
Abstract
Neurogenesis involves the proliferation of multipotent neuroepithelial stem cells followed by differentiation into lineage-restricted neural precursor cells (NPCs) during the embryonic period. Interestingly, these progenitor cells express robust levels of the aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor that regulates expression of genes important for growth regulation, and xenobiotic metabolism. Upon binding 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), a pervasive environmental contaminant and potent AhR ligand, AhR, is activated and disrupts gene expression patterns to produce cellular toxicity. Because of its widespread distribution in the brain during critical proliferative phases of neurogenesis, it is conceivable that AhR participates in NPC expansion. Therefore, this study tested the hypothesis that AhR activation by TCDD disrupts signaling events that regulate NPC proliferation. The C17.2 NPC line served as a model system to (1) assess whether NPCs are targets for TCDD-induced neurotoxicity and (2) characterize the effects of TCDD on NPC proliferation. We demonstrated that C17.2 NPCs express an intact AhR signaling pathway that becomes transcriptionally active after TCDD exposure. 3H-thymidine and alamar blue reduction assays indicated that TCDD suppresses NPC proliferation in a concentration-dependent manner without the loss of cell viability. Cell cycle distribution analysis by flow cytometry revealed that TCDD-induced growth arrest results from an impaired G1 to S cell cycle transition. Moreover, TCDD exposure altered p27 kip1 and cyclin D1 cell cycle regulatory protein expression levels consistent with a G1 phase arrest. Initial studies in primary NPCs isolated from the ventral forebrain of embryonic mice demonstrated that TCDD reduced cell proliferation through a G1 phase arrest, corroborating our findings in the C17.2 cell line. Together, these observations suggest that the inappropriate or sustained activation of AhR by TCDD during neurogenesis can interfere with signaling pathways that regulate neuroepithelial stem cell/NPC proliferation, which could adversely impact final cell number in the brain and lead to functional impairments.
Introduction
N
The developing nervous system is highly susceptible to environmental insults because embryonic brain maturation is dictated by several waves of neurogenesis, which continue into adulthood in certain brain regions [3]. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD or dioxin), a ubiquitous environmental contaminant belonging to a class of structurally related polyhalogenated aromatic hydrocarbons, has been associated with developmental neurotoxicity in humans and experimental animals [4 –8]. However, the precise effects of TCDD on embryonic neurogenesis are largely unexplored. Because TCDD is highly lipophilic, it readily crosses the placenta and can be transferred to the developing offspring during gestation [9 –12]. Moreover, TCDD is resistant to degradation and not readily metabolized or excreted in exposed organisms [13]. Considering these properties, bioaccumulation could allow for low-dose exposures to reach hazardous levels in humans and animals, which could interfere with neurogenesis throughout the lifespan.
Although previous evidence suggests that prenatal TCDD exposure has been connected with neurodevelopmental deficits in humans and experimental animals, the cellular targets and molecular mechanisms of neurotoxicity are poorly understood. Neurobehavioral studies in rodents and primates after developmental TCDD exposure have revealed motor and cognitive deficits, suggesting abnormal brain development [14,15]. For example, maternal TCDD exposure has been linked to deficits in higher brain function in offspring, such as working memory in a radial arm maze [16] and operant response in running wheels or two-lever chambers [15,17]. Further, recent studies revealed that newborn head circumference, an indicator of fetal brain development, was negatively correlated with the concentration of TCDD in human breast milk [18,19]. Smaller head circumference during infancy has been associated with neurodevelopmental disabilities [20]. These observations suggest that TCDD exposure adversely impacts brain maturation and may be a risk factor for neurodevelopmental toxicity and disorders. Therefore, our laboratory is testing the hypothesis that neural progenitors are primary targets for TCDD actions during brain development.
TCDD exerts its toxicity via interaction with the aryl hydrocarbon receptor (AhR), an evolutionary-conserved, ligand-activated transcription factor that regulates expression of xenobiotic metabolizing enzymes and growth regulatory molecules [21]. In its ligand-free state, AhR resides in the cytoplasm bound to several chaperones. After binding TCDD, AhR dissociates from its chaperones [22,23], translocates to the nucleus, and dimerizes with the aryl hydrocarbon receptor translocator (ARNT). The AhR/ARNT complex then binds to aryl hydrocarbon response elements (AhREs) and modulates expression of genes involved in toxicity, adaptive responses, and cell cycle regulation [24]. The AhR is subsequently downregulated via proteasomal degradation [25]. Whereas information regarding AhR has been revealed in numerous TCDD exposure studies, the precise physiological roles of AhR are unclear.
Because AhR activation impacts growth and differentiation in various cell types [26,27], it is reasonable to postulate that exposure to TCDD disrupts these processes during neurogenesis. TCDD, via binding and activating AhR, has been shown to alter expression of genes involved in cell cycle regulation and apoptosis [28 –31]. For example, AhREs are found in the promoter regions of growth regulatory molecules such as p21cip1, p27 kip1, HES1, and Bax [28 –32]. These genes have been shown to be upregulated after polyhalogenated aromatic hydrocarbon exposure, leading to perturbations in cell cycle control mechanisms in various cell types [28 –31,33]. AhR activation by TCDD has also been shown to impact differentiation of keratinocytes, adipocytes, and mammary epithelial cells [34 –37]. We previously demonstrated that TCDD exposure disrupted both proliferation and differentiation of cerebellar granule precursors (GNPs) [31,38]. Moreover, our findings that the cerebellum is smaller and granule neuron differentiation is abnormal in AhR deficient mice [31] suggest that the AhR is important during granule precursor maturation. Considering that AhR and ARNT are expressed in the embryonic neuroepithelium during a peak window of neurogenesis on gestational day 10–13 in mouse embryos [39,40], it is plausible that AhR also participates in embryonic brain development.
Our present study tested the hypothesis that AhR plays a role in regulating NPC proliferation, which is dysregulated after inappropriate activation by dioxin. The multipotent C17.2 neural precursor cell line served as a model to explore the manner in which TCDD may interfere with NPC proliferation. C17.2 cells were originally derived from rapidly dividing neural progenitor cells of the mouse cerebellum and immortalized by the retroviral transfection of v-myc [41]. This NPC model expresses nestin, a marker for neural stem cells and progenitors, and responds to environmental cues resulting in terminally differentiated neurons or glia, thus fulfilling the properties of endogenous neural progenitors [42]. In this study, C17.2 NPCs were shown to express a functional AhR pathway that is responsive to TCDD. Our results suggested that TCDD interacts with the AhR to disrupt NPC proliferation by altering expression patterns of cell cycle regulatory proteins consistent with a G1 to S cell cycle phase arrest. We extended our studies to include primary NPCs derived from the ventral forebrain of mouse embryos. TCDD exposure in primary NPCs also diminished cell proliferation by inducing a G1 to S phase arrest, similar our observations in the C17.2 cells. Together, our findings from both the NPC cell line and primary NPCs provide insight into the cellular mechanisms of the toxic effects of TCDD on NPC proliferation, as well as possible roles for AhR in regulating neurogenesis.
Materials and Methods
Reagents
Mouse C17.2 neural precursor cells were a generous gift from Dr. Evan Snyder (UCSD). TCDD (Cambridge Isotopes) was solubilized in dimethyl sulfoxide (DMSO). DMSO, Triton X-100, phenylmethanesulfonyl fluoride, antiprotease cocktail, 35% bovine serum albumin (BSA) solution, putrescine, progesterone, insulin, transferrin, and sodium selenite were purchased from Sigma. Dulbecco's phosphate-buffered saline (DPBS), 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA), and L-glutamine were purchased from Mediatech. Dulbecco's modified of Eagle's medium (DMEM), Leibovitz's L-15 medium, calcium- and magnesium-free Hanks' balanced salt solution, DMEM/F12 (1:1 mixture), fetal bovine serum, horse serum, normal goat serum, penicillin/streptomycin (100 U/mL each), and EDTA were purchased from Gibco. Collagenase, deoxyribonuclease I, and papain were purchased from Worthington. Human plasma fibronectin was obtained from Millipore. Recombinant human epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) were obtained from Peprotech.
C17.2 cell culture
C17.2 neural precursor cells were maintained in DMEM supplemented with 10% fetal bovine serum, 5% horse serum, 1% L-glutamine (2 mM), and 1% penicillin/streptomycin (100 × stock) and maintained in a humidified atmosphere of 5% CO2 at 37°C. This medium is referred to as the feeding medium. The medium was changed every 2–3 days. Cells were grown in culture dishes to subconfluence before analysis.
