
Abstract
Early germ-like cells (GLCs) derived from human embryonic stem cells (hESCs) have presented new opportunities to study germ cell differentiation in vitro. However, differentiation conditions that facilitate the formation of haploid cells from the derived GLCs have eluded the field. The inability to propagate GLCs in culture is a further limitation, resulting in inconsistent rederivations of GLCs from hESCs with relatively few GLCs in these heterogeneous populations. Here we found in vitro conditions that enrich for DDX4/POU5F1+ GLCs (∼60%) and that has enabled continual propagation for >50 passages without loss of phenotype. Clonal isolation of single GLCs from these mixed cultures generated 3 GLC (>90% DDX4/POU5F1+) and 2 hESC (<0.1% DDX4+) lines that could be continually expanded without loss of phenotype. Differentiation of clonal GLC lines in serum resulted in expression of postmeiotic markers and >11% were haploid, ∼5-fold higher than previous studies. The robust clonal meiotic competent and incompetent GLC lines will be used to understand the factors controlling human germ cell meiosis and postmeiotic maturation.
Introduction
H
hESC-derived GLCs demonstrate significant possibilities; however, previous reports where GLCs are rederived for each experiment resulted in heterogeneous cultures where frequently <20% of cells displaying germ cell character [1 –4]. Even when identical derivation techniques and hESC lines were used GLC derivation cultures were mixed populations with significant differences in (1) gene expression, (2) protein expression, and (3) differentiation potential between and within studies and relative few cells differentiating to haploid cells [1 –8]. The heterogeneity caused by constant rederivation and mixed populations prohibits discerning mechanisms involved in germ cell differentiation. Despite the fact that multiple groups have derived GLCs from hESCs [1 –4], a robust method of generating haploid cells from these cultures have remained elusive. Low numbers of haploid cells have been derived from hESCs when genes DAZL, DAZ, and BOULE are overexpressed in hESC [8]. However, ideally transgene insertion could be avoided since random gene insertion can interrupt or affect important endogenous genes, and the product of uncontrolled overexpression of transgenes can interfere with temporal–spatial events in normal differentiation [9]. Additionally, as hESC lines are not clonally derived, they have been noted to contain subpopulations of GLCs [3,6,7], which inhibits the ability to determine whether a culture condition induced differentiation of hESCs to GLCs or only enriched GLCs that previously existed in hESC cultures. The development of continuous cultures of clonal GLCs and hESCs enabled us to address these issues without artificially induced expression of germ cell genes via genetic manipulation and increased our initial understanding of culture conditions needed to generate haploid cells.
In the current study, hESC-derived GLCs (DDX4/POU5F1+) were continually cultured after initial differentiation, and not only maintained germ cell character, but significantly increasing germ cell gene and protein expression. Three GLC lines were subcloned producing populations where >90% of cells were DDX4/POU5F1+. Upon further differentiation, >71% of GLCs from all 3 lines entered early meiosis with 2 lines expressing postmeiotic germ cell proteins and producing haploid GLCs in the presence of fetal bovine serum (FBS). Two clonal hESC (DDX4−) lines were isolated and were capable of undergoing GLC differentiation, demonstrating de novo GLC differentiation from hESC rather than enrichment of pre-existing subpopulations. Utilizing homogeneous meiotic GLC lines and creating a conducive meiotic microenvironment yielded ∼11% haploid germ cells, compared with the ∼0.5%–2% haploid cells previously reported [8,10].
