantation
Highly concentrated cell suspension (∼10 × 105 in 1 μL) of differentiated and sorted MATH1-GFP+ cells was front-loaded into bevelled glass micropipettes (∼50 μm diameter) that were prefilled with mineral oil and L-15 medium. Micropipettes were connected to a microinjector mounted on a stereotactic apparatus specially adapted for neonatal mice. Three- to 4-day-old CD-1 mice (Charles River Laboratories) were anesthetized by exposure to ∼4°C until pedal reflex was abolished. Anesthesia was maintained by performing surgery on a cold aluminium plate. A total of 10 × 104 cells per mouse in a 100–200 nL volume were injected at the following coordinates from lambda: cerebellum [0.5 mm anterior (A), 0.5 mm lateral (L), 1.0 mm dorsal (D)]. As a control, we injected an equivalent volume of heat-shocked “dead” cells. Grafted pups were returned to their mothers and analyzed after 4 weeks. After this period of time, the pups were deeply anesthetized (Nembutal; Abbot Laboratories) and perfused with 4% paraformaldehyde. We processed the tissue and cut 14-μm cryostat sections, which were immunostained with anti-EGFP antibodies and observed under the confocal microscope. All experimental animals were treated in accordance with the institute's Bioethical Committee.
Results
Differentiation of hESCs
To induce in vitro differentiation, hESC colonies were detached from a feeder layer and cultured in suspension as EBs for 4 days in the hESC growth medium (Fig. 1) following an additional 7 days in the motor induction medium (MIM) medium supplemented with RA and FGF8. In the normal development, the latter induces formation of an ectopic isthmic organizer and isthmocerebellar development via coexpression of OTX2 and GBX2 [23], 2 markers strongly expressed in Stage III (Fig. 2J). Also, after this period, real-time (RT)-PCR analysis showed that common neural markers including NCAM, PAX6, TuJ1, PAX2 (interneuron marker for midbrain and caudal central nervous system [CNS]), and HOXA2 (the marker responsible for delineation of r1, which gives rise to the cerebellum) were expressed (Fig. 2J). The expression of the same markers was found in human developing brain (Fig. 2J). The factors FGF8 and RA induce the expression of common granular cell markers ZIC1, ZIPRO (RU49), and WNT1 but not ATH1, suggesting that FGF8 and RA are sufficient to induce expression of genes that establish the cerebellar anlagen (Fig. 2J). Further positional restriction in the generation of GCP seems to depend on patterning signals well known for their crucial role in establishment of the cerebellar primordium [24]. The expression of these transcription factor genes is under the control of inductive signaling involving different FGF and WNT proteins produced by cells at the mesencephalic/metencephalic boundary. To further characterize the patterns of gene expression and cell movement, we transferred the EBs in an adhesive culture system and chemically defined culture medium containing FGF8, FGF4, and bFGF for 4 days followed by addition of WNT1 and WNT3a for another 5 days (Stage IV). Treatment with FGFs, WNT1, and WNT3a induced the expression of common hindbrain marker EN1 (Fig. 2A, J) as well as transcription factor ATH1, the rhombic lip GCP marker [25,26] (Fig. 2B, J). The function of this gene (together with ZIPRO [27]) is required for cerebellar GC generation and formation of the EGL [26]. Imunocytochemistry analysis after this differentiation stage confirmed the presence of the most important GCP markers. The cells were ATH1+ (46% ± 3%; Fig. 2B, I), NESTIN+ (84% ± 3%; Fig. 2C, I), NEUROD+ (62% ± 3%; Fig. 2D, I), ZIC1+ (75% ± 2%; Fig. 2E, H), ZIC2+ (72% ± 2%; Fig. 2F, I), ZIC3+ (74% ± 3%; Fig. 2G, I), and GBX2+ (65% ± 3%; Fig. 2H, I).

Immunostaining of differentiated hESCs after treatment with FGF8, WNT1, and WNT3A (Stage IV). Differentiated hESCs express different cerebellar markers including EN1
Several studies provide evidence that BMP-mediated inductive signals can promote further formation of granule neuron progenitors in vitro [10,20]. BMPs, with their dorsalizing effects [20,28], further continue the program of GC specification. We observed that treatment of cells only with BMP4 factor did not provide further differentiation of these cells as was the case with mouse ESCs [11] (data not shown). Addition of 4 members of the BMP family, BMP4, BMP6, BMP7, and GDF7, increases the expression of the main GPC markers. The expression of the most important cerebellar genes was maintained or increased and did not reveal the presence of other genes involved in further characterization of differentiated cerebellar cells (Fig. 2J). However, the expression of GBX2 and OTX2 was considerably decreased (Fig. 2J).
