
Abstract
Novel cell-based and gene therapies represent promising approaches for the treatment of incurable diseases, including cancer. Following the success of the hematopoietic stem cell-based transplantation, other populations of adult progenitor cells, including mesenchymal stem cells (MSCs), have been identified as powerful therapeutic tools in humans. The intrinsic capability of MSCs to migrate toward injured tissues emphasizes their suitability to deliver anticancer agents for new clinical applications in addition to the tissue repairing capacity. Here, we revisit the experimental history of MSCs, the most exciting features of their biology in keeping with their promising applications in cell-based therapeutic strategies for cancer treatment.
Introduction
H
On the other hand, a growing body of data also suggest that systemically injected MSCs exert an efficient therapeutic potential through the release of soluble mediators [3] and a constitutive immunosuppressive capacity, whereas undifferentiated MSCs display minimal, if any, immunogenic potential [4] and express both chemokine receptors and adhesion molecules enabling their homing function to injured sites in vivo in response to specific chemokine gradients [5]. Further, interesting but still debated data suggest that undifferentiated MSCs may constitutively show antiproliferative effects against primary tumors of mesenchymal derivation [6]. These observations, together with the development of cell engineering approaches, have provided a new rationale for the use of MSCs as carriers of therapeutic molecules and have definitively extended the MSC-based therapeutic strategies to anticancer programs. The efficacy of MSCs from different tissue sources to deliver anticancer molecules to sites of primary and metastatic tumors has been recently investigated in a number of preclinical models. Several therapeutic molecules, including interferons (INFs), interleukins (ILs), prodrug activating enzymes, apoptosis inducers, as well as oncolytic viruses, have been successfully delivered to tumors by MSCs, resulting in selective tumor suppression with efficient overcoming of the drug resistance to conventional cytotoxic chemotherapies.
Moreover, both poor immunogenicity of MSCs and possibility to plan allogeneic transplantation of engineered MSCs from adult and fetal tissues represent an additional advantage for this type of cell-based cancer gene therapy. However, several questions, including the potential effects of MSCs on tumor growth, their possible contribution to cancer immunotolerance, as well as the safety of their systemic administration, remain controversial and further investigations are required before planning controlled clinical trials in humans.
This review describes major aspects of the MSC biology in conjunction with the most promising applications of MSC-based approaches for cancer treatment recently reported by the medical literature.
Suitability of MSCs for Anticancer Treatments
Sources of MSCs
Availability of great amounts of MSCs is a prerequisite for cell-based gene therapies. This may, thus, represent a major limitation for the development of efficient cellular delivery strategies using autologous MSC preparations for each patient.
Today, variable quantities of MSCs can be easily isolated from a variety of human tissues, albeit the bone marrow (BM) is considered the primary source. Indeed, pluripotent cells that meet the current criteria defining MSCs have been efficiently isolated and expanded from a number of adult tissues including trabecular bone, fat, synovium, as well as skeletal muscle and dental pulpa [7 –12]. Although some of these expanded cultures may produce cell subsets with variable proliferation behaviors during the culture passages, they have been induced to stably produce specific transgenes and, in most cases, to be immortalized as allogeneic cell lines for potential new applications in preclinical settings of cell-based treatments. Recently, cells showing growth kinetics, plasticity, and suitability to genetic engineering, similar to adult MSCs, have been successfully obtained from umbilical cord blood [13], placenta [14], both amniotic fluid and membranes [15], and Wharton's jelly [16]. These tissues are ideal sources for MSC recovery because they include an easily accessible naive stem cell population exhibiting high proliferation rates and negligible immunogenicity and tumorigenicity, with no ethical concerns that would affect their use in both preclinical and clinical settings [17]. Fetal-derived mesenchymal progenitors have been, indeed, successfully utilized in experimental models of cancer treatment, showing a therapeutic potential similar to their adult counterpart. Moreover, recent data suggest that, despite their uniform morphology and immunophenotype, MSCs from different tissues may exert different biologic activities in vivo. This may be consistent with the different cytokine profiles of each MSC population in relation with its tissue origin and may be efficiently employed to optimize the use of certain subsets of MSCs for specific clinical indications.