Materials and Methods
hESC culture conditions
BGO1 (XY) hESC were cultured on mouse feeders (Harlan) inactivated by mitomycin C (Sigma). Cells were cultured in 20% knockout serum replacement (KSR) stem cell medium consisting of Dulbecco's modified Eagle's medium/F12 supplemented with 20% KSR, 2 mM
GLC differentiation and continual culture conditions
As previously described [3] GLCs were differentiated in an adherent culture system by growing them on feeders in 20% KSR medium for 10 days without passaging with medium changes every other day. Postdifferentiation, continual culture, and clonal GLCs were passaged on feeders in 20% KSR medium every 4–6 days to prevent nongerm cell differentiation.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde for 15 min on glass 4-well chamber slides (B D Falcon). Antibodies were directed against POU5F1 (1:500; Santa Cruz Biotechnology), DDX4 (1:200; R&D Systems), MLH1 (1:200; Santa Cruz Biotechnology), SYCP3 (1:200; Santa Cruz Biotechnology), and SOX2 (1:100; R&D Systems) proteins and SSEA4 (1:20; Developmental Studies Hybridoma Bank) and TRA 1-81 (1:100; Millipore) glycolproteins. Alexa Flour 488, 594, and 633 (1:1,000; Molecular Probes). Imaging was done using the Olympus Ix81 with Disc-Spinning Unit and Slide Book Software (Intelligent Imaging Innovations).
Flow cytometry
Cells were fixed in 57%/43% ethanol/phosphate-buffered saline for 10 min. Antibodies were directed against POU5F1 (1:250; Santa Cruz Biotechnology), DDX4 (1:200; R&D Systems), MLH1 (1:200; Santa Cruz Biotechnology), and SYCP3 (1:200; Santa Cruz Biotechnology) proteins. Owing to the presence of feeders, antibodies against Human Nuclei (1 μL per million cells; Chemicon) and/or GFP expression (clonal lines) were used to prevent feeder contamination. Primary antibodies were detected using fluorescently conjugated secondary antibodies Alexa Flour 405, 488, and 647 (1:1,000; Molecular Probes). DNA content analysis was conducted by fixing cells with 70%/30% ethanol/phosphate-buffered saline for 30 min. Cells were treated with RNase A (100 μg/mL) to insure only DNA was stained followed by the addition of propidium iodide (50 μg/mL). Cells were analyzed using a Dakocytomation Cyan (Beckman Coulter) and FlowJo Cytometry analysis software (Tree Star). Significance was determined by 2-way analysis of variance (ANOVA) and Tukey's Pair-Wise comparisons. Treatments where P value was <0.05 were considered to be significantly different.
Lentiviral transduction and fluorescence-activated cell sorting
Mixed GLC cultures were transduced using viPS-EF1 α-TurboGFP (Thermo Scientific) at 20 multiplicity of infection (MOI) in the presence of 1.2% GeneJammer (Stratagene). Single GFP+ cells were fluorescence-activated cell sorted (FACS) using a MoFlo XDP Cell Sorter (Beckman Coulter) into individual wells of 96-well plates, which contained feeders and 20% KSR with 10 ng/mL of basic fibroblast growth factor and 10 μM Y-27632 ROCK inhibitor (Calbiochem). Five clonal lines were selected for further characterization.
Population doubling time was determined by plating cells at a concentration of 250,000 cells with quantification every 12 for 48 h. A nonlinear regression (curve fit) was performed using GraphPad Prism 5 (GraphPad Software) to establish population doubling time. Two-way ANOVA and Tukey's Pair-Wise comparisons were done utilizing SAS, contrasting doubling times between each population (n = 3) with P values <0.05 considered significantly different.
Real-time polymerase chain reaction
RNA was extracted using the Qiashredder and RNeasy kits (Qiagen) according to manufacturer's instructions. RNA quality and quantity was determined using an RNA 600 Nano Assay (Agilent Technologies). RNA was reverse-transcribed using the cDNA Archive Kit (Applied Biosystems Inc.) according to manufacturer's protocols. Quantitative reverse transcription polymerase chain reaction (RT-PCR) (TaqMan) assays were chosen from Assays-On-Demand™ (Applied Biosystems Inc.), a prevalidated library of human-specific quantitative PCR assays, and incorporated into 384-well microfluidics cards and were performed on the ABI PRISM 7900 Sequence Detection System (Applied Biosystems Inc.) following manufacturer's instructions. For calculation of relative fold change values, initial normalization was achieved against endogenous 18S ribosomal RNA using the ΔΔCT method of quantification (Applied Biosystems Inc.) [11]. Average fold changes from 4 independent runs were calculated as 2−ΔΔCT. Significance was determined by running a 2-way ANOVA and Tukey's Pair-Wise (SAS) comparisons for each treatment. P value <0.05 were considered to be significantly different.