Primary neural precursor cell culture
Male and female C57BL/6J mice were obtained from Taconic Farms. Upon arrival, all mice were allowed to acclimate to their home cage for 2 weeks before initiation of experiments. Mice were maintained on a 12-h light/dark cycle with food and water provided ad libitum and kept in accordance with the guidelines set by the University of Rochester University Committee on Animal Resources and the American Association for Laboratory Animal Science. Once acclimated to their home cage, male and female mice were allowed to mate overnight. The day of the plug was designated as embryonic day 0.5 (E0.5). At E12.5, pregnant dams were anesthetized by CO2, and embryos were removed via cesarean section and collected in a Petri dish containing Leibovitz's L-15 medium. The ventral forebrain was carefully dissected using a dissection microscope and transferred to calcium- and magnesium-free Hanks' balanced salt solution where the tissue were subjected to enzymatic dissociation using collagenase, deoxyribonuclease I, and papain. Cells were then mechanically dissociated using a sterile needle attached to a 1 mL syringe to create a single cell suspension. Cells were grown in DMEM/F12 (1:1 mixture) medium supplemented with 100 μg/mL transferrin, 25 μg/mL insulin, 10 mg/mL BSA, 100 μM putrescine, 20 nM progesterone, 30 nM sodium selenite, 5 mg/mL glucose, 2 mM L-glutamine, and 1% penicillin/streptomycin (100 × stock), 20 ng/mL bFGF, and 20 ng/mL EGF. All cell culture dishes were precoated with fibronectin at 1.25 μg/cm2. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2.
Immunoblot analysis
Cells were harvested for protein analysis in ice-cold PBS containing 0.1% Triton X-100, 1% phenylmethanesulfonyl fluoride, 1% EDTA, and 1% antiprotease cocktail. Total protein concentrations were determined by the microBCA assay (Pierce). Proteins (5–25 μg) were fractionated on 10% polyacrylamide gels and transferred to polyvinylidene difluoride membranes (BioRad). Membranes were blocked with 5% powdered milk containing 0.2% Tween-20 and probed for AhR (1:2,000; Biomol), ARNT (1:2,000; Novus), cyclin D1 (1:1,000; Cell Signaling), p27 kip1 (1:1,000; Santa Cruz), nestin (1:1,000, Developmental Studies Hybridoma Bank), or β-actin (1:2,000; Sigma) overnight at 4°C. Membranes were then probed with the appropriate horseradish peroxidase-conjugated secondary antibody (Jackson Immunoresearch Laboratories) for 1 h at room temperature. Proteins were observed with LumiGLO chemiluminescent substrate reagent (Kirkegaard & Perry Laboratories). Densitometric analysis was accomplished with ImageJ software (National Institutes of Health).
Immunocytochemistry
Cells were seeded in 8-well culture chamber slides (Lab-Tek, VWR). After exposure to vehicle (DMSO), 1 or 10 nM TCDD, the medium was removed and cells were rinsed with DPBS and then fixed with 4% paraformaldehyde at room temperature for 30 min. After fixation, cells were blocked in PBS containing 10% normal goat serum and 0.3% Triton X-100 at room temperature for 30 min. Cells were then incubated with an AhR antibody (1:800; Biomol) or nestin antibody (1:100; Developmental Studies Hybridoma Bank) overnight at 4°C, washed, and incubated with the appropriate Alexa Fluor–conjugated secondary antibody (Molecular Probes) for 90 min at room temperature. Nuclei were subsequently stained Hoechst 333452 (1:2,000; Sigma) or DAPI (1:5,000, 1 mg/mL stock; Molecular Probes). Fluorescence was observed using a Nikon Eclipse TS100 inverted microscope and images were taken at 20 × or 40 × magnification with SPOT advanced software. Fluorescence was not detected in cultures incubated with a secondary antibody alone or with a nonspecific IgG.
Electrophoretic mobility shift assay
C17.2 cells were grown to 90% confluence in 100 mm dishes (BD Falcon), exposed for 1 h to DMSO, 1 or 10 nM TCDD, and harvested in ice-cold PBS. Cells were centrifuged and pellets were resuspended in HEDG buffer (25 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM DTT, and 10% glycerol) and NaCl was added to a final concentration of 0.4 M. Cells were then triturated using a 25-gauge needle and incubated on ice for 40 min, with periodic vortexing. After centrifugation at 100,000 g for 50 min, whole cell lysates from the supernatants were stored at −80°C. For gel shift assays, 10–20 μg of protein was used in HEDG buffer and equilibrated in 0.5 μg of herring sperm DNA and 20 mM DTT. About 2.5 × 10 4–5.0 × 10 4 cpm of radiolabeled 32P-AhRE and unlabeled AhRE oligonucleotides were then added to the mixture for 20 min at room temperature. Protein–DNA complexes were resolved on a 4% native polyacrylamide gel. AhR/ARNT-AhRE bands were quantified using a PhosphorImager (Molecular Dynamics).
Transient transfections
C17.2 cells were seeded in 24-well plates (BD Falcon) at a density of 2 × 10 4 cells per well. When the cells reached 60% confluency, 0.3 μg of the p2dLuc DNA plasmid was transiently transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. As an internal control, 1 ng of Renilla luciferase (pRL-tk) was cotransfected along with p2dLuc. Twenty-four hours after transfection, cells were exposed to DMSO, or 1 nM or 4 nM TCDD for 24 h. Cells were harvested using 1 × passive lysis buffer (Promega) and relative luciferase activity was measured using the Dual-Luciferase Reporter assay system (Promega). Normalized luciferase activity is reported as a ratio of the p2dLuc firefly luciferase to the Renilla luciferase.
3H-Thymidine incorporation
C17.2 cells were seeded in 96-well plates (BD Falcon) at a density of 2.5 × 10 3 cells per well in the feeding medium. When they reached 50% confluency they were transferred to serum-free DMEM for 15 h. After serum starvation, cells were exposed to 3 μg/mL sonic hedgehog (Shh; a generous gift from Dr. Kenney, Memorial Sloan Kettering Cancer Center, NY), 25 ng/mL EGF, and 25 ng/mL insulin-like growth factor 1 (IGF-1; R&D Systems) with and without 0.1–10 nM TCDD for 24 h. For primary NPCs from mouse embryos, E12.5 ventral forebrain cells were seeded in 96-well plates previously coated with 1.25 μg/cm2 fibronectin at a density of 5 × 10 4 cells per well. Cells were grown with or without 20 ng/mL EGF and 20 ng/mL bFGF in the absence or presence of 1 nM TCDD for 24 h. C17.2 and E12.5 cells were then labeled with 1 μCi of methyl-3H-thymidine (Perkin Elmer) during the last 6 and 12 h of incubation, respectively, and harvested onto filter paper using a Skatron cell harvester. After the filters dried, the amount of incorporated radioactivity was quantified by liquid scintillation counting.
Alamar blue reduction assay
Cells were seeded and treated as described for the 3H-thymidine incorporation experiments. Alamar blue (BioSource) was added directly to the culture medium at 10% of the medium volume during the last 10 h of the 24 h exposure period. Absorbance was measured spectrophotometrically at 570 and 600 nm using a microplate reader.
Flow cytometry for cell cycle analysis
C17.2 and E12.5 ventral forebrain cells were seeded in 100 × 20 mm cell culture dishes (Corning, Inc.) and treated as described for the 3H-thymidine incorporation experiments. Cells were then harvested by trypsinization 24 h after TCDD exposure, washed with PBS containing 0.5% BSA, fixed by the dropwise addition of ice-cold 70% ethanol, and incubated at least overnight at −20°C. After permeabilization, cells were treated with 1 mg/mL RNase A (Roche Diagnostics) and DNA was stained with 50 μg/mL propidium iodide (Molecular Probes). Cells were acquired with a FACS CantoII flow cytometer using a 488 nm argon blue laser (Becton Dickinson). A minimum of 50,000 events was collected per sample. Cell cycle analysis was performed using FlowJo software (Tree Star).
Cell survival assay
Neuron viability was assessed with the Live/Dead Cytotoxicity Kit (Molecular Probes). Calcein-AM and ethidium homodimer were used to distinguish dead and living cells. C17.2 and E12.5 ventral forebrain cells were seeded in 24-well plates (BD Falcon) at 2 × 10 4 cells per well and 3 × 10 5 cells per well, respectively, and then exposed to TCDD as described for the 3H-thymidine incorporation experiments. After the 24 h exposure period, cells were rinsed with DPBS and incubated for 30 min at 37°C in serum-free DMEM containing ethidium homodimer and calcein-AM. Fluorescence was observed using a Nikon Eclipse T100 fluorescent microscope (20 × magnification). Approximately 800 cells from 3 randomly selected fields for each exposure condition were counted using Image-Pro Plus software Version 6.2 (Media Cybernetics) with the experimenter blinded to the treatment conditions. The percentage of live cells was determined as the ratio of calcein-AM-stained cells over the total cells stained for calcein-AM and ethidium homodimer. Cells grown in their standard feeding medium were used as positive control for calcein-AM staining. Cells treated with 70% methanol for 30 min were used as positive control for ethidium homodimer staining.