Immunologic and homing properties of MSCs
As an attractive option for a wide range of anticancer therapies, MSCs from different sources have been accurately assessed in their differential phenotypes as surface molecules and their sensitivity to soluble factors that respectively activate their immunologic and homing functions.
Indeed, it is currently accepted that adult MSCs show poor immunogenicity and concomitant immunosuppressive activity that emphasize their suitability for transplantation approaches [4]. This “immune-privileged” status has been related to the poor surface expression of MHC class I antigens as well as to the lack of both MHC class II determinants and the costimulatory molecules CD80, CD86, CD40, and CD40L. Thus, based on the putative absence of MHC antigens, the mesenchymal progenitors are able to completely escape the natural killer (NK) cell-mediated cytotoxicity in vitro, suggesting that no interactions between NK cells and MSCs may occur in hosts transplanted with either autologous or allogeneic MSCs [18]. Moreover, MSCs express a number of surface molecules including CD90 (Thy-1), CD106 (vascular cell adhesion molecule-1), intercellular adhesion molecule 1 (ICAM-1), ICAM-2, and CD166 activated leucocyte cell adhesion molecule (ALCAM), which have cognate ligands on T cells, and they also constitutively produce variable amounts of immunologically active soluble factors, including transforming growth factor-β (TGF-β), hepatocyte growth factor (HGF), IL-2, IL-8, IL-10, as well as indolamine 2,3-dioxygenase, nitric oxide (NO), and prostaglandin E2 [19]. Thus, although the mechanisms underlying the interplay between MSCs and immune cells are mostly unknown, it is conceivable that they are mediated by both direct cell-to-cell interactions and soluble factors. In this context, current evidence emphasizes the immunomodulatory activity of MSCs [20] and indicates that they can exert multiple effects on immune cells both in vitro and in vivo.
BM-MSCs have been described to inhibit T-cell proliferation, cytokine secretion, and cytotoxicity [21,22]. Therefore, MSCs fail to elicit a proliferative response in alloreactive T lymphocytes, even prolonging the skin graft survival in mice [23]. Moreover, Le Blanc and colleagues demonstrated that IFN-γ–induced MSCs increase their class II antigen expression [24], but fail to promote the proliferation of allogeneic T cells. Similarly, MSCs, induced by viral transduction to express costimulatory molecules such as CD80 or CD86, also fail to generate appropriate T-cell response [25], whereas parallel in vivo studies proved that MSC-precultured memory T-cells show a weak response against specific antigens [26]. Similarly, MSCs are able to impair functions of other immune cells, including antibody production by B lymphocytes [27], as well as maturation, cytokine production, activation, and antigen presentation of dendritic cells (DCs) [28]. Taken together, these biological properties make the MSCs particularly suitable for transplantation in allogeneic recipients, although further in vivo studies are needed to definitively address the issue of immunoregulatory and anti-inflammatory activities of MSCs. Several microenvironmental cues may affect, indeed, the final outcome of MSC-mediated immunomodulation, as proved by the variable interaction of MSC with DCs and NK cells in the presence of different concentrations of endogenous IFN-γ [4].
Similarly to adult MSCs, fetal-derived mesenchymal progenitors maintain their low immunogenicity in conjunction with the immunomodulatory effect as supported by the loss of both human leucocyte antigen (HLA) class II antigens and costimulatory molecules, as well as by their inhibitory activity on allogeneic peripheral blood mononuclear cell proliferation and suppression of mature DC functions [29,30]. Therefore, human fetal-derived MSCs represent a considerable and virtually unlimited source of stem cells for future allogeneic applications, including the cell-mediated anticancer drug delivery.
Besides their immunologic features, the intrinsic capability of MSCs to both reach and survive within several organs such as BM, spleen, liver, and lung after systemic infusion may significantly influence their efficacy in delivering genes of interest [31]. However, MSCs also show a peculiar sensitivity to microenvironmental cues of damaged or inflamed tissues in relation to the local higher levels of chemokines, growth factors, and other soluble mediators [32]. Preclinical observations provide evidence on the efficiency of MSC engraftment after systemic infusions into several injured sites including stroked brain [33], ischemic myocardium [34], and dystrophic muscles [35]. In these instances, the MSC recruitment appears similar to that of leucocytes because, once activated by gradients of soluble factors, they extravasate for the expression of specific integrins and roll across the endothelium [36]. Indeed, under stimulation of specific inflammatory cytokines such as tumor necrosis factor-α (TNF-α), TGF-β, and IL-1β, MSCs also release matrix metalloproteinases and acquire the ability to cross the basement membranes to finally infiltrate the injured site [37].