RT-PCR amplification was performed using GoTaq Green Master Mix (Promega) following manufacture's instructions. Primers used for RT-PCR were as follows: acrosin forward 5′-ACTGCCATTCTGCTGGTCTT-3′ and reverse 5′-CACACATTGGTTGGCTGAAC-3′; haprin forward 5′-CCCTCGTTTGTTCCGTTAGA-3′ and reverse 5′-GGTAGCCTCGTCTCCAATCC-3′; protamine 1 (PRM1) forward 5′-CAGAGTTCCACCTGCTCACA-3′ and reverse 5′-GGATGGTGGCATTTTCAAGA-3′; PRM2 forward 5′-CAGTAACACCAAGGGCAGGT-3′ and reverse 5′-TTTCGGGCGACTTTTTCTTA-3′; transition protein 2 (TNP2) forward 5′-CACCCACACTCAGCTCCATA-3′ and reverse 5′-AGTGTTGCGTAGAAATCACCA-3′; ring finger protein 17 (RNF17) forward 5′-GCGGTTGTTCCAGAAGAAAG-3′ and reverse 5′-TCCAGCCATTGGGTAGTAGC-3′; GAPDH forward 5′-GAGTCAACGGATTTGGTCGT-3′ and reverse 5′-TTGATTTTGGAGGGATCTCG-3′. PCRs were performed by initially denaturing cDNA at 95°C for 3 min followed by 35 cycles of denaturing at 95°C for 60 s, annealing at 60°C for 30 s, polymerization at 72°C for 30 s, and a final 10 min extension.
Results
Continual culture of hESC-derived germ cells
BG01 hESCs were differentiated for 10 days in 20% KSR medium on feeders without passaging and with medium changes every other day to enhance germ cell differentiation signaling [3,7]. Day 10 GLCs morphologically resembled cultured PGCs [12,13] displaying colonial growth patterns, cobblestone morphology, a high nuclear-to-cytoplasmic ratio, and large nucleolus (Fig. 1D) similar to hESCs (Fig. 1A). However, day 10 GLCs expressed both the definitive germ cell marker DDX4 [14] (Fig. 1F) and the pluripotency marker POU5F1, unlike the DDX4− POU5F1+ hESCs (Fig. 1C). The DDX4/POU5F1+ population (18.7%) under these conditions was greater than that found in hESC cultures (4.0%; P < 0.05; Fig. 1G). GLCs were then serially passaged on feeders for 20 passages resulting in a