SHH signaling controls the development of cerebellum, induces the differentiation of Bergmann glia [29], and is required for the proliferation of GCPs. The GCP differentiation depends on ATH1, which acts by regulating the level of multiple components of the Notch signaling pathway [30]. To assess the mitogenic effects of SHH and Notch signaling in a further cerebellar phenotype, we cultured differentiating hESCs after Stage V in a medium supplemented with SHH and JAG1 in addition to the factors used in the previous step (Fig. 1). We included also BDNF and NT3, 2 neurotrophins involved in axonal growth of developing CG cells [31]. The outcome was a very rapid proliferation of differentiated cells, which is the most important characteristic of EGL cells in vivo after birth. The cell proliferation analysis revealed an important population of differentiated Ki67+cells (22% ± 1%; Fig. 3K, R). The RT-PCR analysis showed that the applied factors induce expression of CYCLIND2, which is expressed in neonatal GCPs in the EGL (Fig. 2J). Continued expression of specific GCP markers including ATH1, ZIC1-3, ZIPRO1, and PAX6 as well as PAX2 was also observed (Fig. 2J). The cells also expressed TAG1, an axonal glycoprotein that positively regulates, besides tangential migration of cortical GABAergic interneurons, a subset of precerebellar neurons in the caudal hindbrain [32]. GLI1 was expressed in Stage VI (Fig. 2J), confirming that the expression of the Shh target gene GLI1 is restricted to proliferating GCP and Bergmann glia [33]. Finally, the majority of differentiated cells were ATH1+ (39% ± 2%; Fig. 3A, B, R), MEIS1+ (53% ± 2%; Fig. 3B, R), ZIPRO1+ (74% ± 2%; Fig. 3C, R), ZIC1+ (73% ± 2%; Fig. 3D, R), ZIC2+ (76% ± 3%; Fig. 3E, R), ZIC3+ (77% ± 2%; Fig. 3F, R), NEUROD+ (76% ± 2%; Fig. 3G, R), PDE1C+ (49% ± 2%; Fig. 3H, R), PAX6+ (20% ± 2%; Fig. 3R), and GABAα6R+ (47% ± 1%; Fig. 3R). Cerebellar granule neurons normally express GIRK, which is involved in the neurotransmitter regulation of the excitatory input to the Purkinje fibers of the cerebellum [34], and DOUBLECORTIN, a marker for migrating GCs [35]. We observed that about 41% (44% ± 6%) of the cells were GIRK2+ (KIR3.2, Fig. 3I, R) and DOUBLECORTIN+ (Fig. 3J). An inhibitory neurotransmitter that can activate GIRK in CG neurons is GABA. About 36% (36% ± 4%) of cells were GABA+ (not shown) and about 61% (61% ± 3%) of differentiated cells were glutamatergic (Fig. 3L, R).