Chemokines belong to the family of small, chemotactic cytokines originally identified on the basis of their ability to regulate the trafficking of immune cells. Their essential role, however, has been shown in hematopoiesis, in the embryologic development and angiogenesis, as well as in stem cell trafficking and tissue localization. Thus, the high expression levels of chemokine receptors by MSCs imply that freshly derived MSCs are able to exert higher migration and tissue homing capacity when compared with the cultured counterpart [38]. In contrast to hematopoietic stem cells (HSCs), whose mobilization and homing to the BM are mediated by the axis chemokine C-X-C motif receptor 4 (CXCR-4)/stromal cell-derived factor-1 (SDF-1), MSCs express lower levels of CXCR4 with respect to the intracellular content [39]. However, the surface expression of this chemokine may increase in relation to the activation state of MSCs. To this, Shi and coauthors reported the reinforcement of CXCR4 expression by MSCs in the presence of inflammatory cytokines [40].
The MSC migration capability in vitro may also be influenced by other functional axes including chemokine receptor (CCR1)/macrophage inflammatory protein 1-α, CCR2/macrophage chemoattractant protein 1, CCR4/chemokine ligand (CCL22), CCR7/CCL19–CCL21, and CXC-chemokine receptor (CXCR5)/B-cell attracting chemokine (BCA-1). Both chemokines, including CCL5/RANTES, SDF-1, and macrophage-derived chemokine (MDC)/CCL22, and growth factors, including HGF, platelet-derived growth factor, and insulin-like growth factor-1, exert efficient chemoattraction on MSCs, whereas after TNF-α stimulation, cultured MSCs enhance the expression of matrix metalloproteinases in parallel with their migratory activity. This is primarily dependent on the reinforced CXCR4 signal transduction rather than the membrane molecular expression. Thus, it is conceivable that both recruitment and homing of MSCs toward injured tissues may depend on both general and local inflammatory states that strengthen the propagation of the MSC intracellular signaling [5]. Further, adhesion molecules such as P-selectin, vascular cell adhesion molecule-1, ICAM-1, ALCAM, and other integrins including α3, α5, α6, β1, β2, and β3 subunits are closely involved in both MSC mobilization and homing and are strongly upregulated both in vitro and in vivo in response to inflammation stimuli [36] (Fig. 1). Thus, as the tumor microenvironment resembles the milieu of inflamed tissues [41], high levels of chemokines and soluble factors released by both malignant and normal cells drive a prompt homing of the MSCs to the tumor sites in relation to the high membrane expression of functional receptors. Indeed, increasing data in literature underline the ability of systemically injected MSCs to migrate to both primary and metastatic foci of solid tumors, including gastric and ovarian cancers, and to cross the blood–brain barrier toward gliomas and glioblastomas [42 –44].

MSC migration and homing to inflamed sites are mediated by chemokines, receptors, and cell membrane molecules. Chemokine receptors (typed in purple) expressed by MSCs promote their sensitivity to chemokines within the sites of injury and inflammation. Those with highest effect on MSCs are indicated in red. MSCs are also able to express, adhesion molecules (blue), and growth factor receptors (black) in keeping with chemoattractive molecules (red). MSCs, mesenchymal stem cells; MCP-1, macrophage chemoattractant protein 1; MIP, macrophage inflammatory protein; RANTES, regulated upon activation, normal T-cell expressed and secreted; SDF-1, stromal cell-derived factor-1; EGFR, epidermal growth factor receptor; HGFR, hepatocyte growth factor receptor; PDGFR, platelet-derived growth factor receptor; VEGFR, vascular endothelial growth factor; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1.
Based on such a specific property of MSCs to reach the tumor sites once systemically injected, their genetic modification to release tumor-suppressive molecules appears a promising therapeutic approach to defeat a number of cancers.
