
Abstract
Most stem cell therapies involve direct, intraparachymal placement of neural progenitor cells. These cells provide physical support to the endogenous neuronal population and may be engineered to provide in situ growth factor support. Insulin-like growth factor-I (IGF-I) has potent neurotrophic and neuroprotective properties and is expressed by human neural stem cells (hNSCs). IGF-I is implicated in multiple aspects of cell behavior, including proliferation, differentiation, and survival. Enhancing hNSC function through IGF-I overexpression may increase the benefits of stem cell therapy. As a first step to that goal, we examined the direct effects of IGF-I on hNSC behavior in vitro. We demonstrate that IGF-I treatment enhances both the number and length of hNSC neurites. This is correlated with a decrease in proliferation, suggesting that IGF-I promotes neurite outgrowth but not proliferation. While IGF-I activates both AKT and MAPK signaling in hNSCs, we demonstrate that IGF-I-mediated neurite outgrowth is dependent only on AKT signaling. Finally, we demonstrate that IGF-I is neuroprotective after glutamate exposure in a model of excitotoxic cell death.
Introduction
H
IGF-I is a potent trophic factor essential for both neuronal development and normal function [5,6]. IGF-I signaling through the type I IGF receptor (IGF-IR) stimulates proliferation, differentiation, neurite outgrowth, and survival of multiple cell types, including MNs and sensory neurons, oligodendrocytes, and Schwann cells [7 –16]. Several groups have demonstrated the protective capacity of IGF-I in multiple cell types, including MNs [11,15,17,18]. Vincent et al. demonstrated that IGF-I protects primary MNs against glutamate excitotoxicity [11,19]. In vivo, IGF-I signaling is required in the developing cortex for axonal outgrowth [20]. When IGF-I-producing cells were grafted into the spinal cord after injury, in situ IGF-I production was sufficient to protect against neural degeneration and promote partial re-growth [21].
Advances in cellular therapies have led to the development of combinatorial strategies. Donor cells engineered to produce additional growth factors provide both cellular and trophic support as potential therapeutics [22 –25]. For multifaceted neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) and Alzheimer's disease, cellular therapies may provide a possibility for therapeutic development circumventing unknown cellular mechanisms. Recent studies demonstrate the increased potency of neural precursors in conjunction with GDNF for MN support in ALS models [26,27]. Initial development of hNSC treatment for both spinal cord trauma and ALS has demonstrated their beneficial potential as a treatment option [2 –4]. Currently, the hNSCs described here are the first cellular therapy approved for Phase 1 human trials in ALS due to their innate ability to protect MNs and effectively incorporate into the spinal cord. Enhancing hNSC function by adding additional trophic factor production may result in greater therapeutic potential.
As a first step toward studying the combination of IGF-I with hNSC therapy for neuronal injury, we investigated the direct effects of IGF-I on hNSC in chemically defined culture. We demonstrate that IGF-I has positive effects on both neuronal survival and axonal outgrowth without affecting proliferation or differentiation of hNSCs. Our findings suggest that a combination of IGF-I overexpression and hNSC may enhance stem cell therapy for the treatment of neurodegenerative diseases and neuronal injuries.
Materials and Methods
Cell culture
All culture reagents were obtained from Sigma unless otherwise noted. hNSCs were prepared from a cervical–upper thoracic region of spinal cord tissue obtained from a single 8-week human fetus after an elective abortion. The fetal tissue was donated by the mother in a manner fully compliant with the guidelines of NIH and FDA and approved by an outside independent review board as described [28]. Cell culture was carried out as described previously [1,3]. Briefly, flasks were prepared by incubation for 24 h at room temperature with 100 μg/mL poly-
Immunocytochemistry
Immunocytochemistry (ICC) was performed as previously described [29]. Briefly, hNSCs were grown on poly-
To measure proliferation, cells were grown as above and cells were incubated with 10 μM 5′-ethynyl-2′-deoxyuridine (EdU) for 2 h before fixation and processing following the manufacturers' protocols for the Click-It EdU kit (Invitrogen). We measured EdU incorporation by fluorescence and images were captured using an Olympus BX-51 microscope equipped with a digital camera. For neural index measurements, TuJ1-labeled images and their corresponding DAPI images were opened in MetaMorph. Cell number was counted using the “count nuclei” plug-in, and manual adjustment was made to correct for any miscounted cells. To measure the area covered with neuritis, images were thresholded and the area covered by neurites was measured using region statistics. Neural index is expressed as neurite area/cell (μm2/cell). Measurements represent a minimum of 2,000 cells from 5 images in 3 independent experiments for a total of at least 6,000 cells.
RNA isolation and real-time polymerase chain reaction
Total RNA was extracted from hNSCs cultured in the presence or absence of IGF-I using an RNeasy Kit (Qiagen) according to the manufacturers' instructions. Reverse transcription was performed using the iScript cDNA Synthesis Kit (Bio-Rad). Real-time polymerase chain reactions (PCRs) were carried out in 96-well 0.2 mL PCR plates sealed with iCycler Optical Sealing tapes (Bio-Rad). PCR primers are listed in Table 1. Growth factor primers were previously described [1]. The PCR amplification profile was as follows: 95°C for 5 min, 40 cycles of denaturation at 95°C for 30 s, annealing at 58°C for 1 min, and extension at 72°C for 30 s, and a final phase of 72°C for 5 min. The fluorescence threshold C t value was calculated by iCycler iQ system software and the levels were first normalized to the endogenous reference actin (ΔC t), followed by relation to the control (ΔΔC t) and expressed as 2−ΔΔCt. PCR product levels were expressed as mean ± SEM and a two-sample equal variance t-test was performed (N = 3 replicates over 3 independent experiments).