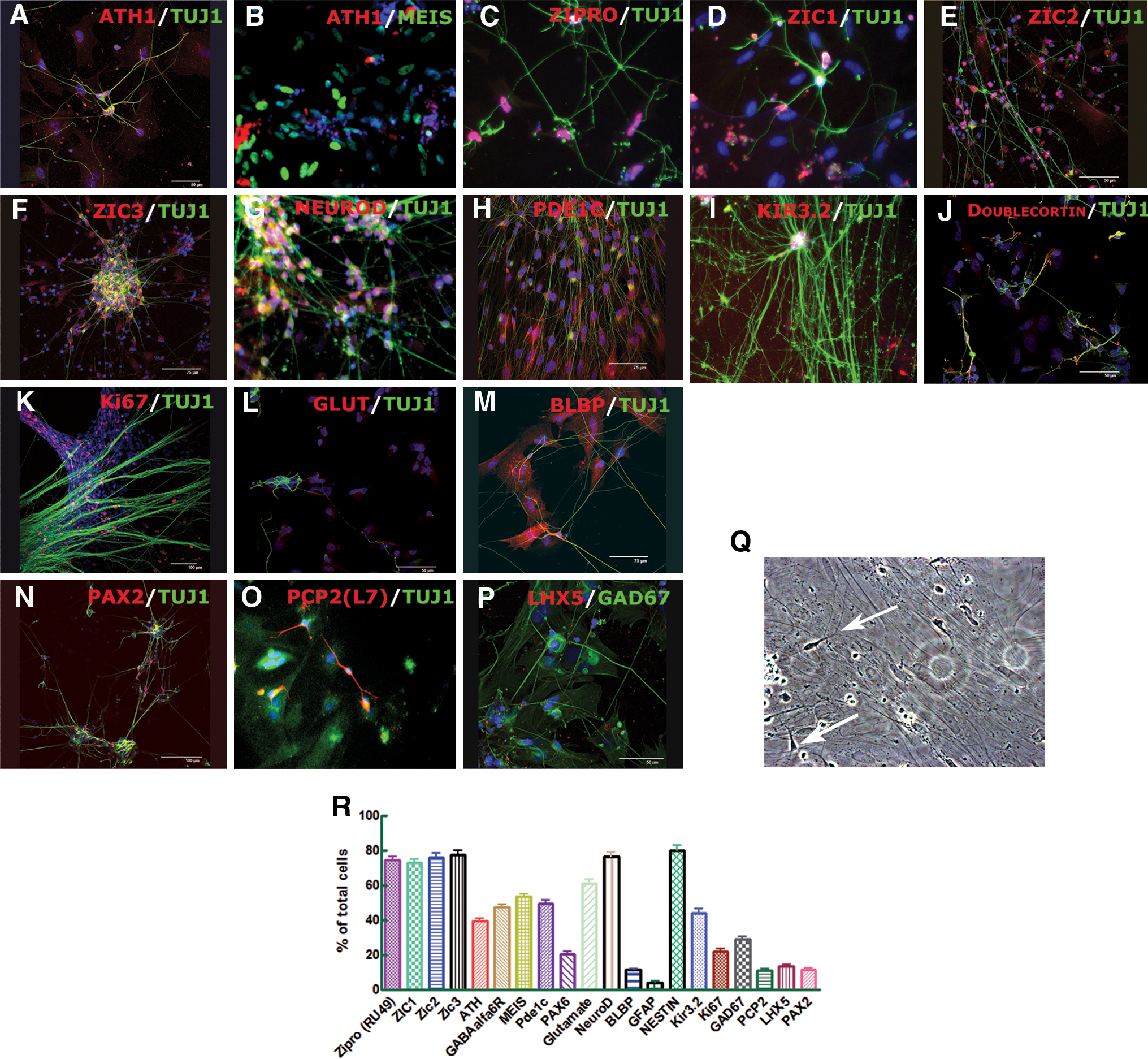
The immunocytochemical analysis of differentiated cells after 7–8 days of culture with BMP4, BMP6, BMP7, GDF7, SHH, JAG1, BDNF, and NT3 (Stage VI). Differentiated hESCs coexpressed the granule neuron marker ATH1, with TUJ1
A very exciting result was the formation of “T-shaped” axons characteristic for GC neurons [36] (Fig. 3Q), which suggests that cells, under our culture conditions, express genes and use signaling pathways required for the development of the polarity of wild-type CG neurons [37]. About 11% ± 1% of cells also expressed the Bergman cell marker BLBP (Figs. 2J and 3M) and interneuron marker PAX2 (Figs. 3N, R). Immunocytochemical assays showed very weak expression of the astroglial marker GFAP (4% ± 1%; Figs. 2J and 3R) but also the presence of Purkinje cells expressing PCP2 (L7; 11% ± 1%; Fig. 3O, R), GAD67 (29% ± 1%; Fig. 3P, R), and LHX5 (13% ± 1%; Fig. 3P, R), which is a considerably higher yield compared with other protocols using murine ESCs [11]. High expression of CALB1 (Fig. 2J) confirms the efficiency of our protocol to generate Purkinje cells. To confirm the regional specificity of generated cells, RT-PCR analysis showed that the cells did not express other CNS markers such as HB9 (spinal motoneurons), ASH1, NG2 (oligodendrocytes, not shown), and NKX2.2 (motoneurons) (Fig. 2J). We also observed continual decrease of main pluripotent markers (OCT4 and REX1) throughout the protocol. The specificity of the protocol was additionally confirmed by the absence of cells of endodermal and mesodermal trophoblast lineage (not shown). Usage of other differentiation methods, such as medium conditioned by rat GCP, induced generation of cells that displayed expression phenotypes characteristic of mostly GCP cells and interneurons; however, the presence of high OCT4 expression indicates that this treatment does not eliminate the undifferentiated cells (Supplementary Data and Supplementary Fig. S1, available online at
Electrophysiology
Cells visually scored as neurons, with a mean capacitance of 9.5 ± 4.2 pF (n = 89), were recorded by the patch-clamp whole-cell method to characterize differentiated cultures. Neurons analyzed by electrophysiology represent the whole neuronal population in the mixture of generated cells. When held in current clamp configuration at V h = −70 mV and a depolarizing current step of 40–60 pA for 150 ms was applied, virtually all cells (n = 80) showed 1 or more action potentials, and 30 of 80 cells showed 2 or more action potentials (Fig. 4A). The action potential spikes could be effectively and reversibly blocked by the application of the specific voltage-dependent Na+-channel blocker TTX at 1 μM (n = 6). Voltage-dependent Na+ currents were recorded in the voltage-clamp configuration upon a depolarizing voltage step of −10 mV (n = 80), which was readily blocked by TTX at 1 μM (n = 13). Voltage-dependent K+ currents (n = 80) were measured at +60 mV, and 66% ± 7% was blocked with 2 mM 4-aminopyridine (n = 5) (Fig. 4B).