Statistical analyses
Data are expressed as means ± standard error of the mean from a minimum of 3 independent experiments. Sample sizes are indicated in the figure legends. Statistical analyses were performed with either 1- or 2-way analysis of variance (ANOVA) where appropriate (GraphPad Prism 4.0). Tukey's or Dunnett's (for 1-way ANOVA), and Bonferroni's (for 2-way ANOVA) post hoc comparisons were used to compare select groups. The type of statistical test performed is indicated in the figure legends. P values of <0.05 were considered statistically significant.
Results
TCDD activates the AhR in C17.2 neural precursor cells
The first objective of the study was to confirm that C17.2 NPCs express an intact AhR pathway that is responsive to TCDD. To determine if C17.2 cells express the proteins that comprise an intact AhR pathway, several protein concentrations (5–25 μg) of C17.2 lysate were analyzed for AhR and ARNT protein content by immunoblot analysis. These studies indicate that C17.2 cells robustly express AhR and ARNT proteins (Fig. 1A). Mouse hepatoma cell lysate (Hepa) served as a positive control.
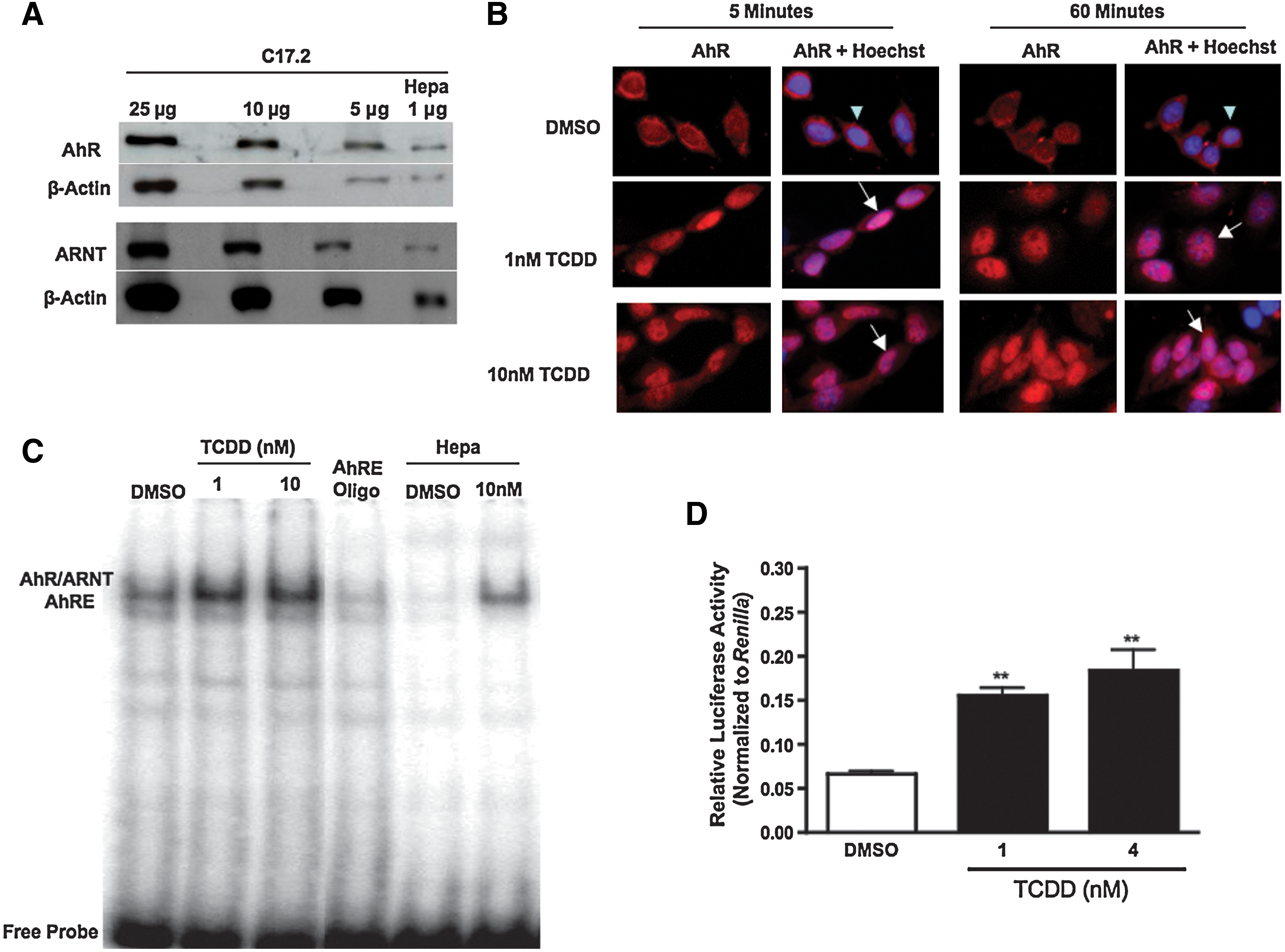
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) activates aryl hydrocarbon receptor (AhR) in C17.2 neural precursor cells.
To establish that TCDD activates AhR in C17.2 cells, cellular localization of AhR was determined by immunocytochemistry before and after TCDD exposure at specific time intervals. AhR was primarily localized to the cytoplasm in vehicle-treated cells, with minimal staining in the nucleus (Fig. 1B, top panels). There was no difference in AhR localization in the untreated cells compared to vehicle-treated cells (data not shown). After 1 and 10 nM TCDD exposure, AhR expression colocalized with the Hoechst 33342 nuclear stain within 5 min and remained in the nucleus after 60 min (Fig. 1B, bottom panels).
The DNA binding capacity of the AhR/ARNT complex to AhREs after TCDD exposure was next evaluated by an electrophoretic mobility shift assay (EMSA). EMSA analysis in cells exposed to vehicle (DMSO) exhibited no difference in the baseline levels of AhRE binding capability compared to cells not treated with vehicle (data not shown). After exposure to 1 and 10 nM TCDD for 1 h, there was a notable increase in AhR/ARNT binding with AhRE, respectively (Fig. 1C). Unlabeled AhRE-oligonucleotide confirmed the specificity of AhR/ARNT-AhRE binding. Mouse hepatoma cells (Hepa) exposed to vehicle or 10 nM TCDD served as negative and positive controls, respectively.
To confirm that the AhR/ARNT complex becomes transcriptionally active after AhRE binding, C17.2 cells were transiently transfected with a firefly luciferase plasmid containing 2 AhREs upstream of the luciferase promoter [43]. TCDD exposure elicited an increase in AhRE-driven luciferase activity after 24 h compared to the control (Fig. 1D), with 1 and 4 nM TCDD exposures inducing 2.3- and 2.75-fold increases in reporter activity, respectively, over the control. Together, these studies confirm that AhR becomes transcriptionally active after TCDD exposure in C17.2 neural precursor cells.
AhR protein levels decline after TCDD exposure
Several studies report that AhR levels rapidly decline after ligand binding [25,44]. AhR downregulation appears to be ubiquitin-mediated, as demonstrated by studies in which inhibition of the 26S proteasome results in enhanced AhR activity and the absence of ligand binding-induced reduction of AhR protein levels [25,44,45]. To determine if AhR mRNA levels were altered after TCDD exposure, C17.2 cells were exposed to vehicle (DMSO) or TCDD for 6–48 h, and mRNA expression was evaluated. AhR mRNA transcript levels were not altered after TCDD exposure (data not shown). AhR protein levels were then evaluated after cells were exposed to vehicle, 1, or 10 nM TCDD and harvested at various time intervals for immunoblot analysis. Differences in AhR protein levels were not detected in vehicle-treated cells compared to untreated cells (data not shown). After TCDD exposure, there was a marked reduction in AhR protein as early as 4 h after TCDD treatment and was sustained through 24 h postexposure (Fig. 2A). This result indicates that receptor levels may be diminished by post-translational mechanisms such as ubiquitination and degradation via the proteasome. Receptor levels returned to baseline levels 72 h after TCDD exposure and remained at baseline levels (Fig. 2B, C).