BDNF, brain-derived neurotrophic factor; GDNF, glial-cell-derived neurotrophic factor; IGF-1, insulin-like growth factor-I; IGF-IR, type I IGF receptor; Syn, synaptophysin; VEGF, vascular endothelial growth factor.
Western blotting
Western blotting was performed as previously described [30]. hNSC lysates were prepared by lysing cells in modified RIPA buffer [20 mM Tris (pH 7.4), 150 mM sodium chloride, 1 mM ethylenediaminetetraacetic acid, 0.1% sodium dodecyl sulfate, 1 mM sodium deoxycholate, 1% Triton X-100, 0.1 trypsin units/L aprotinin, 10 mg/mL leupeptin, and 50 mg/mL phenylmethylsulfonyl fluoride (PMSF)]. Equal amounts of protein were loaded in each lane of a polyacrylamide gel, with gel percentages (7.5%, 12.5%, or 15%) dependent on the size of the protein of interest. Polyvinylidene fluoride membranes were incubated with primary antibody overnight at 4°C and incubated with appropriate horseradish-peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) for 1 h at 22°C. Primary antibodies used are from Cell Signaling unless otherwise indicated: phospho-p44/42 (Thr202/Tyr204) MAPK; phospho-Akt (Ser473); phospho-IGF-IRβ (Tyr1135/1136); p-mTOR (Ser2448); glyceraldehyde 3-phophate dehydrogenase (GAPDH; Chemicon). Antibody binding was developed with LumiGLO Reagent and Peroxide (Cell Signaling) and exposed to Kodak BioMax XAR film (Sigma). Each experimental paradigm was tested on 3 separate occasions using different cultures or tissue samples. Membranes were re-probed for GAPDH to confirm equal protein loading.
TUNEL analysis
hNSCs were grown and treated on glass coverslips followed by fixation in 4% PFA for 5 min. TUNEL was used to detect DNA fragmentation as an indication of apoptotic death. hNSC plating densities were equal between treatment conditions. TUNEL analysis included blinded counting of at least 10 representative fields per condition in at least 3 independent experiments per our published protocol for an average total of ∼1,000–2,000 hNSCs per condition. Samples were labeled with digoxygenin-dUTP and then stained with horseradish-peroxidase-conjugated anti-digoxygenin antibody using the ApopTag Plus In Situ Apoptosis Peroxidase Detection Kit (Chemicon). Alternatively, fluorescent TUNEL processing was carried out as described above with a fluorescein-labeled conjugate using the ApopTag Plus In Situ Apoptosis Fluorescein Detection Kit (Chemicon). Fluorescent signal was detected and recorded using an Olympus BX-51 microscope.
Results
hNSC differentiation and growth factor production
Previous studies documented the differentiation pattern and growth factor profile of hNSCs at extended differentiation time points [1]. Our goal was to examine hNSCs over a time period when they were plastic, that is, leaving their proliferative stage and entering terminal differentiation, a period when production and influence of endogenous growth factors were likely to be the most profound. mRNA was collected for quantitative real-time PCR from hNSCs that were either undifferentiated (UD) or had differentiated for either 3 or 7 days. Differentiation was initiated by removal of FGF, the sole mitogen required to maintain hNSC proliferation, from the medium. UD hNSCs were grown for 24 h before mRNA collection. Day 3 (D3) and day 7 (D7) hNSCs were cultured in the differentiation medium containing no FGF. UD hNSCs exhibited baseline levels of IGF-I, VEGF, BDNF, or GDNF (Fig. 1A). At D3, an increase in the expression level of both IGF-I and VEGF was detected (Fig. 1B). By D7, there was a 2.5-fold increase in IGF-I gene expression with continued low-level expression of VEGF (Fig. 1C). Baseline levels of IGF-IR were detectable in UD hNSCs, and increased at both D3 and D7 (Fig. 1D). These data demonstrate that IGF-I and VEGF are endogenously produced during early differentiation.