MSCs to Treat Solid Cancers
A controversial aspect related to the potential anticancer utilization of MSCs is that, once localized within tumor niche, MSCs may also promote cancer progression. Subcutaneous parallel injections of both cancer cells and MSCs in mice have been proved, indeed, to prime the development of tumors by increasing the vascular network and the invasive tumor tendency when compared with mice treated with malignant cells alone [45]. Moreover, the in vitro proliferation of breast cancer cells can be strongly enhanced by adding MSCs to the culture [46].
Despite such a potential of MSCs that primes tumor progression [47 –49], other studies emphasize their intrinsic powerful anticancer activity. Intravenously injected BM-MSCs, in fact, dramatically inhibit by themselves the tumor growth in animal models of Kaposi's sarcoma, Lewis lung carcinoma, and B16 melanoma as well as in rats bearing colon carcinoma or gliomas [6,50 –52]. On these bases, a number of subsequent investigations have demonstrated that genetically modified mesenchymal progenitor cells can be successfully used as vehicles delivering therapeutic molecules such as modulators of the immune system, oncolytic and antiangiogenetic factors, as well as prodrug-activating enzymes. Preclinical outcomes have been achieved in terms of therapeutic response primarily depending on the different transduction approaches used and on appropriateness of the transgene choice exploited for treating each tumor model. An incomplete list of anticancer treatments with MSCs in different animal models is shown in Table 1.
INF, interferon; IL, interleukins; CD-UPRT, cytosine deaminase-uracilphosphoribosiltransferase; TK, thymidine kinase; UCb, umbilical cord blood; BM, bone marrow; MSCs, mesenchymal stem cells; AD, adipose tissue; I.t., intratumoral; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
MSCs transduced with the IFN-β gene and systemically injected into mice bearing melanoma, breast cancer, glioma, and lung metastasis of prostate cancer resulted in significant inhibition of tumor growth, angiogenesis, and metastasis spreading [53 –56]. In addition, mesenchymal progenitors delivering IFN-α in murine models of lung metastatic melanoma exerted powerful suppression of both tumor vasculature and growth [57]. Using viral-mediated transfections, a number of ILs have been successfully delivered by MSCs within the tumor sites, leading to efficient cytotoxic effect on target tumor cells in vivo [58 –63]. For instance, in a recent study, Duan and colleagues evaluated the therapeutic efficacy of autologous BM-derived MSCs genetically modified to express IL-12, which was systemically injected into Ewing sarcoma–bearing mice. Interestingly, immunohistochemical analyses revealed that, once selectively homed to the tumor, IL-12–releasing MSCs were capable to induce a significant suppression of the cancer growth when compared with controls [58]. These data not only underline the usefulness of MSC-based therapies in cancer, but also emphasize their potential safety even for treatment of mesodermal-derived tumors. Similarly, systemic injections of IL-12–transfected MSCs were successfully exploited to treat a mouse xenograft model of renal cell carcinoma, whereas intratumor administrations of IL-18–engineered MSCs to malignant glioma rat models significantly prolonged their survival with respect to controls [62].
Other studies showed that targeted transgene delivery can also be supported by MSCs genetically induced to deliver oncolytic viruses, as well as to release antiangiogenic factors and express prodrug-converting enzymes combined with their chemical cytotoxic substrates. To this regard, MSC-based systemic transfer of the herpes simplex virus-thymidine kinase gene in combination with intraperitoneal injected ganciclovir showed a significant antitumor activity in mice models of pancreatic cancer [64]. Also, adult MSCs used to deliver the antagonist of HGF, NK4, for treatment of colon cancer lung metastases in vivo [65] have been shown to induce a significant prolongation of mice survival consistently with high rates of malignant cell apoptosis via inhibition of the tumor-associated angiogenesis. Alternatively, mesenchymal progenitor cells have been employed to deliver the enzyme cytosine deaminase, followed by systemic administration of the prodrug 5-fluorocytosine, to experimental tumors of both colon and prostate as well as to melanoma-bearing mice. In all instances, engineered MSCs significantly suppressed the cancer growth and prolonged the overall survival of the animals with these tumor models [66 –70].