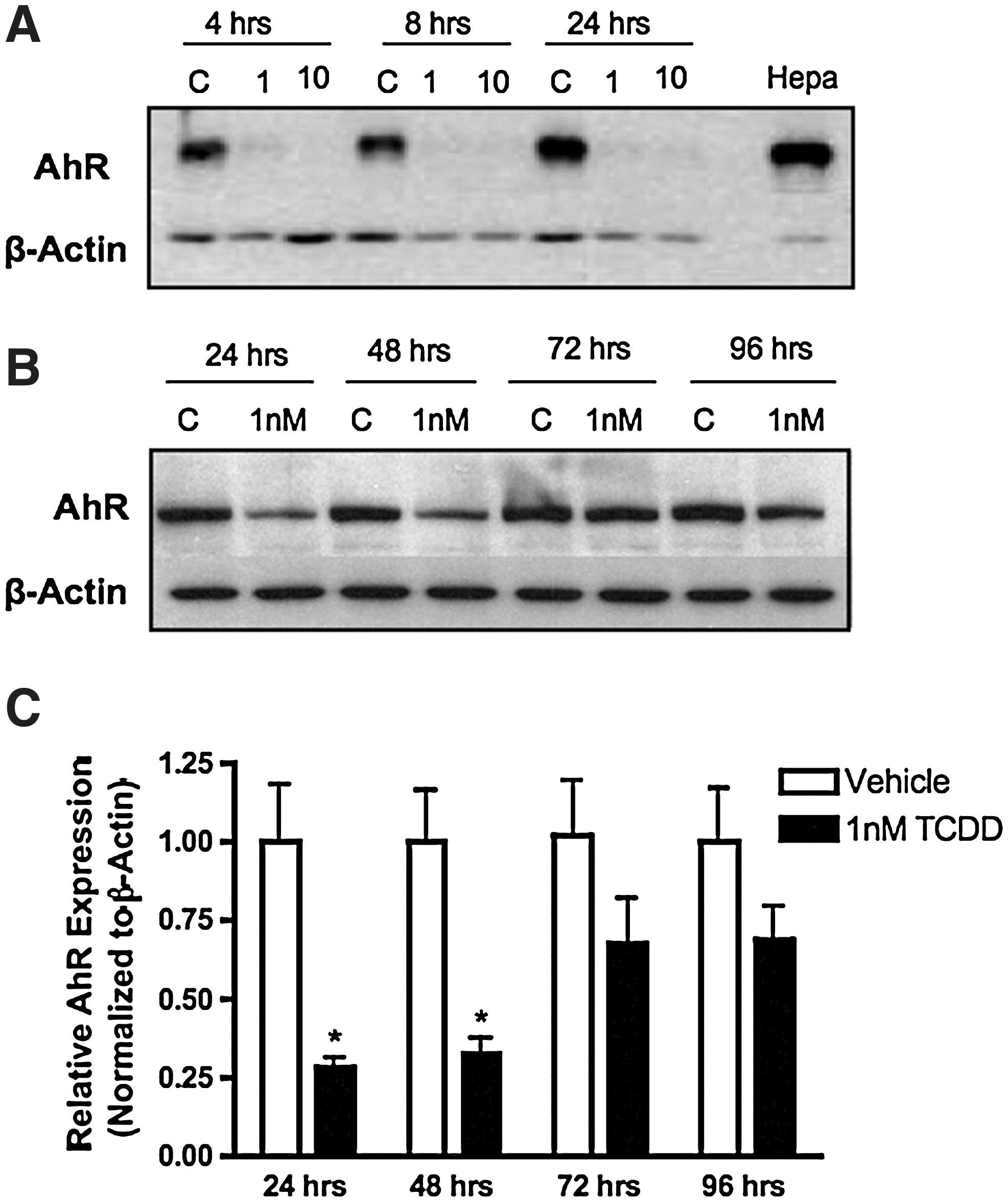
AhR protein levels decline after TCDD exposure. C17.2 cell lysate (10 μg) were examined for AhR protein content by immunoblot analyses. Cells were treated with vehicle (DMSO), 1 or 10 nM TCDD for 4–24 h
C17.2 cells respond to known NPC mitogens by increasing DNA synthesis and cell growth
To study the impact of TCDD on NPC proliferation, it was necessary to determine whether C17.2 progenitor cells respond to mitogens in ways similar to their in vivo counterparts. One morphogen that is critical for ventral midline (floorplate) development and for stimulation of neuronal precursor proliferation is Shh [46 –48]. In addition to Shh, molecules such as EGF and IGF-1 have been shown to regulate NPC proliferation [49 –51]. Therefore, we evaluated the mitogenic potential of Shh, EGF, and IGF-1 in C17.2 precursor cells. Before assessing proliferation, C17.2 cells were serum-starved to synchronize them in the G0/G1 phase of the cell cycle. Cells were then exposed to vehicle, Shh, EGF, or IGF-1 for 24 h. DNA synthesis was evaluated by measuring 3H-thymidine incorporation during the last 6 h of incubation. Cell growth was also examined by quantifying alamar blue reduction. The results demonstrated that Shh, EGF, and IGF-1 stimulated NPC proliferation by increasing DNA synthesis (Fig. 3A) and cell growth (Fig. 3B). Shh was the most efficacious mitogen, inducing an ∼3-fold increase in both DNA synthesis and cell growth compared to cells treated with a vehicle. Because C17.2 cells respond to Shh, EGF, and IGF-1 in a similar manner as native NPCs, it strengthens the use of the C17.2 cell line as an appropriate model to study NPC proliferation. Cell survival was not enhanced after treatment with any of the mitogens, confirming that Shh, EGF, and IGF-1 function as mitogens to induce cells to re-enter the cell cycle, rather than as trophic factors to promote cell survival (Fig. 3C).
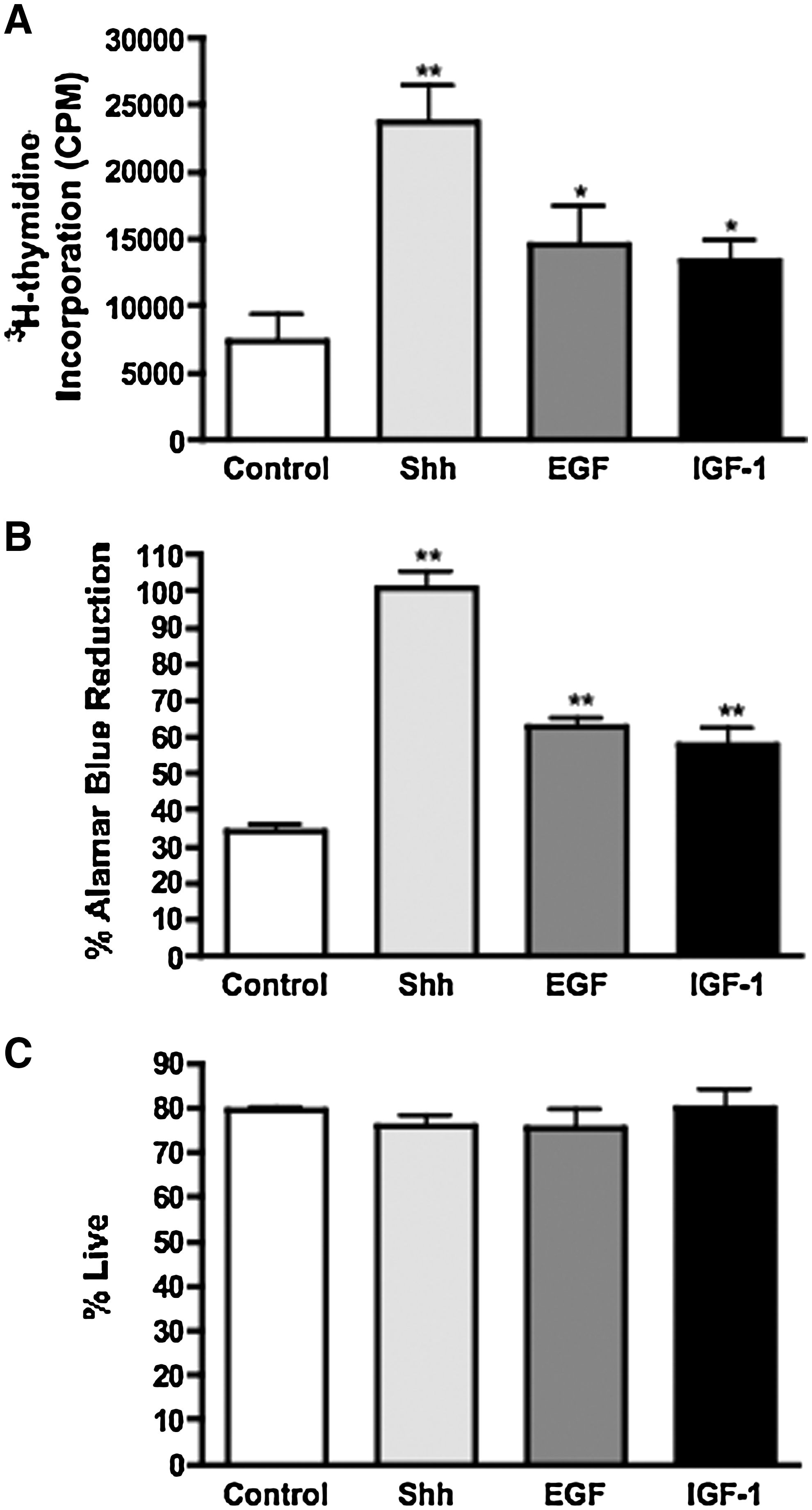
Mitogenic factors stimulate C17.2 DNA synthesis and cell growth without altering cell survival. After serum starvation, C17.2 cells were exposed to 3 μg/mL sonic hedgehog (Shh), 25 ng/mL epidermal growth factor (EGF), or 25 ng/mL insulin-like growth factor 1 (IGF-1) for 24 h and were assessed for DNA synthesis by measuring 3H-thymidine incorporation
TCDD suppresses mitogen-induced DNA synthesis and cell growth in a concentration-dependent manner
To investigate the effects of AhR activation by TCDD on cell proliferation, C17.2 cells were exposed to Shh, EGF, or IGF-1 with or without 1 nM TCDD for 6 and 24 h, and then assayed for DNA synthesis and cell growth. TCDD did not significantly alter DNA synthesis or cell growth in quiescent cells at either time point (Fig. 4A–C). In contrast, when cells were stimulated to proliferate with Shh, EGF, or IGF-1, coexposure to 1 nM TCDD for 6 and 24 h suppressed the mitogen-induced DNA synthesis (Fig. 4A, B) and cell growth (24 h only, Fig. 4C). TCDD did not significantly alter cell survival compared to the control (Fig. 4D).