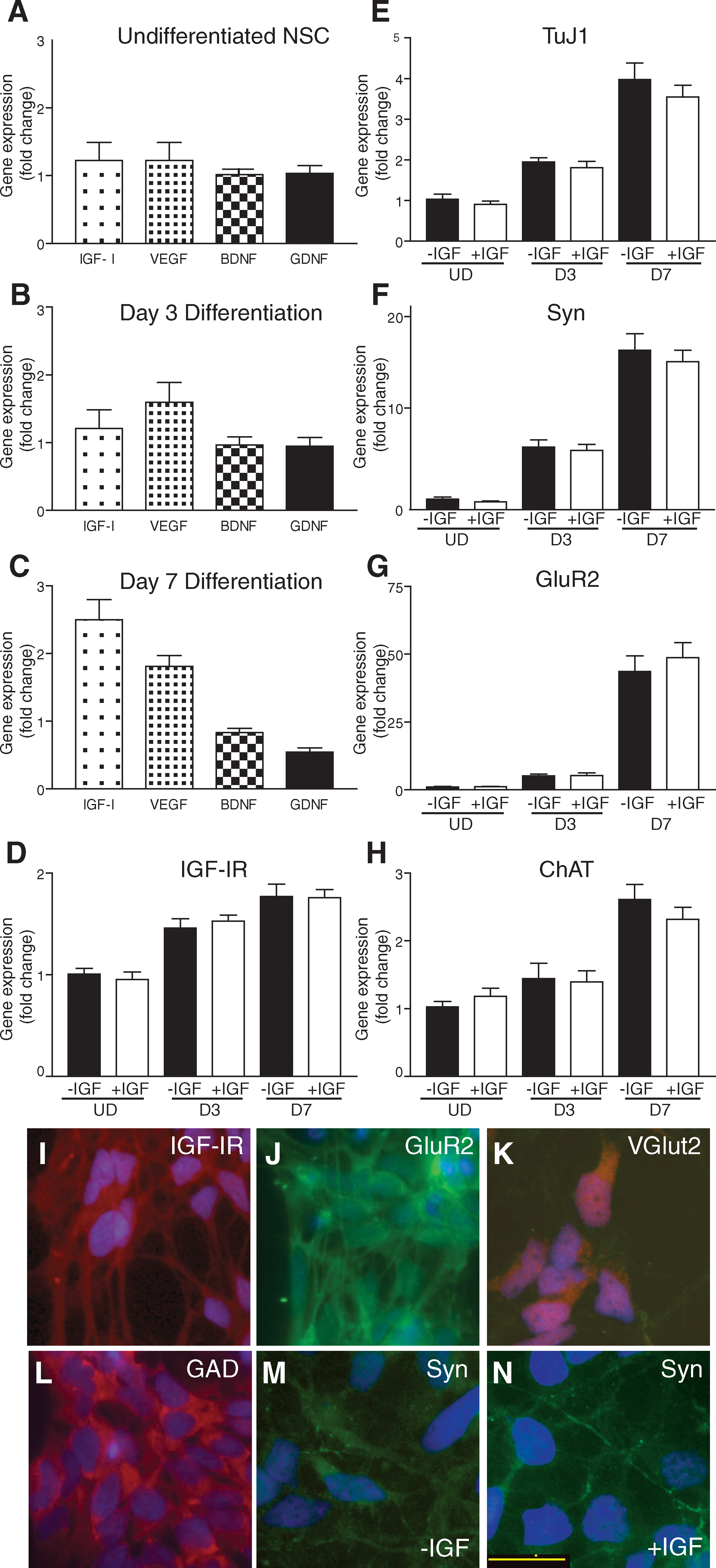
Growth factor and differentiation makers were measured by polymerase chain reaction analysis of hNSCs over the first 7 days of differentiation. (
We next examined whether the addition of IGF-I during the early differentiation process alters neurotransmitters or maturation markers in hNSCs using quantitative real-time PCR. Over the course of 7 days of differentiation, we observed a temporally appropriate increase in TuJ1 (an early marker of neurons) and Syn (a synaptic marker) compared to UD cells (Fig. 1E, F). We also observed a 48-fold increase in GluR2 expression. After the addition of IGF-I, we did not detect significant changes in gene expression for any of the examined genes (Fig. 1D–H). Likewise, there was no change on growth factor production (data not shown). These data demonstrate that additional IGF-I has no effect on the expression profile during hNSC differentiation.
To confirm the identity and maturation of hNSCs into neurons at D7, we next investigated protein localization by ICC labeling. We observed expression of IGF-IR along the cell surface and neurites of hNSCs at D7 (Fig. 1I). The striking increase in expression of GluR2 at D7 was reflected by ICC and we detected extensive localization on the cell body and along neurites (Fig. 1J). Two neurotransmitter markers of cell fate expressed by hNSCs, VGlut2, and GAD [1] were highly expressed in all hNSCs at D7 (Fig. 1K, L). At D7 we observed no expression of astrocyte or oligodendrocyte markers (GFAP and Olig2, respectively; data not shown). Finally, we examined the localization of Syn along neurites in culture. Syn-positive terminals were detected on a small subset of neurites at D7 indicative of synapse formation (Fig. 1M, N). These data support the extensive neural differentiation and maturation of hNSCs by D7 of culture.
IGF-I effects on neurite outgrowth and proliferation
IGF-I promotes axonal outgrowth [29,31 –33]; therefore, we next assessed the effect of IGF-I on hNSC neurite outgrowth as an early indicator of NSC neuronal differentiation [34]. We cultured hNSCs as a monolayer on glass coverslips in the presence or absence of IGF-I for the first 7 days of differentiation. Both the number of hNSCs with neurites and neurite length were quantified for UD hNSCs after 24 h of IGF-I, and D3 hNSCs cultured for 72 h in IGF-I. hNSCs were immunolabeled with NF to observe primary neuronal processes, and 500–700 individual cells were counted per condition over 3 experiments. Neurites were assessed as projections from the cell body ≥2 cell diameters. Under UD hNSC conditions after 24 h of IGF-I treatment, we detected an increase in the number of hNSCs with neurites; however, this did not reach statistical significance. By D3, IGF-I treatment resulted in ∼20% of hNSCs extending primary neurites compared to only 5% of untreated hNSCs (Fig. 2A). We next measured neurite length using ImageJ [35]. Neurite length of UD hNSCs was not significantly increased by IGF-I treatment. At D3, IGF-I-induced neurite growth was significantly increased from 191 to 233 nm (P < 0.01; Fig. 2B). By D7, hNSCs require a high plating density for survival precluding the low culture density conditions necessary for observing single neurites. To examine the effects of IGF-I at D7, we relied on image analysis to measure a neural index. The neural index is expressed as the complete neuronal area divided by the number of nuclei, and results are presented as neurite area per cell (μm2/cell). hNSCs were immunolabeled by TuJ1 (neurites) and stained with DAPI (nuclei), which allowed us to take into account any variability in density across experiments. For validation, 6 images per condition were counted from 3 individual experiments representing over 6,000 DAPI-labeled cells. DAPI-positive cells were counted using MetaMorph image analysis software, and the corresponding TuJ1-positive image was inclusively thresholded to measure the total area covered in TuJ1-positive neurites (Fig. 2C). This method does not measure pixel intensity, only the area covered in neurites. Using this method, D3 samples treated with IGF-I demonstrated a significant increase in the neural index compared to untreated cells (10 μm2/cell vs. 4.7 μm2/cell, respectively; P < 0.001; Fig. 2F). The D3 data confirmed that this method was sensitive enough to detect small changes in neurite outgrowth. At D7, IGF-I treatment for the duration of culture significantly increased the neural index from 70 to 91 μm2/cell (P < 0.02; Fig. 2I), indicating a sustained effect on neurite length at D7. These data demonstrate that IGF-I stimulates hNSC neurite outgrowth and enhances NSC maturation over the first 7 days of differentiation.