significant (P < 0.05) increase in DDX4/POU5F1+ cells at passage 5 (58.0%), relative to hESCs and day 10 controls (Fig. 1G), and maintained at passages 10 (58.7%) and 20 (60.7%). Previously, we demonstrated that DDX4/POU5F1+ cells could not be maintained for 30 days, a point that temporally corresponds with serial passage 5, under differentiation conditions without passaging [3]. Similarly, <1.0% of the population in this study was DDX4/POU5F1+ at day 30 in nonpassage cultures (Fig. 1G), which was significantly (P < 0.05) less than day 30 continual culture cells (58.0%), hESCs (4.0%) and day 10 controls (18.7%; Fig. 1G). These results suggest that GLCs can be propagated for at least 20 passages and that passaging is essential to maintain a germ cell phenotype. Additionally, we were able to freeze and thaw GLCs without loss of DDX4 and POU5F1 expression (Fig. 1H) and maintain normal karyotype (Supplementary Fig. S1; Supplementary Data are available online at
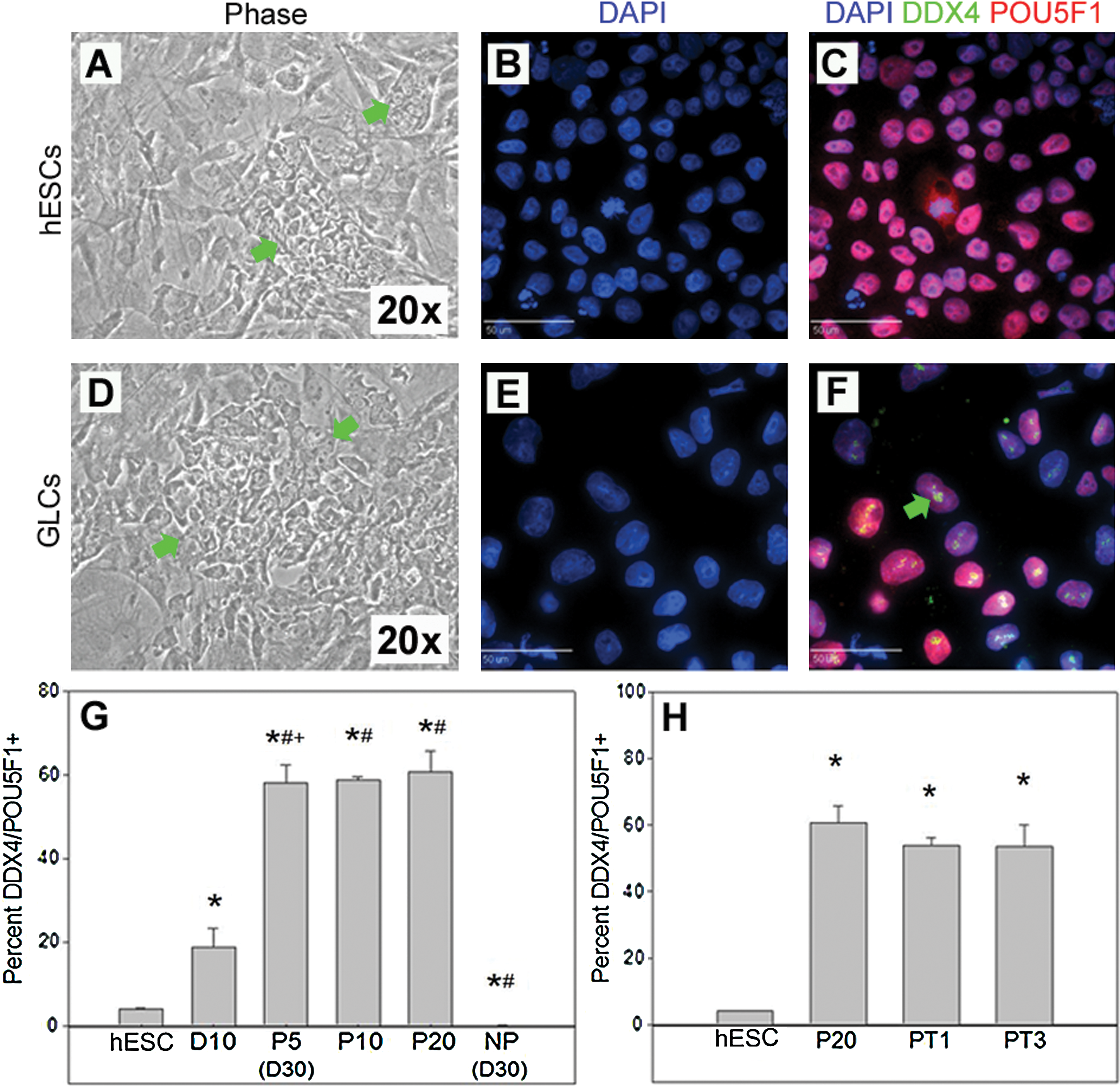
Human embryonic stem cell (hESC)-derived germ-like cells (GLCs) can be continually propagated without loss of germ cell phenotype. DDX4− POU5F1+
Gene expression in propagated GLC cultures
Propagation of DDX4/POU5F1+-enriched cultures demonstrated significant (P < 0.05) upregulation of the premigratory and migratory genes IFTIM3, POU5F1, NANOG, and DDX4 after 10 days of differentiation relative to hESCs (Fig. 2). This upregulation was maintained for 5, 10, and 20 passages for genes IFITM3, POU5F1, NANOG, and DDX4. The premigratory genes DPPA3 and KIT did not show upregulation after initial differentiation. However, these genes showed significant (P < 0.05) increases in gene expression after 5 passages relative to hESCs (Fig. 2). This increase in gene expression was maintained for 10 and 20 passages.