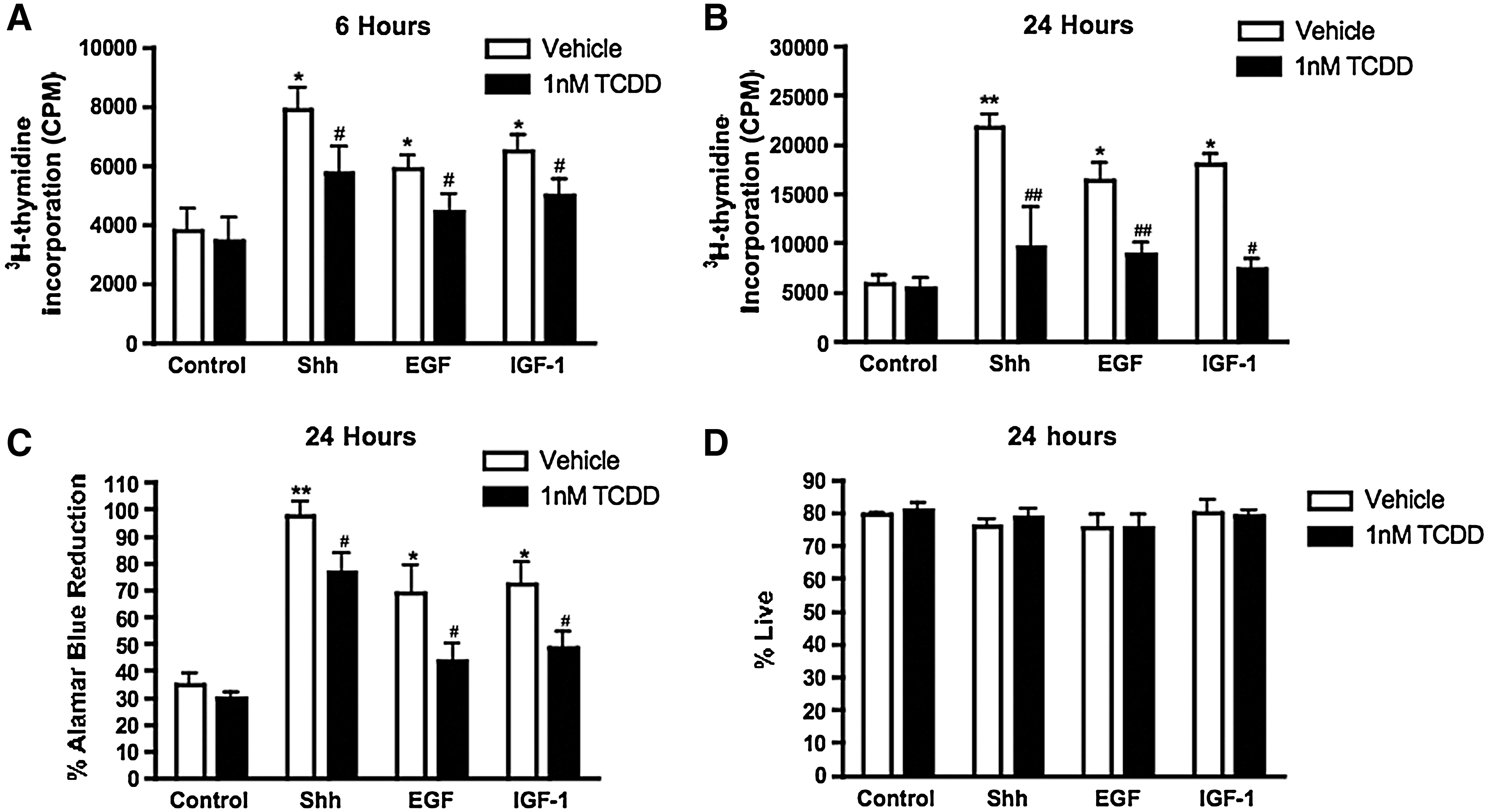
TCDD suppresses mitogen-induced DNA synthesis and cell growth without altering survival. C17.2 cells were exposed to a vehicle, Shh, EGF, or IGF-1, in the presence or absence of 1 nM TCDD for 6 or 24 h and assayed for DNA synthesis
To ascertain whether TCDD attenuated C17.2 proliferation in a concentration-dependent manner, cells were exposed to 0.1–10 nM TCDD in the presence or absence of Shh for 6 or 24 h. TCDD disrupted Shh-mediated proliferation in a concentration-dependent manner at both time points (Fig. 5A–C). C17.2 survival was only modestly reduced after exposure to 10 nM TCDD (Fig. 5D). Together, these observations indicate that TCDD reduces mitogenic actions in C17.2 cells, which leads to diminished growth rather than cell death.
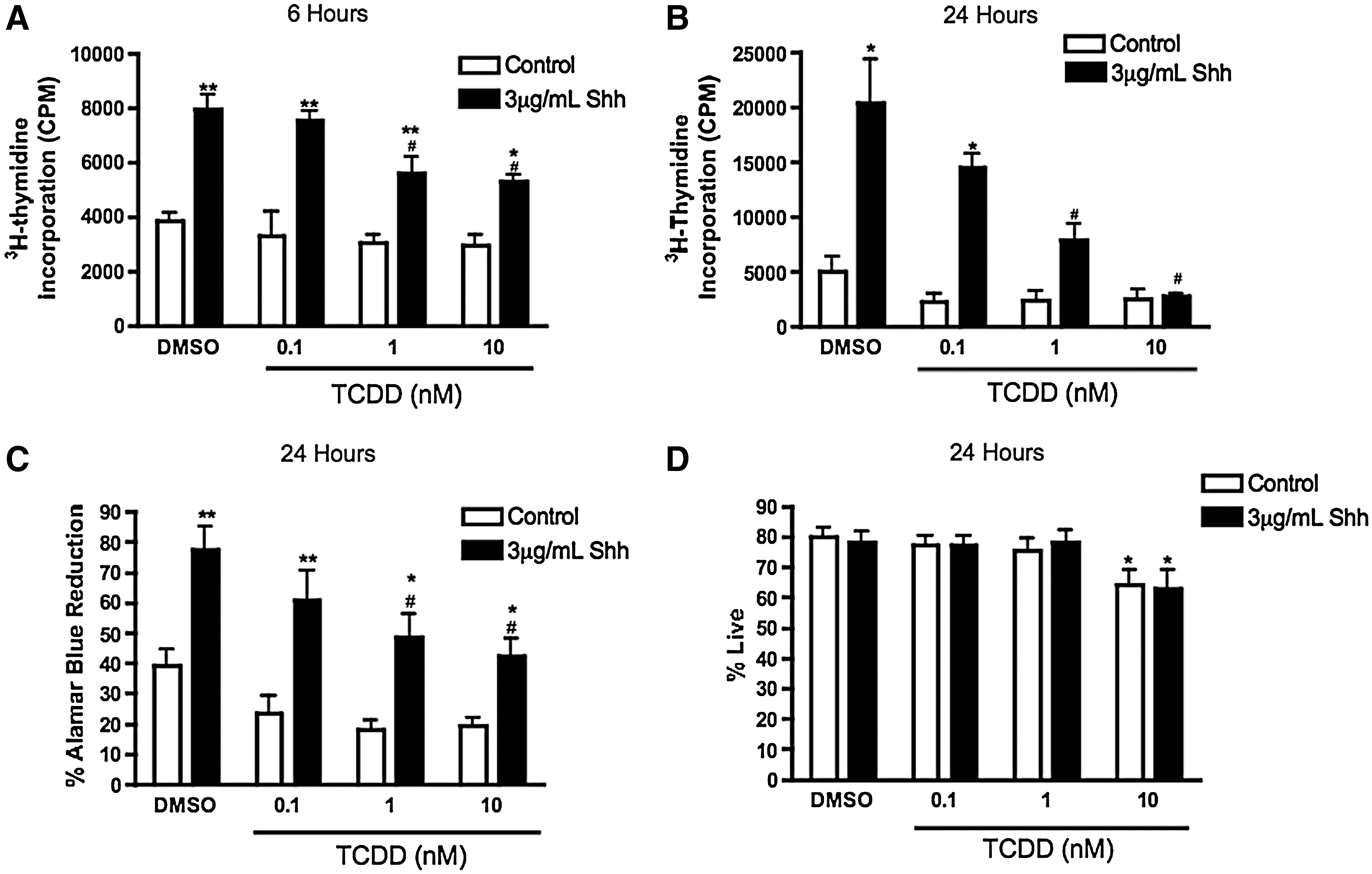
TCDD disrupts Shh-mediated proliferation in a concentration-dependent manner without significantly altering cell survival. C17.2 cells were exposed to a vehicle or 3 μg/mL Shh with 0–10 nM TCDD for 6 or 24 h and assayed for DNA synthesis
TCDD causes an accumulation of C17.2 cells in the G0/G1 stage of the cell cycle
C17.2 cells were exposed to vehicle, Shh, EGF, or IGF-1, with or without 1 nM TCDD, for 24 h and were then subjected to flow cytometric analysis to investigate alterations in cell cycle distribution. The Watson Pragmatic algorithm was used to determine the percentage of cells in the G0/G1, S, and G2/M phases of the cell cycle based on PI fluorescence. When C17.2 cells were stimulated to proliferate in the presence of each of the 3 mitogens, there was a significant decrease in the percentage of cells in the G0/G1 phase, which coincided with a significant increase in the percentage of cells in S phase. This led to a significant decrease in the G0/G1:S ratio, signifying that these mitogens were effective in stimulating the cells past the G1/S phase checkpoint and into mitosis (Table 1). As expected, exposure to 1 nM TCDD for 24 h did not significantly alter the cell cycle phase distribution of quiescent cells (Table 1). In contrast, TCDD caused a 20% increase (P < 0.05) in the percentage of cells in the G0/G1 phase and a 40% decrease (P < 0.05) in the percentage of cells in the S phase when cells were induced to proliferate in the presence of Shh, EGF, or IGF-1 (Table 1). TCDD did not elicit any change in the percentage of cells in the G2/M phase. However, the G0/G1:S ratio was increased by ∼50% in cycling cells, but not in noncycling cells, when exposed to TCDD. These results suggest that TCDD reduces NPC proliferation by causing an accumulation of cells in the G0/G1 phase, thereby interfering with the G1 to S phase transition.