We classified neuronal populations using neurotransmitter sensitivity to GABA, Glu, acetylcholine (Ach), and Glu plus glycine (Glu + Gly) (Fig. 4C–E). All the recorded neurons showed a marked response to GABA (n = 44), with a current density of 270 ± 155 pA/pF. In contrast, 34% of cells (10/29 cells) showed sensitivity to Glu + Gly, with a current density of 7.8 ± 6.4 pA/pF. Both N-methyl-D-aspartic acid (NMDA) and non-NMDA receptors contributed to the glutamatergic-induced currents because the NMDA receptor antagonist AP5 reduced the inward current generated by extracellular perfusion with Glu + Gly by ∼50% (from 8.4 ± 5.4 to 4.2 ± 4.2 pA/pF, n = 9), whereas combined application of AP5 with the non-N-methyl-D-aspartic acid (NMDA) receptor antagonist 6-cyano-7-nitroquinoxaline 6 blocked ∼71% of the current (from 10.9 ± 9.9 to 3.2 ± 2.2 pA/pF, n = 8). Interestingly, 48% (14/29 cells) contained nicotinic Ach receptors, which were readily blocked by the specific antagonist α-tubocurarine (n = 4).
MATH1-GFP construct
To monitor expression of a GCP-specific gene at each of the differential stages, we generated hESCs transfected with a GFP reporter gene expressed under control of the MATH1 enhancer. We sorted MATH1-GFP+ cells by fluorescence-activated cell sorting in each differentiation step (Fig. 5A). Approximately 3% (3% ± 1%) of cells after adding FGF8 and RA were GFP+, but about 19% (19% ± 3%), 15% (15% ± 5%), and 23% (23% ± 5%) (Fig. 5B, C, E) were in Stages IV–VI, respectively. Untreated MATH-hESCs showed GFP expression (Fig. 5A, E), but the cells were not positive for any of the GC markers (ATH, ZIC1, ZIC2).

Sorting and analysis of MATH1-GFP+ cells. Typical sorting profile of 2 different fractions (GFP + and GFP−)

The RT-PCR analysis of the cells generated at the end of our protocol showed that sorted MATH1-GFP+ cells express several crucial GCP markers including ATH, ZIC1, ZIPRO1, TUJ1, TAG1, and CYCLIND2, but not PAX2, which is characteristic for interneurons (Fig. 5F). The lack of expression of specific sequence for VENUS-GFP construct in MATH1-GFP− cells provides the evidence that sorting was successful. After culturing of the sorted cells on laminin/fibronectin for 2 days, immunocytochemistry analysis confirmed some of the results obtained with RT-PCR. The majority of the cells (>99%) were ATH+ and ZIC+ (Fig. 5D). Granule neurons in vitro do not survive without supporting cells; therefore, these cells were used for cell transplantation.