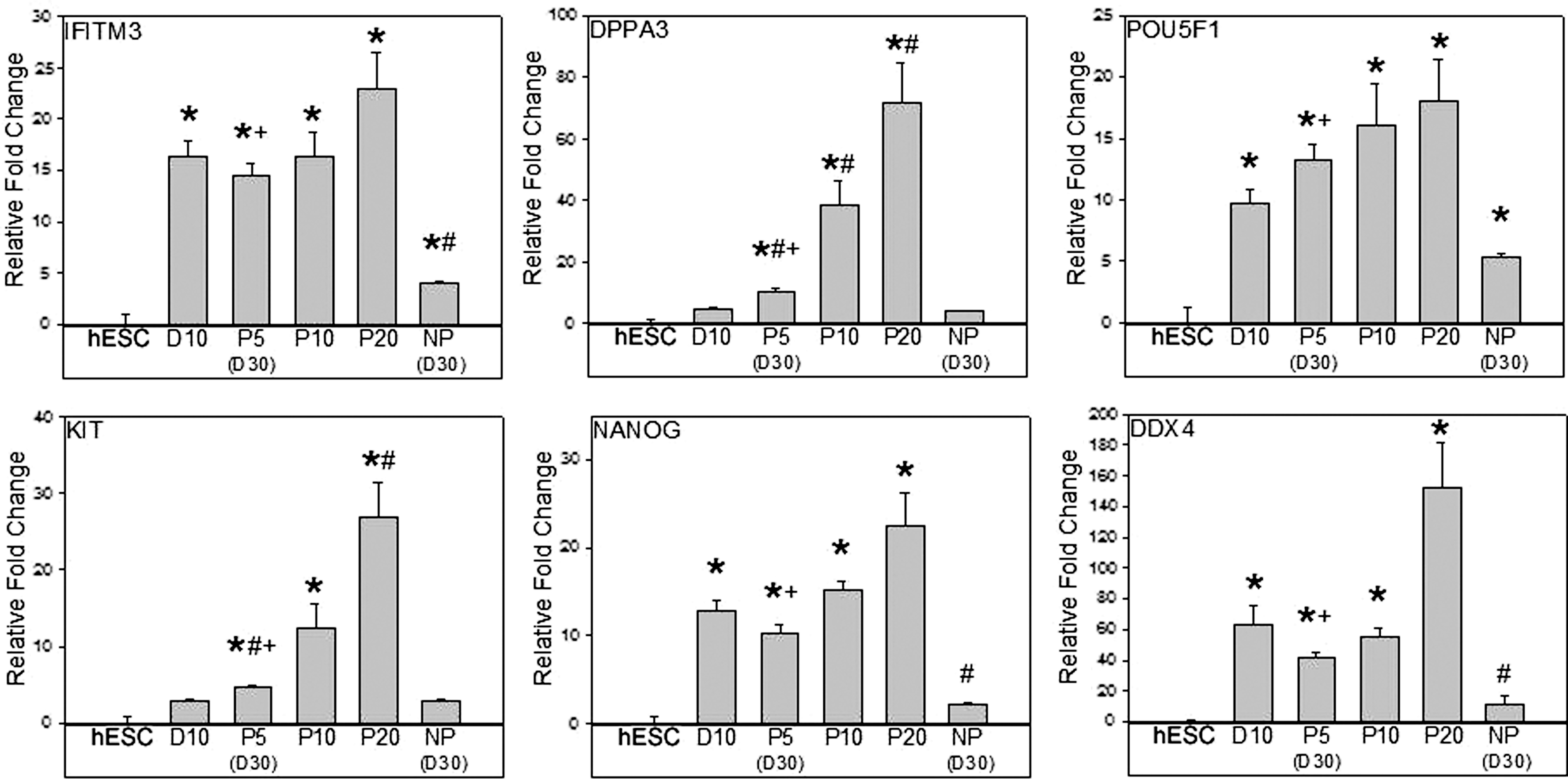
Continually cultured GLCs maintained premigratory and migratory germ cell gene expression. Significant (*P < 0.05) upregulation of premigratory and migratory genes IFTIM3, POU5F1, NANOG, and DDX4 was observed after 10 days (D10) of differentiation relative to hESCs, which was maintained or significantly increased (# P < 0.05) at 5 (P5), 10 (P10), and 20 (P20) passages. Premigratory genes DPPA3 and KIT were not upregulated after initial differentiation; however, these genes were increased (*P < 0.05) after 5 passages relative to hESCs and was maintained for 10 and 20 passages. Comparison of day 30 (or P5) continually propagated cultures and day 30 NP cultures demonstrated that passaged cultures possessed upregulation (+ P < 0.05) of premigratory and migratory genes.
Postmigratory genes DAZL, PUM2, and NANOS1 and the meiotic genes MLH1, SYCP1, and SYCP3 were more highly expressed after 10 days of differentiation relative to hESCs (P < 0.05; Fig. 3). SYCP3 expression was maintained at passages 5, 10, and 20. However, DAZL, PUM2, NANOS1, MLH1, and SYCP1 all showed further