C17.2 cells were exposed to a vehicle, Shh, EGF, or IGF-1, with or without 1 nM TCDD for 24 h. Twenty-four hours after exposure, C17.2 cells were harvested into a single cell suspension, permeabilized, and stained for PI. Cells were subsequently acquired on a FACS CantoII flow cytometer and data were analyzed using the Watson Pragmatic cell cycle algorithm to determine the percentage of cells in the G0/G1, S, and G2/M phases. Data represent means ± standard error of the mean from 4 independent experiments and analyzed by 2-way analysis of variance with Bonferroni post-test for select comparisons.
Statistically significant from control without TCDD.
Statistically different from respective mitogen alone. P < 0.05.
EGF, epidermal growth factor; IGF-1, insulin-like growth factor 1; Shh, sonic hedgehog; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
TCDD alters cell cycle regulatory protein expression consistent with G0/G1 phase arrest
Protein content for cell cycle regulators that govern the progression of a cell through the G0/G1 phase were examined to explore intracellular molecular signaling targets that may be responsible for the accumulation of cells in the G0/G1 phase of the cell cycle after TCDD exposure. In these experiments, C17.2 cells were exposed to a vehicle, Shh, EGF, or IGF-1 in the presence or absence of 1 nM TCDD for 6 and 24 h. Cyclin D1 and p27 kip1 protein content was quantified by immunoblot analysis. In the absence of mitogens, TCDD alone did not significantly alter cyclin D1 expression (Fig. 6A) but significantly increased p27 kip1 expression (Fig. 6B). After exposure to Shh, EGF, or IGF-1 at all time points, there was elevated cyclin D1 expression; coexposure to 1 nM TCDD significantly attenuated this effect (Fig. 6A). Cells exposed to 1 nM TCDD in the presence of a mitogen expressed significantly more p27 kip1 than cells exposed to the respective mitogen alone (Fig. 6B). Collectively, these results suggest that the upregulation of p27 kip1 protein levels by TCDD is one possible mechanism for the G0/G1 phase arrest.
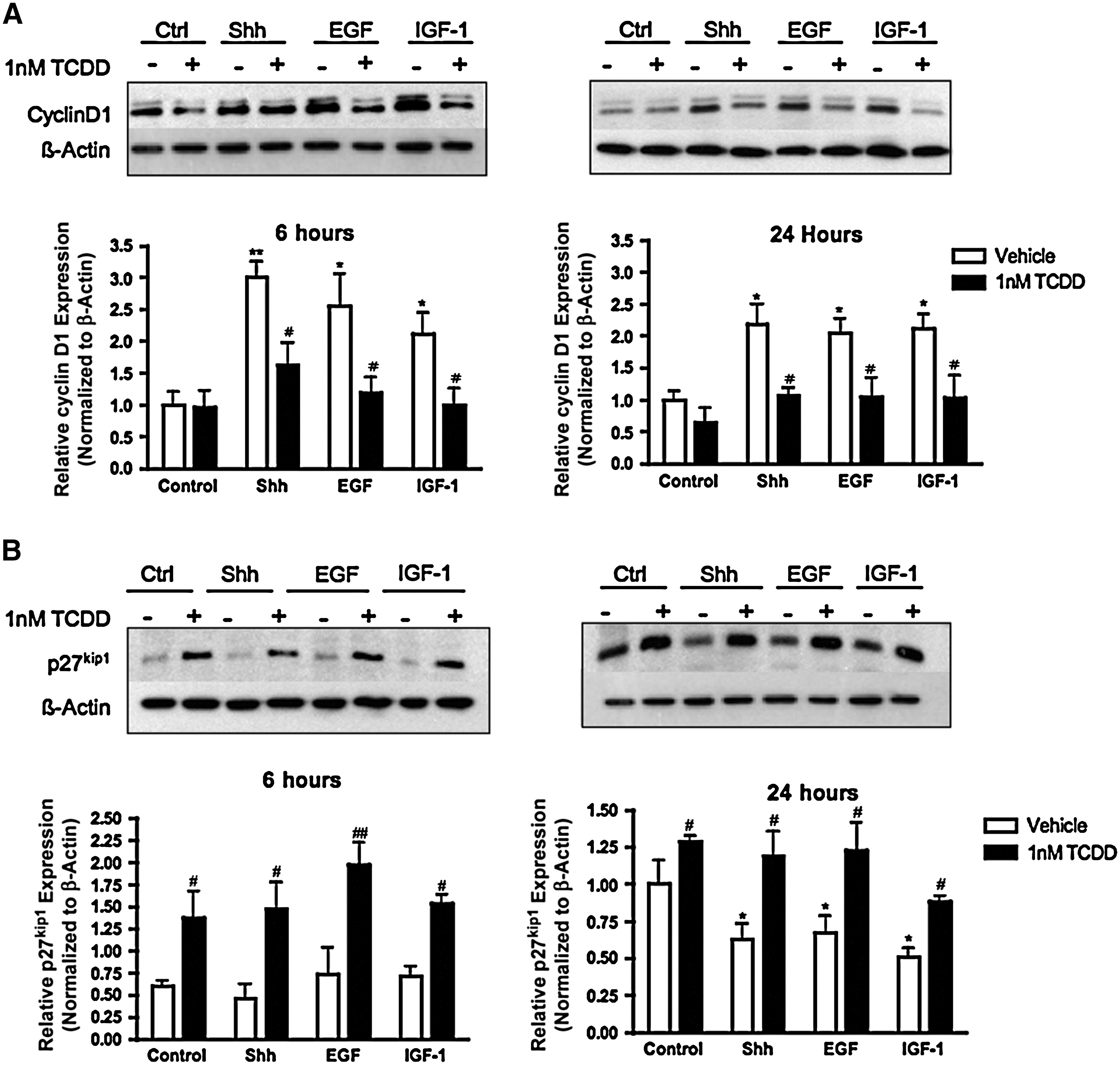
TCDD alters cyclin D1 and p27kip1 expression patterns consistent with G0/G1 phase arrest. C17.2 cells were exposed to a vehicle, Shh, EGF, or IGF-1, with or without 1 nM TCDD for 6 and 24 h. C17.2 lysates (10 μg) were analyzed for cyclin D1
AhR is expressed and responsive to TCDD in neural precursor cells derived from ventral forebrain of mouse embryos
Our studies were extended to evaluate AhR expression activation by TCDD in primary NPCs isolated from the ventral forebrain of E12.5 mouse embryos. We isolated NPCs at E12.5 because AhR and ARNT expression in the embryonic neuroepithelium peaks during E10-13 in mouse embryos [39,40]. Immunoblot analysis indicated that cultured NPCs robustly express AhR and nestin (Fig. 7A). In contrast to C17.2 cells, immunocytochemical studies revealed that AhR was localized in both the cytoplasm and nuclei of vehicle-treated cells (Fig. 7B), which is consistent with other studies reporting nuclear expression of AhR in the absence of a known exogenous ligand [52]. After TCDD, there was an increase in nuclear-localized AhR (Fig. 7B), demonstrating that AhR is responsive to TCDD.