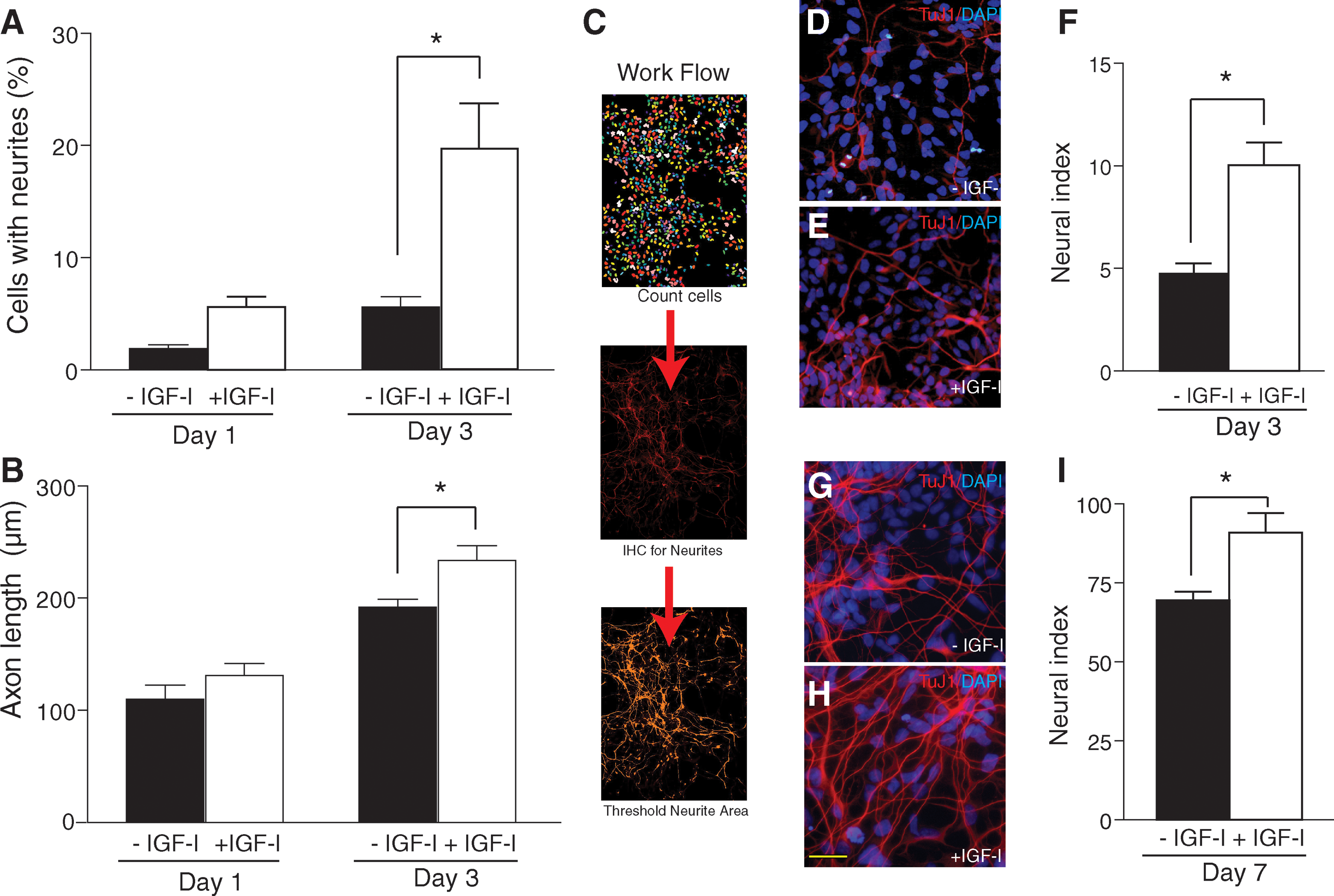
IGF-I increases neurite outgrowth in differentiating hNSC. (
Neurite outgrowth normally occurs in postmitotic neurons. We next examined the effects of IGF-I on cell proliferation. hNSC cultures at UD, D3, or D7 were incubated with EdU for 2 h before fixation. Approximately 1,000 cells were counted per experiment for the UD samples (N = 3). For D3 and D7 hNSCs, which are plated at higher density, a minimum of 3,000 cells were counted per experiment (N = 3; Fig. 3B). Approximately 27% of untreated UD hNSCs were EdU positive (Fig. 3A; Control UD). When UD hNSCs were treated with IGF-I, we detected a small reduction in EdU incorporation (25%; Fig. 3A). At D3, the effect of IGF-I treatment was significant with a reduction in EdU-positive nuclei from 17% to 14% (P < 0.02; Fig. 3A). We did not observe any change at D7, suggesting that the effects on proliferation are only associated with the earlier time points (Fig. 3A). These data demonstrate that treatment of hNSC cultures with IGF-I does not promote or maintain proliferation during initial stages of differentiation.