In line with these results, TNF-related apoptosis-inducing ligand (TRAIL) was delivered by MSCs to solid tumors such as gliomas and lung carcinoma [71,72]. TRAIL is a protein that induces apoptosis in cancer cells while sparing normal cells, thus representing a new ideal candidate for tumor therapy (Fig. 2). Although, because of its short pharmacokinetic half-life, the recombinant soluble protein needs high dose and repeated infusions to be effective, this may, however, result in heterotopic cytotoxic effects as those detected in both hepatic and central nervous system sites [73].
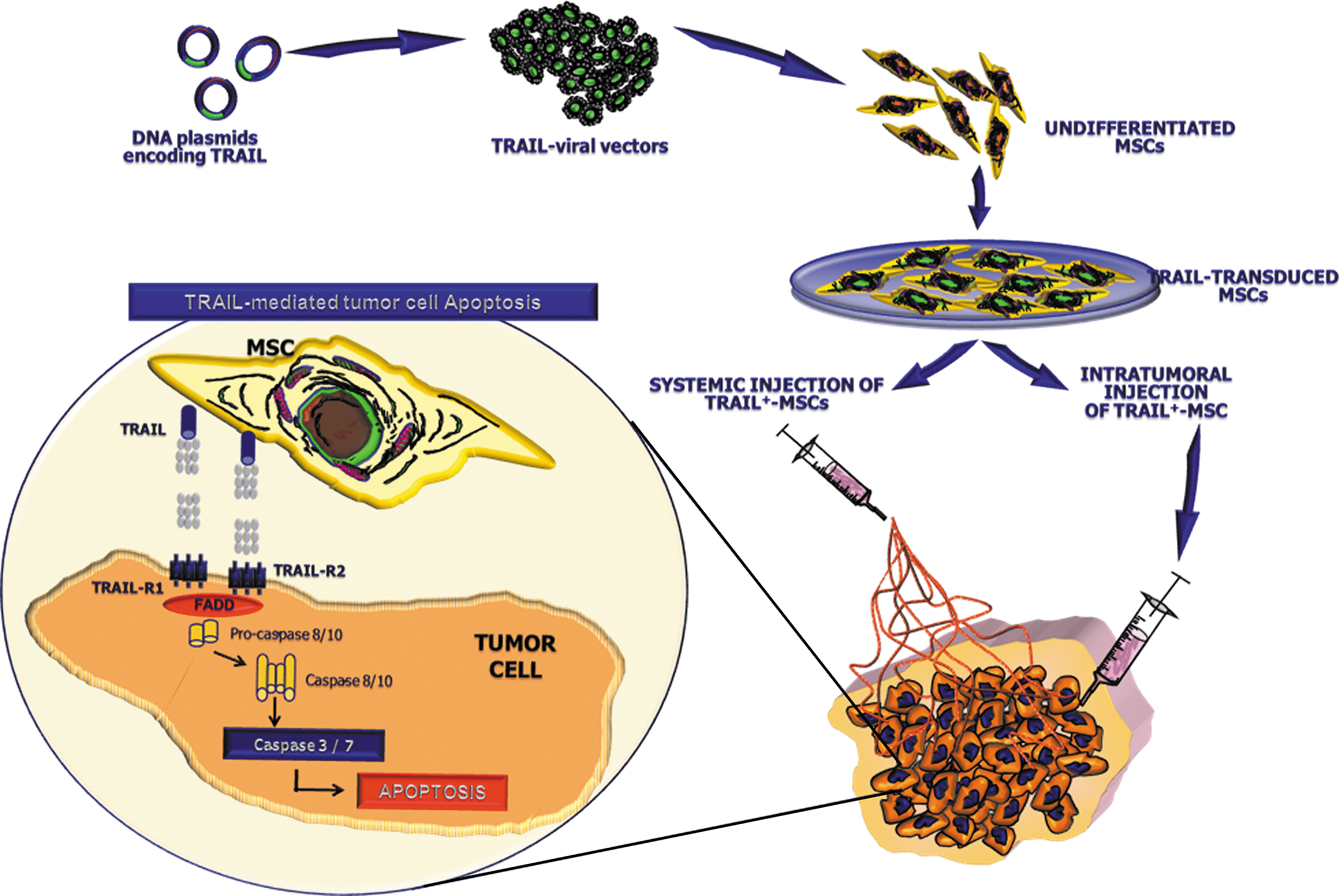
Therapeutic potential of TRAIL-engineered MSCs. TRAIL-expressing viral vectors can be employed to engineer adult MSC populations isolated from multiple sources. Once in vitro expanded, large amount of MSCs, capable to express both membrane-bound and soluble forms of the apoptotic inducer, can be used to treat cancers by either systemic or intratumoral injection. After their selective homing within the tumor site, TRAIL, exposed by MSCs, engages its cognate receptors R1 and R2 on tumor cells and triggers their apoptotic pathway by activation of both caspase-8 and -10. TRAIL, tumor necrosis factor-related apoptosis-inducing ligand. FADD, Fas-associated death domain.