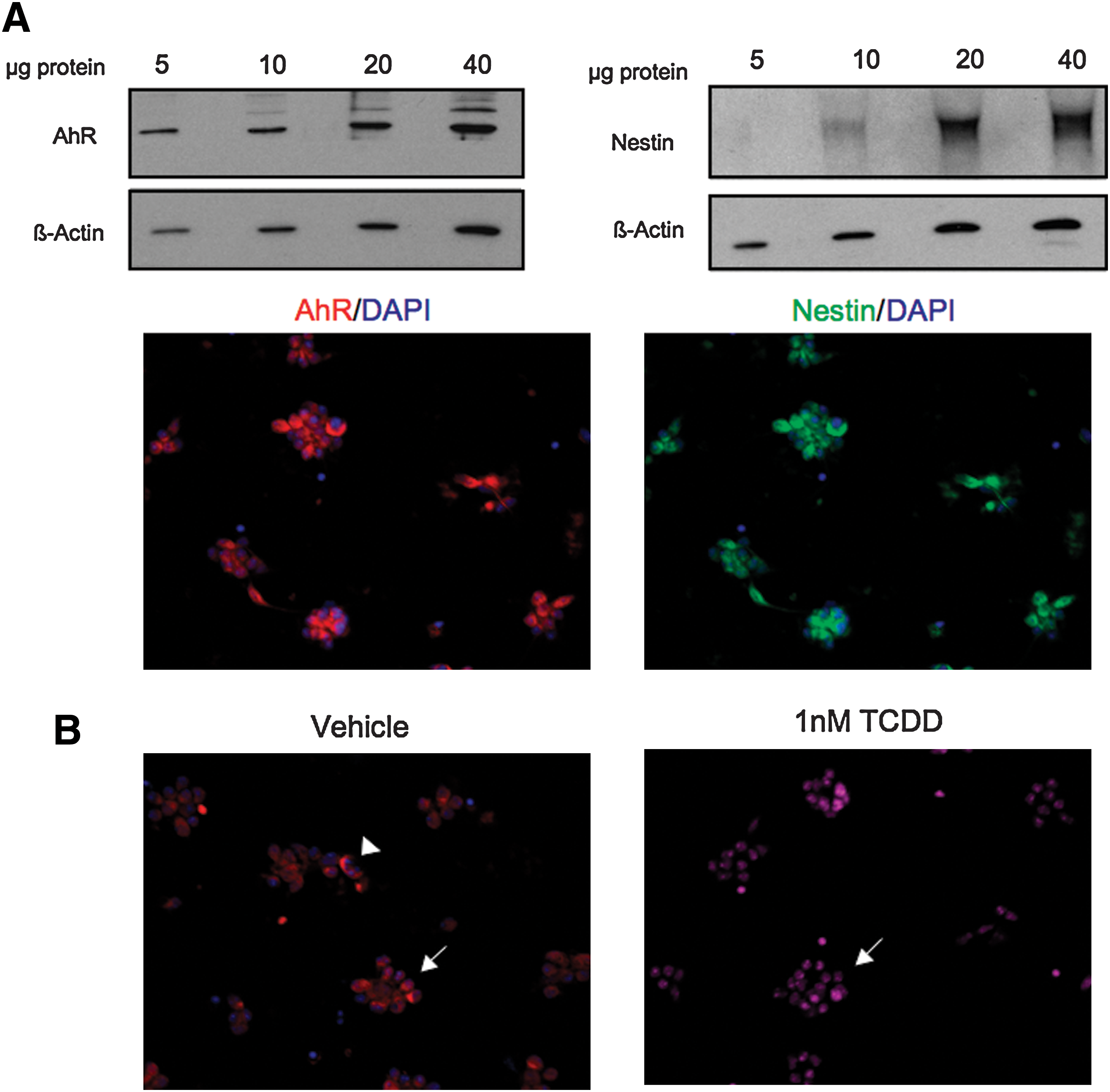
Primary neural precursor cells (NPCs) from the forebrain of mouse embryos express nestin and AhR. (
TCDD reduced cell proliferation and leads to a G1 phase arrest in primary NPCs without altering survival
To investigate the effects of AhR activation by TCDD on cell proliferation, primary NPCs were cultured with or without EGF and bFGF in the absence of presence of 1 nM TCDD for 24 h then assayed for DNA synthesis. TCDD did not significantly alter DNA synthesis in cells grown without mitogenic factors (Fig. 8A). When cells were stimulated to proliferate with EGF and bFGF, coexposure to 1 nM TCDD for 24 h suppressed EGF- and bFGF-mediated proliferation (Fig. 8A) without the loss of cell viability (Fig. 8B).
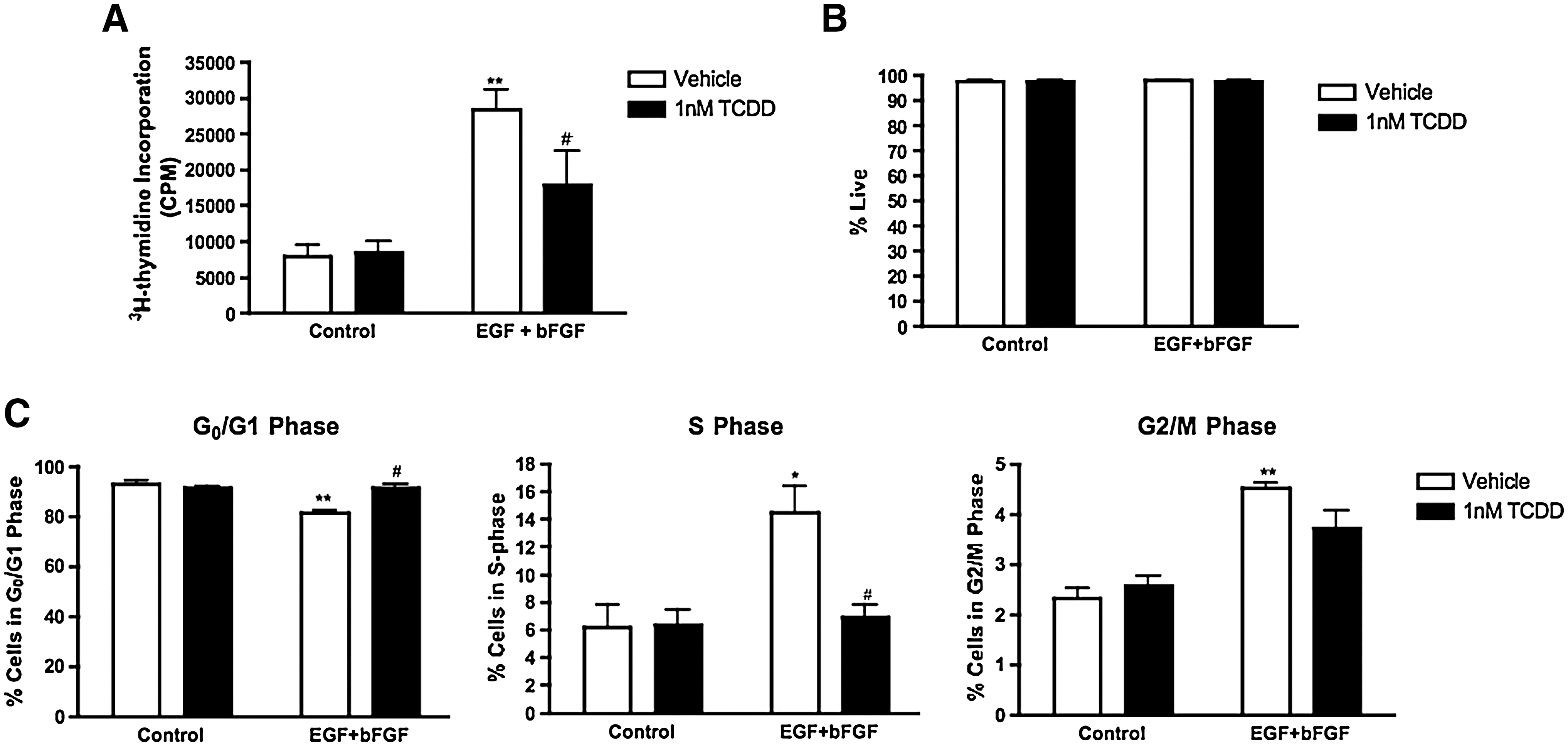
TCDD suppresses proliferation in primary NPCs forebrain without causing cell death.