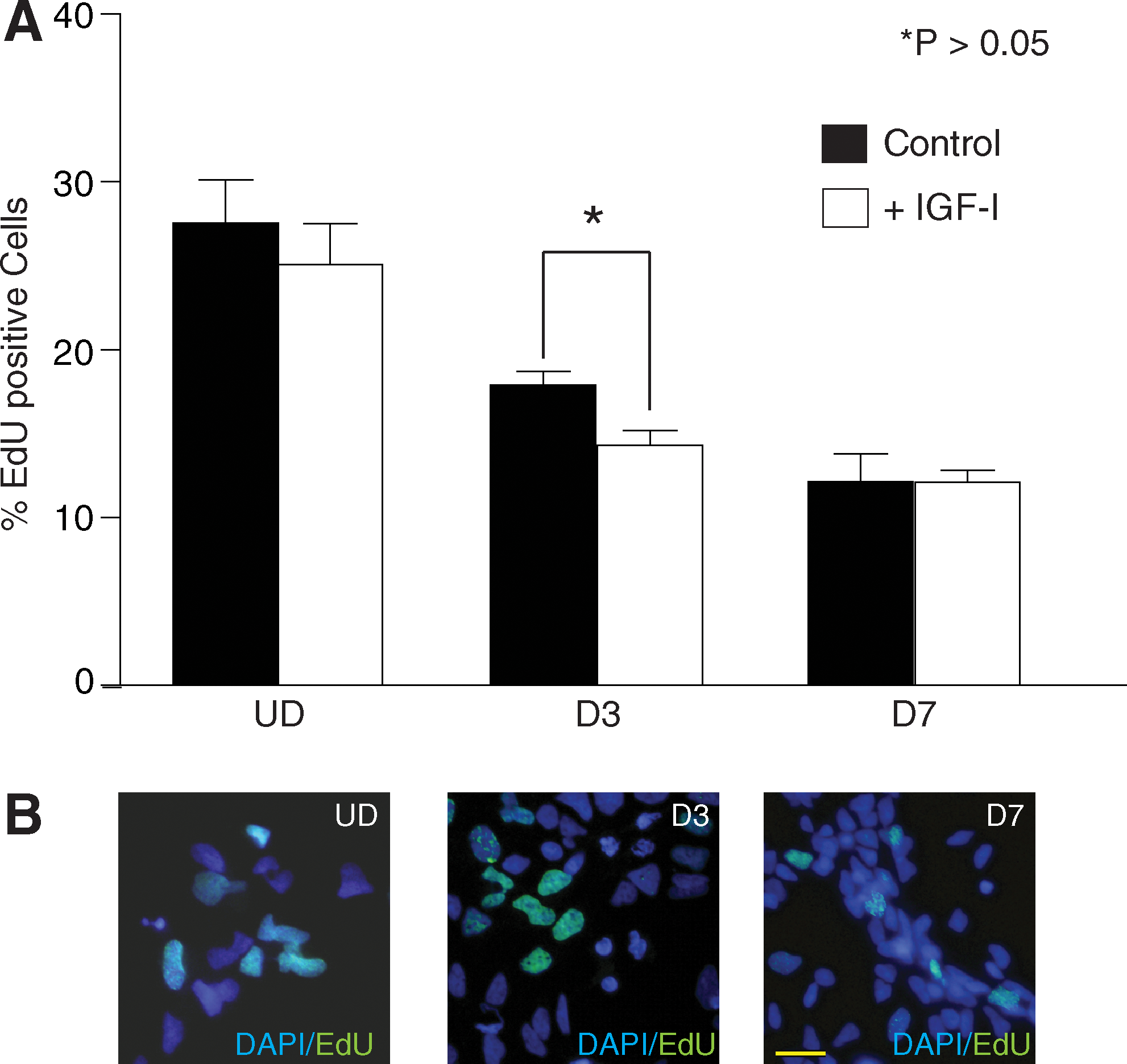
Effects of IGF-I on hNSC proliferation. (
IGF-I-mediated signaling
IGF-I signaling stimulates neurite outgrowth via either AKT or MAPK [8,29,33,36 –38]; therefore, we investigated which signaling mechanisms are active in hNSCs and responsible for IGF-I-mediated neurite outgrowth. hNSCs were treated with IGF-I and the activation was examined for up to 2 h to examine the time course of down stream signaling. The specificity of IGF-I signaling was assessed using pathway-specific inhibitors of MAPK (U0126; U) or AKT (LY294002; LY), or with the IGF-IR inhibitor NVP-AEW541 (NVP). For all hNSCs (UD, D3, and D7), we detected IGF-I activation of IGF-IR within 5 min of stimulation (Fig. 4A–C). Levels of IGF-IR were considerably lower in UD samples, requiring additional amplification with a stronger enhanced chemiluminescence (ECL) reagent to observe receptor phosphorylation (Fig. 4A). With amplification, we did observe the same pattern of IGF-IR activation at D3 and D7. IGF-IR signaling was abolished in the presence of IGF-IR antagonist, NVP. In UD hNSCs, we observed low levels of basal AKT activity and rapid activation of AKT signaling within 5 min of stimulation. AKT activation returned to near baseline 2 h after stimulation. The same pattern was observed at D3 and D7 (Fig. 4B, C). We next examined mTOR, a downstream effector of AKT signaling associated with enhancing NSC differentiation [39]. We detected little mTOR phosphorylation in UD hNSC samples (Fig. 4A). At D3 and D7, we observe phosphorylation of mTOR in response to IGF-I signaling (Fig. 4B, C). In UD hNSCs, MAPK signaling was activated by 5 min and maintained for up to 30 min. Under basal conditions, we detected low levels of MAPK signaling at D3 and D7. After IGF-I stimulation, there was a robust increase in MAPK signaling by 5 min, which was maintained over the course of the experiment. hNSCs treated with NVP and IGF-I or NVP alone at D7 exhibited a reduction in MAPK activation below control conditions. When MAPK signaling was inhibited we observed increased AKT phosphorylation, suggesting that inhibition of IGF-I-mediated MAPK signaling is compensated for by AKT. The reverse, however, does not occur, and attenuation of AKT signaling did not result in increased MAPK activation (Fig. 4A–C). Inhibitor-mediated changes in IGF-I signaling were maintained under all experimental conditions (Fig. 4A–C).