BM-MSCs have been modified by lentiviral vectors to produce both bioluminescent markers and sTRAIL, thus allowing a direct monitoring of their migration and delivery as well as their fate and therapeutic effect in mouse models of glioma [74]. Further, TRAIL-producing MSCs have been shown to survive longer in diseased mice than in mice with normal brain and to efficiently migrate toward gliomas and exert dramatic antitumor effects in vivo [75]. In a recent work by Loebinger and coworkers, transduced BM-MSCs producing high quantities of TRAIL have been also demonstrated to exert high in vitro cytotoxic activity against lung, breast, squamous, and cervical cancers in a similar manner as in murine models of lung metastatic disease after subcutaneous injection. In these animals, the TRAIL-expressing MSCs reached the lung metastases, released the apoptogenic molecule, and restrained the metastatic tumor burden [76].
To emphasize the effectiveness of this approach of cell-based gene therapy, human adipose tissue–derived MSCs (AD-MSCs) have been recently armed with TRAIL by retroviral transduction and successfully used in vitro to kill multiple tumor types including human cervical carcinoma, pancreatic and colon cancers, and, in combination with bortezomib, breast cancer. Genetic modification of AD-MSCs allowed high rate of TRAIL expression as either surface or soluble form. Importantly, given their poor availability of the membrane TRAIL receptors death receptor (DR)4 and DR5, no cytotoxicity was reciprocally observed against MSCs themselves, thus making them fully feasible for cancer gene therapy approaches. Thus, when cocultured with TRAIL-producing AD-MSCs, all the tumor cell lines underwent apoptosis in a time-dependent fashion, whereas in 24–48 h, TRAIL released by AD-MSCs induced cancer cell apoptotic rates even superior than the equivalent amounts of the recombinant TRAIL used as control. Further, the specificity of this strategy was confirmed by the high levels of caspase-8 activation observed in cancer cells. In this experiment, bortezomib was used as inducer of TRAIL receptors and it increased the responsiveness of TRAIL-resistant breast cancer cells to this cell-based treatment. Finally, TRAIL-engineered AD-MSCs showed high in vivo antitumor potential after both intratumor and systemic inoculation in a subcutaneous model of cervical carcinoma [77]. In parallel with these encouraging results, Kim et al. successfully demonstrated that umbilical cord blood–derived MSCs can be engineered by adenoviral vectors to express TRAIL. Similar to MSCs from other sources, these transduced cells displayed definite migratory ability toward the tumor cells in vitro and, after direct intratumor injection, induced significant inhibition of tumor growth with considerable prolongation of the overall survival in glioma-bearing mice, when compared with control animals [78].