Primary NPCs were next subjected to flow cytometric analysis to investigate if the reduced proliferation was also due to a G1 to S phase arrest that was observed in C17.2 cells. The percentage of cells in the G0/G1, S, and G2/M phases of the cell cycle was determined in the same manner as for C17.2 cells. When E12.5 NPCs cells were stimulated to proliferate with EGF and bFGF, there was a significant decrease in the percentage of cells in the G0/G1 phase, which coincided with a significant increase in the percentage of cells in S phase (Fig. 8C). As expected, exposure to 1 nM TCDD for 24 h did not significantly alter the cell cycle phase distribution in cells growing without EGF and bFGF. In the presence of TCDD there was a 10% increase (P < 0.05) in the percentage of cells in the G0/G1 phase and an 8% decrease (P < 0.05) in the number of cells in the S phase when cells were induced to proliferate in the presence of EGF and bFGF (Fig. 8C). TCDD did not elicit any significant change in the percentage of cells in the G2/M phase. Our observations in primary NPCs provide additional evidence that TCDD reduces mitogenic actions in neural precursor cells by arresting cells in the G1 phase of the cell cycle.
Discussion
In this study TCDD served as an environmentally relevant chemical to test the hypothesis that inappropriate activation of AhR interferes with neurogenesis by disrupting signaling pathways important for NPC proliferation. The effects of AhR activation by TCDD on NPC proliferation were investigated in the C17.2 cell line model and in NPCs derived from the mouse embryonic forebrain. Whereas TCDD-induced G1 arrest in cell proliferation has been previously observed in the SK-N-SH neuroblastoma cell line [33], derived from a neuroendocrine tumor that originates from the neural crest of peripheral nervous system, this model is not reflective of a neural precursor in central nervous system. In contrast, our study is the first to report that TCDD interferes with the proliferation of multipotent NPCs in 2 different cell models derived from the developing brain. Although C17.2 cells are derived from progenitor cells of a mouse cerebellum, they are predominately used as a model for multipotent stem cells [53 –55]. C17.2 precursor cells possess distinct characteristics from cerebellar granule cell precursors [56] and have the unique ability to self-renew and generate both neurons and glia in vitro [57]. We determined that AhR was robustly expressed in C17.2 cells. Moreover, studies with TCDD demonstrated that C17.2 NPCs contain an intact and active AhR pathway, as evidenced by nuclear localization of AhR, AhRE binding, and an increase in AhRE-driven luciferase activity after TCDD exposure. C17.2 NPC proliferation was disrupted after TCDD exposure, suggesting that untimely or sustained AhR activation could impede neurogenesis. Studies in primary NPCs prepared from mouse embryos confirmed reduced cell proliferation and a G1 phase arrest after dioxin exposure, which could have long-term consequences on final cell number in the brain. Collectively, our observations revealed that NPCs are novel targets for the neurotoxic actions of TCDD and suggest potential roles for AhR during neurogenesis. Moreover, studies confirming TCDD effects on proliferation in primary NPCs validate the C17.2 model for future mechanistic studies that require large numbers of cells, which are challenging to isolate in sufficient quantities from embryonic brain tissue.
Since neurogenesis is mediated by a tightly controlled spatiotemporal program that includes proliferation, differentiation, and apoptosis [3,58], there are likely to be precise windows that may be vulnerable to environmental insult. For example, it has been shown that the nervous system is particularly sensitive to antimitotic agents such as methylazoxymethanol when proliferation is actively occurring in a given region. However, when cell division ceases, the brain is more resilient [59]. Proliferative cells in the brains of experimental animals have also been shown known to be specifically targeted by exposure to other teratogens, such as ethanol [60,61], methyl mercury [62], and organophosphorous pesticides [63,64]. Because proliferating NSCs/NPCs robustly express AhR in vivo [39], the maturing brain is likely to be more susceptible to TCDD exposure during critical periods of embryonic development. In addition, we have previously demonstrated that the developing brain is also vulnerable to TCDD exposure during critical periods of postnatal neurogenesis [31]. For these reasons, TCDD can be added to the growing list of environmental contaminants that impair nervous system development, which could ultimately hinder proper brain function.
Our observations suggested that the susceptibility of NPCs to dioxin exposure may be dependent on the specific neurodevelopmental event occurring at the time of exposure. When cell cycle progression was initiated with growth factors, TCDD reduced the mitogenic effects in both the C17.2 cell line and primary NPCs. TCDD was found to impede proliferation in dividing NPCs by suppressing DNA synthesis and cell growth. Because NPC proliferation is disrupted after AhR activation by TCDD, it suggests that AhR may be involved in regulating NPC proliferation.
Further examination of the NPC growth arrest revealed that TCDD exposure interfered with the G0/G1 to S phase transition in mitogen-stimulated cells. One mechanism of G0/G1 phase arrest can be associated with a reduction in G1 phase cyclins. Cyclin D proteins are G1 phase regulators that act as mitogen sensors and their activation allows noncycling cells to reenter the cell cycle. A reduction in cyclin D levels can hinder the ability of cells to respond to extracellular mitogens and consequently inhibit cell cycle activity [65]. While TCDD did not alter cyclin D1 levels in quiescent C17.2 cells, our studies demonstrated that TCDD exposure in C17.2 cells dividing in the presence of a mitogen had significantly less cyclin D1 protein levels compared to nonexposed cells. Because cyclin D1 has not been identified as an AhR target gene, this observation suggests that while TCDD may not directly target cyclin D1, exposure may interfere with the ability of cells to respond to extracellular mitogens and hence their ability to initiate cell cycle progression. This finding is consistent with other reports demonstrating that TCDD interferes with the G0/G1 to S transition in proliferating, but not quiescent, cells by modulating expression of regulatory proteins controlling cell cycle progression [29].
An additional mechanism for G0/G1 phase arrest could involve enhanced levels of antimitogenic regulatory proteins such as the cdk2 inhibitor p27 kip1 [28]. Cdk2 activity is necessary for entry into the S-phase and its inhibition by p27 kip1 stalls the cell cycle in the G0/G1 phase. In addition to drug metabolizing enzymes such as cytochrome p450s, p27 kip1 is known to be a direct target of AhR-mediated transcriptional regulation [21,28]. Therefore, directly targeting p27 kip1 is a feasible mechanism by which inappropriate activation of AhR by exogenous ligands may interfere with the G0/G1 to S phase transition and consequently induce cell cycle arrest. Our studies demonstrated that exposure of quiescent, nonproliferating C17.2 cells to TCDD stimulated p27 kip1 expression, which is in contrast to our cyclin D1 results. Cells exposed to TCDD in the presence of a mitogen also had significantly higher p27 kip1 expression than cells exposed to the respective mitogen alone [65]. Because p27 kip1 is an AhR target gene and its expression is elevated in nonproliferating cells, it is possible that AhR activation by TCDD may first directly upregulate p27 kip1 and contribute to cyclin D1 downregulation, which instigates G0/G1 phase arrest. Supporting this idea, studies in the chick embryonic spinal cord have demonstrated that forced expression of Cip/Kip cell cycle inhibitors in proliferating progenitor cells blocks cell cycle progression by antagonizing cyclin D1 activities, suggesting that an upregulation or overexpression of Cip/Kip inhibitors can subsequently downregulate cyclin D1 activity [66]. Taken together, our observations suggest that TCDD exposure may disrupt the dual regulation of p27 kip1 and cyclin D1 to suppress NPC proliferation at the G1 to S phase transition.
The principal hypothesis of our study was that TCDD targets NPCs by interfering with AhR-mediated signaling events that are vital for NPC development. Neurogenesis is a multifaceted process and NPC proliferation is crucial in setting the stage for proper nervous system development. As a result, disrupting NPC proliferation by TCDD may have adverse consequences on nervous system development and function. From our findings, we propose that dioxin exposure suppresses NPC cell proliferation, suggesting that perturbations of normal AhR-mediated signaling events may impinge on regulatory mechanisms that exist to modulate neuronal development. However, additional studies are necessary to gain further understanding of the adverse impact of TCDD on NPCs during critical developmental periods in vivo, as well as to elucidate the biological roles of AhR during neurogenesis.
Footnotes
Acknowledgments
This research was supported by the National Institutes of Health (R21 ES013512, R01 ES016357, P30 ES01247, and T32 ES07026). The authors gratefully acknowledge Bryan Thompson for technical assistance, Dr. Michael Laiosa for assistance with flow cytometry analysis, and Dr. Michelle Janelsins for helpful discussions. The authors also thank Dr. Loretta Collins for her comments during the preparation of this article.
Author Disclosure Statement
No competing financial interests exist.