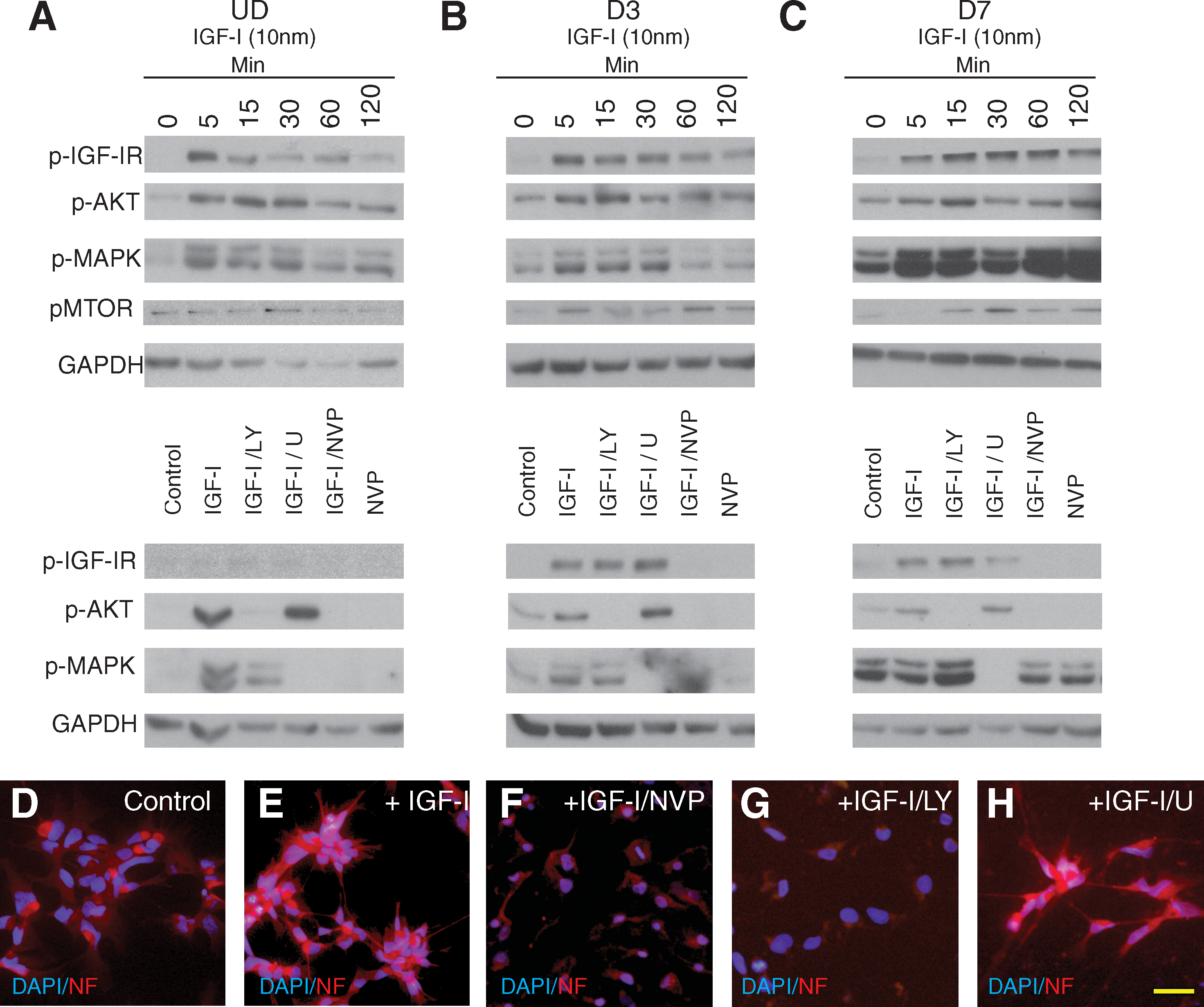
IGF-I-mediated signaling in hNSC. Western blot analysis of IGF-I signaling of UD, D3, and D7 hNSC. hNSC treated with either a time course of IGF-I (10 nM) stimulation between 0 and 120 min, or an inhibitor panel of LY, U, or NVP followed by 30 min IGF-I stimulation. (
We then investigated which downstream signaling pathway was required for IGF-I-mediated neurite outgrowth. hNSCs were treated with IGF-I with or without inhibitors for 24 h before fixation and ICC for NF to assess primary neurite outgrowth. By 24 h, IGF-I promotes increased neurite outgrowth compared to the control that remain rounded (Fig. 4D, E). When IGF-I signaling was inhibited with either NVP or LY, IGF-I-induced neurite outgrowth was lost and the hNSCs maintained their round UD morphology (Fig. 4F, G). When MAPK signaling was inhibited with U, IGF-I-mediated neurite outgrowth was maintained (Fig. 4H). Neurite growth in the presence of the inhibitors for 3 or 7 days resulted in increased cell death, suggesting that over a longer period both MAPK and AKT are required for survival and growth. Together these data demonstrate that IGF-I-induced neurite outgrowth is mediated via AKT but not MAPK in this system.