Cell transplantation
To explore whether hESC-derived MATH1-GFP+ cells can survive and integrate in the postnatal CNS, we implanted sorted and previously treated MATH-hESCs with the entire protocol (Stage VI). The cells were implanted in the mouse neonatal cerebellum of 7-day-old CD-1 pups at the level of EGL. The majority of surviving cells (14%–18% of total injected cells survived) did not reveal typical morphology for mature GCs (Fig. 6), consistent with the previous studies of cerebellar transplantation [10,11,20]. GFP staining revealed that cells migrated across the molecular layer, past the Purkinje cell layer, and settled in the internal granular layer, which is supposed to be the final destination of mature GCs. The presence of teratoma or other signs of tumorigenicity in any of the animals grafted were not observed.
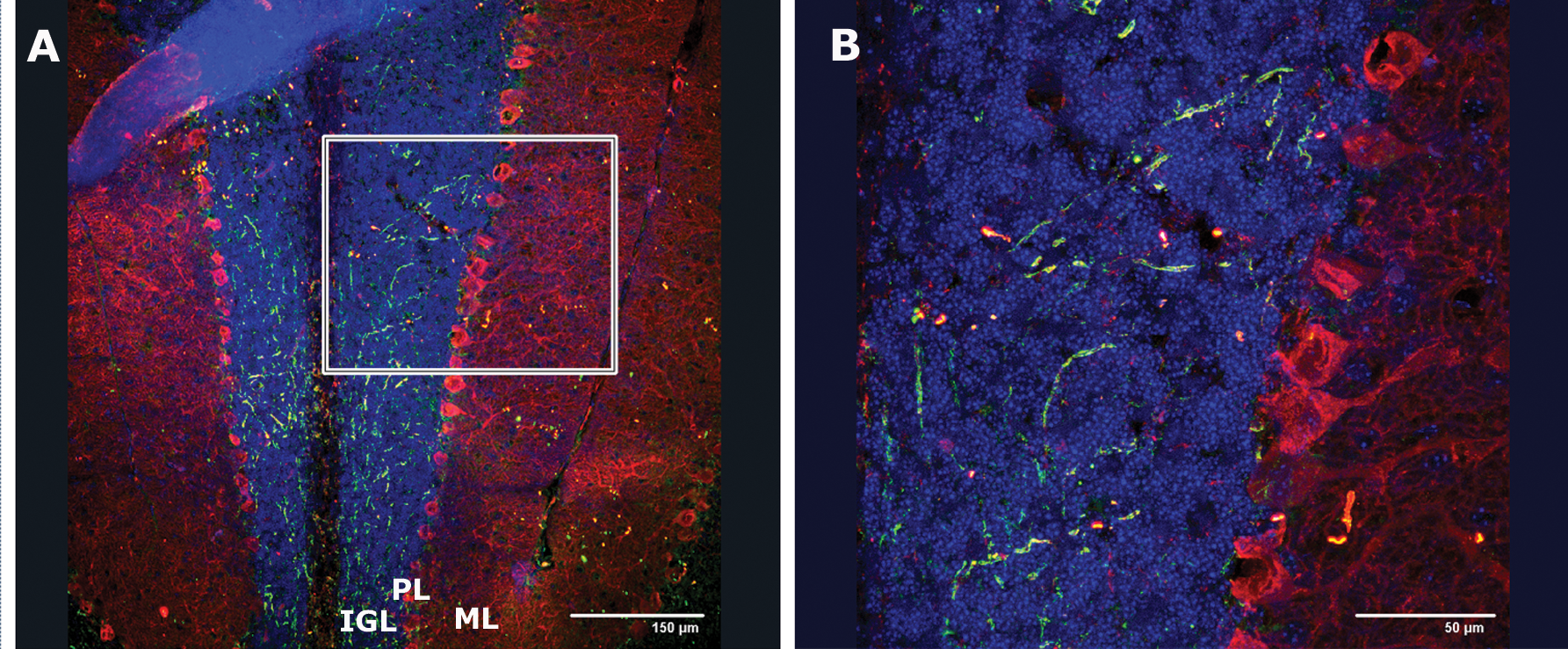
Incorporation of differentiated hESCs into neonatal cerebellum. To test whether differentiated hESCs incorporate into cerebellar cortex, exit the cell cycle, and migrate into the IGL, either differentiated hESCs or granule cell progenitors from Stage VI differentiated from MATH1-GFP+ hESCs and sorted were implanted in mouse pups. Two weeks after implantation, differentiated hESCs migrated and were detected by immunohistochemistry in the deeper layers of the developing cerebellar cortex. Postmigratory hESC-derived cells (GFP+ cells) are seen in the IGL and PL (Purkinje cells were immunostained with Calbindin-red), where terminal differentiation occurs.