Stem Cell-Based Gene Therapy for Hematologic Malignancies
The successful preclinical results in solid tumors with “armed” stem cells have suggested similar approaches for treating hematologic cancers, particularly those growing within defined microenvironments such as lymphomas and multiple myeloma (MM). Indeed, neoplastic lymph nodes and BM, more than the stroma of solid tumors, represent preferential sites of stem cell recruitment. This is primarily due to the innate high tropism of stem cells for either secondary lymphoid organs or BM as sites highly rich in chemotactic growth factors (epidermal growth factor, fibroblast growth factor, insulin-like growth factor, HGF, vascular endothelial growth factor), inflammatory molecules (TNF-α, ILs, macrophage inflammatory protein-1α), and chemokines (SDF-1, macrophage chemoattractant protein-1), resulting from tight interactions between the tumor and the resident cells [79]. Therefore, CD34+ cells engineered to express a membrane-bound form of TRAIL were shown to kill either TRAIL-sensitive or -resistant lymphohematopoietic cells in vitro [80], as well as to significantly reduce the tumor growth in a mouse model of MM [81]. Given the established procedures of HSC transplantation in the clinical setting, the use of “armed” hematopoietic progenitors appears a suitable approach. To date, however, no data are available on a similar use for MSCs. Therefore, further studies are needed to address whether or not, although engineered to express killing molecules, MSCs can anyhow contribute to the growth of certain hematologic malignancies, such as those which are strictly dependent on SDFs [79,82]. Investigation comparing the efficacy of different cell carriers will thus provide a useful suggestion concerning the utilization of specific stem cell populations for distinct hematologic tumors. To this end, recent observations supported the use of MSC transplantation procedures to repair bone loss associated to different osteotropic tumors. Solid neoplasms, as well as hematologic malignancies, including MM, commonly metastasize to the skeleton and produce severe osteolytic lesions. In this context, BM-MSCs transduced to express osteoprotegerin, a factor involved in the physiology of bone turnover, have been described to exert therapeutic effects in a preclinical model of tumor osteolytic disease [83]. Similarly, in a recent work by Mukherjee et al., animal models of MM have been implanted with osteogenic MSCs and subsequently treated with bortezomib. After 4 weeks, mice restored the bone mineralization without evidence of disease progression, thus providing a promising model of MSC-based therapy for the treatment of MM and the related bone loss [84]. On these bases, it is conceivable to plan future studies to assess whether MSCs, even from tissue sources other than BM, can effectively deliver apoptotic molecules or other drugs to target myeloma cells, thus providing new exciting perspectives to treat incurable diseases such as MM.
Concluding Remarks
The beginning of the MSC era was traced more than 40 years ago with the first evidence of a stromal stem cell-like precursor within the BM. In the wake of results obtained in the field of HSC-based treatments, a multitude of studies have been devoted to improve knowledge of MSC biology, as well as to emphasize their prospective use in a wide range of new cell-based therapies. MSC plasticity, low immunogenicity, and suitability for genetic manipulation have represented the rationale for the employment of these cells in the emerging field of gene therapies. In addition, the simple methods of isolation and expansion of MSC pools from various postnatal tissues beyond the BM have further encouraged the study of MSCs and their applicability to overcome the ethical and political concerns that, at present, may restrain the experimental and clinical use of embryonic stem cells.
On the other hand, great enthusiasm has stemmed from studies using genetically manipulated MSCs to deliver anticancer drugs to tumor sites in vivo. In this context, MSCs could be engineered to survive in vivo for individual applications and for the specific pharmacokinetics of the molecules to be delivered, thus potentiating both tumor selectivity and effectiveness of some well-known anticancer drugs. However, high variability of antitumor effects has been reported, depending on both the type of MSCs used and the tumor cells to be targeted in each study. In addition, it is unclear how long the engineered MSCs may survive in vivo because definitive data on their long-term fate after transplantation are presently unavailable. Finally, although much work emphasizes the absence of spontaneous in vitro transformation of human MSCs, as well as no modification of their replicative and differentiative behavior after genetic manipulation, this topic is still under debate and further investigation is necessary to explore unknown risks of the MSC use in vivo.
Overall, the high therapeutic potential of MSCs implies the removal of skepticism arising from a few open questions by the scientific community, thus making the utilization of these cells a real hope for new anticancer treatments.
Footnotes
Acknowledgments
This work was supported in part by the Italian Association of Cancer Research (AIRC) and Ministero Istruzione Università e Ricerca-PRIN 2006 and PRIN 2008. The authors are grateful to Rita Bussolari, Monica De Matteo, Giulia Grisendi, and Stefania Stucci for their excellent assistance in preparing the manuscript.
Author Disclosure Statement
The authors declare no conflicts of interest related to the present manuscript.