IGF-I-mediated neuroprotection of hNSCs
Glutamate excitotoxicity is one proposed pathophysiological mechanism for neurodegeneration in a variety of diseases such as stroke and ALS; therefore, we examined the effects of excitotoxicity on hNSC and the neuroprotective potential of IGF-I [40]. We first assessed the susceptibility of D7 hNSCs to glutamate excitotoxicity by treating hNSCs with increasing concentrations of glutamate from 1 to 200 μM. The hNSCs were fixed and cell death measured by assessing DNA fragmentation via TUNEL labeling. TUNEL-positive cells were counted and expressed as a percentage of the total cells. With increasing concentrations of glutamate, there was a dose-dependent increase in cell death (Fig. 5A). Maximal cell death was observed at 100 μM glutamate, which resulted in ∼28% cell death. To investigate the ability of IGF-I to protect against glutamate-induced cell death, D7 hNSCs were cocultured with 100 μM glutamate and 10 nM IGF-I. After 24 h, hNSCs were fixed and assessed for cell death. IGF-I reduced the level of hNSC death back to baseline in the presence of 100 μM glutamate (P < 0.001; Fig. 5B). These data suggest that IGF-I is protective against glutamate excitotoxic stress in neurons differentiated from hNSCs.

IGF-I-mediated neuroprotection of hNSC. Cell death after glutamate exposure was assessed by DNA fragmentation (TUNEL). (
Discussion
hNSCs in combination with IGF-I may be a more potent therapy than either alone for the treatment of both neurodegenerative diseases and neuronal injuries. In this study we demonstrated the positive effects of IGF-I on hNSC differentiation. We report that IGF-I increases neurite outgrowth, decreases proliferation, and is neuroprotective against glutamate excitotoxicity. Our findings demonstrate for the first time that IGF-I has a beneficial effect on the differentiation and survival of hNSCs and a proliferative effect on UD hNSCs.
In this study we focus on the initial 7 days of differentiation as we speculate that this is a critical period when hNSCs are plastic, that is, leaving the proliferative phase of their development and beginning terminal differentiation. hNSCs are directed along differentiation pathways by extracellular signaling molecules, including IGF-I [39,41,42]. We examined expression of 4 growth factors by PCR: GDNF, BDNF, VEGF, and IGF-I (Fig. 1). UD hNSCs display baseline expression of all 4 of these growth factors. On D3 we observe an increase in IGF-I and VEGF expression. By D7, there are higher levels of IGF-I than VEGF. These findings are consistent with previous studies demonstrating that by D14 of differentiation, IGF-I is the most highly represented of these 4 growth factors [1]. In UD hNSCs, we detect low levels of IGF-IR expression by PCR (Fig. 1). Given the changes at the gene expression level do not necessarily reflect changes in protein expression or functional signaling, we assessed expression and activation of IGF-IR. Western blotting reveals phosphorylation of both AKT and MAPK (Fig. 4), indicating functional receptor expression and activation of IGF-IR signaling pathways. At D3 and D7, differentiating hNSCs exhibit consistent activation of MAPK and AKT in response to IGF-I stimulation. Incubation of hNSCs with the IGF-IR blocker NVP abolished IGF-I activation of both pathways at all time points. Interestingly, incubation with NVP alone at D7 without the addition of IGF-I also reduced MAPK below baseline, suggesting endogenous IGF-I autocrine stimulation of MAPK signaling (Fig. 4). VEGF is expressed by hNSCs at D3 and D7 and we have previously shown that VEGF activates MAPK signaling in primary neurons [43]. Endogenous IGF-I and VEGF production and signaling may account for the basal MAPK activation at these time points (Fig. 1). Together these findings demonstrate that hNSCs are an early source of endogenous growth factors and that they respond to IGF-I stimulation, with activation of both AKT and MAPK signaling.
We next examined if IGF-I treatment directed early differentiation of hNSCs. We detected a temporal increase in expression of several markers associated with neuronal differentiation and maturation, including the neuronal markers neuron-specific tubulin (TuJ1) and Syn (Fig. 1). Surprisingly, we did not observe any changes at the transcriptional level of either of these markers after IGF-I treatment, suggesting that IGF-I does not affect neuronal fate or rate of maturation per se. By D7, we did observe Syn-positive synaptic terminals along neurites, indicating that hNSCs are beginning to mature and form synaptic connections. PCR results indicate that, as hNSCs differentiate, there is an increasing level of Syn. By ICC we also observe that D7 hNSCs are interconnected by Syn-positive terminals (Fig. 1). These data demonstrate that by D7, hNSCs are maturing and creating neural networks; however, IGF-I does not affect differentiation at a transcriptional level of the markers we assessed.