Discussion
Our results show that application of inductive signals involved in the early patterning in the cerebellar region of the neural tube (FGF and WNT signaling and RA) followed by the factors involved in early GCP development (BMP4/6/7 and GDF7), mitogens (SHH and JAG1) and neurotrophins (BDNF and NT3), directs hESC differentiation into functional cerebellar cells (Figs. 1 –3). The generated progenitors showed a GC phenotype expressing both markers of dorsal neurons (MATH1 and ZIC1) and markers specific for GCs (ZIPRO1, EN1, ZIC1-3, MEIS1, PDE1c, PAX6, TAG1, CYCLID2, and GABAα6R). In addition, cerebellar-like neurons expressed the T-shaped polarity phenotype of granule neurons (Fig. 3). We also observed the expression of the most important markers for Purkinje cells (Fig. 3): PCP2 (L7), LHX 5, GAD67, and CALBINDIN.
The molecular mechanisms underlying human development are complex and largely undefined. However, undifferentiated and differentiated hESCs offer an excellent in vitro tool to recapitulate mechanisms activated during early development. Previous developmental studies provided evidence that the formation of the cerebellar primordium depends on inductive signaling at the mes/met boundary [10,14,20,24]. Rhombic lip GCPs express the transcription factors MATH1 [25,26] and ZIPRO1(RU49) [27], and MATH1 function is required for GC generation and EGL formation [26,27]. Early cerebellar development also depends critically on inductive signaling involving canonical Wnt signaling [21] and FGF proteins secreted by cells of the isthmic region at this midbrain–hindbrain junction. Indeed, the inductive signals of FGFs, RA, and WNT used sequentially in our study were crucial for expression of human ATH1 in cerebellar EGL progenitors. These factors are well known for their crucial role in early neural development, where also RA and FGF8 signals induce differentiation of cells along the rostral/caudal axis [38,39]. Also, all these factors were required to induce the expression of EN1 in differentiated hESCs, a gene required for the formation of the mes/met domain of the neural tube. Further differentiation of GCP progenitors and the generation of other cerebellar cell types, such as deep nuclei and Purkinje cells, depends on BMP-mediated dorsalizing signals [10,11,20]. GCPs expressing MATH1 arise close to the metencephalic roof plate, a source of BMP6, BMP7, and GDF7. Also, the dorsalizing activity of BMP4 on developing CNS tissue or differentiation of murine ESC toward GCP has been reported [20,28]. Our results show that treatment of differentiated hESCs with BMP4, BMP6, BMP7, and GDF7 is sufficient for the maintenance and increased expression of all cerebellar markers previously activated, but it is not crucial for further regional specification of cerebellar cells. By assessing the expression of Ki67 we found that differentiated cells showed high proliferation probably induced by SHH, JAG1, and Notch signaling, which plays instructive roles in the proliferation of granule neuron precursors and promotes the identities of several types of glial cells, including Bergmann glia [40].
Highly mitogenic factors BDNF and neurotrophins stimulate migration of individual GCs in vitro. In contrast, NT3 alters the morphology of outgrowth. High axonal growth observed in hESC-derived cerebellar cells confirms that regulation of neurotrophin receptors during cerebellar development is important for the timing and morphology of axonal growth [31,41].
With respect to efficiency, the induction of Purkinje cells, namely the yield of PCP2 (L7)+ neurons, was higher when compared with another protocol using murine ESCs [11]. Very high expression of GAD67 as well as GABA itself indicates an early acquisition of this transmitter expression by cerebellar neurons, far in advance of their final positioning and establishment of synapses, confirming previous in vivo findings [42]. In our study, cells positive for Purkinje markers did not coexpress CALBINDIN, which corroborates with a previous in vivo work [43] in which Purkinje cells began to exit the cell cycle before expressing the classical postmitotic Purkinje marker CALBINDIN.