Neurite outgrowth is one of the earliest indicators of postmitotic hNSCs becoming terminally differentiated [34,44]. We demonstrate that IGF-I increases the number and length of neurites in hNSCs by D3 of differentiation and IGF-I-mediated axon outgrowth is maintained at D7 using a measure of neural index (Fig. 2). Using other cell types, we and others have shown that IGF-I directly impacts cytoskeletal rearrangement and enhances axon outgrowth [20,32,45,46]. Using signaling pathway-specific inhibitors, we observe that the IGF-I-mediated neurite outgrowth is mediated by AKT signaling (Fig. 4). These findings are consistent with IGF-I-mediated outgrowth in neuroblastoma cells, corticospinal neurons, and hippocampal neurons [20,33,46]. As hNSCs already express IGF-I, it may be possible that endogenous levels are insufficient to elicit maximal outgrowth. We do not detect changes in expression of different neural markers and suggest that IGF-I is enhancing and maintaining the development of hNSCs rather than directing differentiation. We anticipated a dual role for both AKT and MAPK signaling for neurite outgrowth based on previous studies on IGF-I action in both neuroblastoma and corticospinal neurons, hypothesizing that a common mechanism for signaling via MAPK would be required for outgrowth [20,32,46]. In these experiments, however, MAPK was not required for neurite outgrowth. mTOR mediates IGF-I-directed differentiation of forebrain-derived neural progenitor cells in vitro [39]. Consistent with these findings, we detected low levels of mTOR activation in UD hNSCs, while at later time points, especially at D7, we observed a robust activation of mTOR in response to IGF-I (Fig. 4). Together these findings highlight AKT signaling as the mechanism for IGF-I-mediated neurite outgrowth in hNSCs.
On the basis of these findings demonstrating the positive effects of IGF-I on neurite outgrowth, we hypothesized that IGF-I may enhance neural development of hNSCs. IGF-I is known to promote proliferation downstream of both AKT and MAPK signaling [47,48]. We wanted to examine whether IGF-I maintained proliferation of hNSCs. One limitation of cellular therapies based from stem cell technology is the potential for aberrant proliferation and formation of teratomas [49]. We observe that across all time points IGF-I does not maintain or promote proliferation of hNSCs (Fig. 3). Both of these findings regarding neurite outgrowth and differentiation at the expense of proliferation are supported by results in hNSCs of oligodendrocyte fate, where IGF-I increases the proportion of hNSCs differentiating concurrent with a decrease in proliferation [42]. These results demonstrate that combining IGF-I with hNSCs as a combined therapy would not promote proliferation and lead to unwanted propagation.
The capacity of IGF-I to prevent programmed cell death has been demonstrated in many neural cell types under a variety of conditions, including MNs and sensory neurons [10,11,15,17,50 –55]. One mechanism associated with MN cell death is via glutamate excitotoxicity [40,56,57]. We exposed hNSCs to glutamate to assess if hNSCs are susceptible to excitotoxic stress, and if so, is IGF-I neuroprotective. We observed 27% cell death after treatment with glutamate (Fig. 5). This is in contrast to primary MN that, under the same conditions, display up to 60% cell death [40]. These findings suggest that at D7, hNSCs are resistant to glutamate-induced cell death potentially due to an early neuronal differentiation state. Further differentiation of hNSCs, however, may confer increased sensitivity and therefore make them more susceptible to glutamate-induced cell death. The high levels of GluR2 expression (Fig. 1) may contribute to the increased excitotoxic resistance of hNSCs, given that the GluR2 subunit of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor regulates susceptibility to excitotoxic stress [56,58,59]. Yan et al. observed production of IGF-I up to the first month of differentiation before the level started to decrease; therefore, IGF-I may be protective in vivo [1].
Conclusions
Our findings demonstrate that IGF-I is produced early in hNSC differentiation. Direct treatment with IGF-I does not change the expression profile of hNSCs as they are differentiating, suggesting that IGF-I does not affect lineage selection, but rather enhances neural development and neurite outgrowth. Signaling via AKT, but not MAPK, mediates IGF-I-stimulated neurite outgrowth. hNSCs are more resistant to glutamate excitotoxicity than mature MNs. As such, hNSCs may be less susceptible to pathogenic factors while they mature; however, IGF-I remains a potent neuroprotective factor for excitotoxic stress in hNSCs. Finally, IGF-I does not promote proliferation of hNSCs. Our data support the idea that upregulation of IGF-I production in hNSCs may offer additional therapeutic benefits when hNSCs engineered to overexpress IGF-I are transplanted into the nervous system of animal models and humans with neurological disorders.
Footnotes
Acknowledgments
This work was supported by the A. Alfred Taubman Medical Institute and the Program for Neurology Research and Discovery. The authors thank Drs. Stacey Sakowski and Kelli Sullivan for scientific input and editorial assistance.
Author Disclosure Statement
Dr. Karl Johe has a financial interest in Neuralstem, Inc. The remaining authors would like to state that they have no commercial associations that might create a conflict of interest in connection with this article.
This work was presented at the Society for Neuroscience Meeting, 2009, Chicago, under the title of “Functional Effects of IGF-I During the Differentiation of Human Spinal Cord Stem Cells.”