The results in the present study are consistent with the studies done in mouse ESCs reported by Su et al [11] and Salero and Hatten [10], who introduced factors involved in the mechanisms of cerebellar development under in vitro conditions. Our results confirm that the same in vitro neural differentiation mechanisms can be applied using hESCs, but RT-PCR analysis revealed different timing of expression of some cerebellar markers during the differentiation of hESCs compared with murine counterparts. With this study we extend their previous findings showing expression of 2 additional GCP markers ZIC3 and GIRK3 and analyzing the functional electrophysiological activity of generated cells. We showed that differentiated neurons generated overshooting action potentials in response to depolarizing current injection. Further characterization of voltage-gated channels revealed the presence of Na+ and K+ channels as physiological markers of differentiation. Moreover, all recorded neuron-like cells responded to GABA, whereas only one-third responded to glutamate. When stimulated with GABA or Glu, newborn rat cerebellar explants of granule and Purkinje neurons elicit currents. In newborn mouse cerebellar slices, whole-cell recordings from Purkinje cell somata showed α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor–mediated spontaneous excitatory postsynaptic currents and GABAA receptor–mediated spontaneous inhibitory postsynaptic currents [44]. Similarly, in GCs, a part of the NMDA and non-NMDA excitatory components [44] showed GABAA receptors to mediate tonic inhibition [45]. Interestingly, nicotinic Ach receptors in ∼50% neurons correlate with reports that link cerebellar nicotinic receptors with GABA and Glu neurotransmitter release [46] and therefore to influence neuronal activity like in autism [47].
Finally, we show that the previously designed MATH1 enhancer–directed GFP vector [11] could be a very useful tool to sort and purify the MATH1+ cells (Fig. 5). As a result, we have isolated MATH1-GFP+ cells (>99% of sorted GFP cells were ATH+) as GCPs. This approach allowed us to isolate hESC-derived cerebellar progenitors, removing the population of undifferentiated hESC, as reflected in the absence of expression of the pluripotent markers OCT4 and REX1 in the cells from the sorted pool. This fact is one of the most important features of this system for its putative clinical application. Although this approach allowed us to reliably prepare and purify ATH+ neurons from undifferentiated and other hESC-derived cells, the efficiency of this procedure may yet be significantly improved. Designing the genetic system that includes human ATH enhancer could be an approach for the future medical application of generated neurons. Nevertheless, these results suggest the utility of enhancer-based fluorescence for the isolation of specific phenotypes from differentiated hESC populations as a mean of purifying clinically appropriate cells.
We also successfully grafted MATH1-GFP+ hESC-derived cerebellar cells in the postnatal mouse cerebellum, which resulted in cell survival and migration toward the region where the granular cells exert their function despite the cross-species nature of the grafts (Fig. 6), demonstrating the vitality of the cells and the robustness of our protocol, but further in vivo functionality analysis has to be performed.
Our study extends our knowledge about early human development and shows that it is possible to recapitulate human cerebellar development by using hESCs and human differentiation factors. For the first time, demonstration of the functional activity of differentiated hESC by patch-clamp analysis confirmed the cerebellar phenotype of generated neurons, which is a prerequisite for future applications in preclinical models of cerebellum-related diseases. Most importantly, improvement of this protocol for derivation of Purkinje cells is an important challenge for cell therapy for spinocerebellar atrophy diseases [48]. Although the road to the clinical application of hESC-derived cerebellar precursors remains extremely challenging, the ability to generate unlimited numbers of pure granular or Purkinje cell progeny and the capacity for in vivo survival and integration in the developing and adult cerebellum are important first stages in this journey.
Footnotes
Acknowledgments
The authors are thankful to Dr. Majlinda Lako, Dr. Lyle Armstrong, and Richard Griffeth for critical reading; Laura Nuñez Arrufat, Maria Amparo Pérez-Aragó, and Maria Gomez Lopez for their excellent technical support. This work was supported by funds for research in the field of regenerative medicine from the Regional Government Health Department (Generalitat Valenciana) and the Instituto Carlos III belonging to the Spanish Ministry of Health and Consumer Affairs, and through grants from the Spanish Ministry of Science and Innovation (SAF2007-63193) and La Marató (TV3 070330).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.