increases (P < 0.05), relative to day 10 differentiation cultures, after 20 serial passages. In addition, direct comparison of day 30 (passage 5) continually passaged cultures and day 30 cultures that were not passaged demonstrated that passaged cultures possessed significant (P < 0.05) upregulation of all observed germ cell genes (Figs. 2 and 3). These results indicate that serially passaging enriched DDX4/POU5F1+ cultures can maintain or even enhance germ cell character with increased levels of early and late germ cell gene expression.
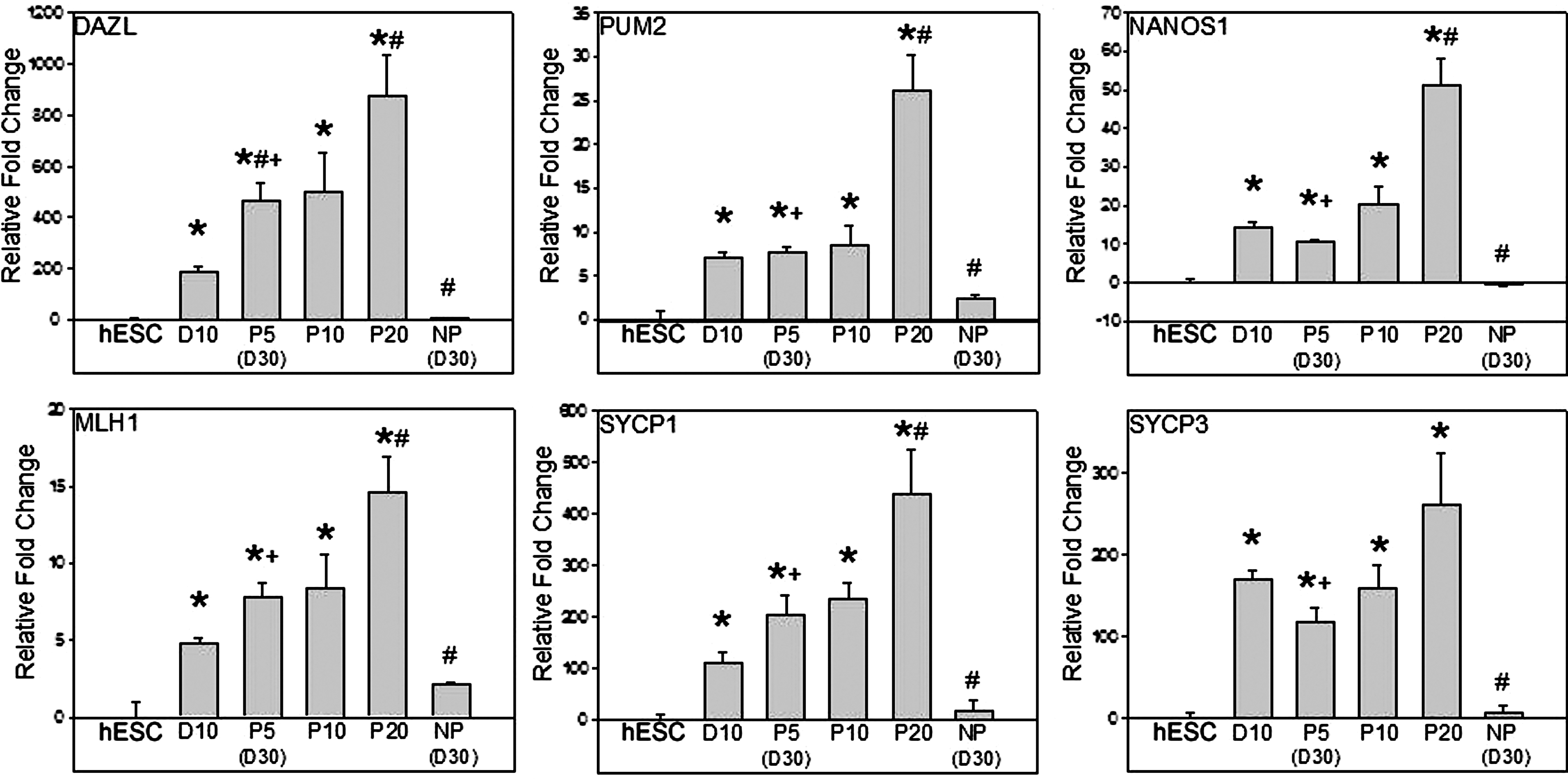
Propagated GLCs demonstrated continually increasing expression of postmigratory and meiotic germ cell genes. Expression of postmigratory genes DAZL, PUM2, and NANOS1 and the meiotic genes MLH1, SYCP1, and SYCP3 were increased (*P < 0.05) after 10 days of differentiation relative to hESCs. SYCP3 expression was maintained at passages 10 (P10) and 20 (P20); however, continual passaging of GLC cultures resulted in increased (# P < 0.05) DAZL, PUM2, NANOS1, MLH1, and SYCP1 expression in passage 20 (P20) cultures relative to D10 differentiation cultures. At 30 days, all germ cell genes were more highly expressed in passaged cultures [passage 5 (P5)] versus NP cultures (+ P < 0.05).
Isolation of clonal GLC lines
To isolate pure GLC lines, continually cultured cells were transduced using an EF1-α promoter–driven turboGFP construct packaged in lentivirus particles with GeneJammer. Transduced cells were FACS (Fig. 4A), and highly GFP+ single cells were plated into individual wells of a 96-well plate. Only a single colony was derived from each well (Fig. 4B, C). FACS resulted in 41 (42.7%) GFP+ clonal cell populations of which 5 lines were randomly selected and expanded for further characterization. Clonal cell lines 1–3 expressed both DDX4 and POU5F1 (Fig. 4D) in >90% (91.5%–92.6%) of cells (Fig. 4E), whereas <2% of clone 4 and 5 cells and 3.8% of hESCs were DDX4/POU5F1+ (Fig. 4D, E). In addition to differences in DDX4 expression, the population doublings (Fig. 4F) for clonal GLC lines were significantly (P < 0.05) different, with clonal GLCs (31.3–34.8 h) exhibiting slower doubling rates than hESC clones (28.2–28.5 h) and hESCs (28.3 h).
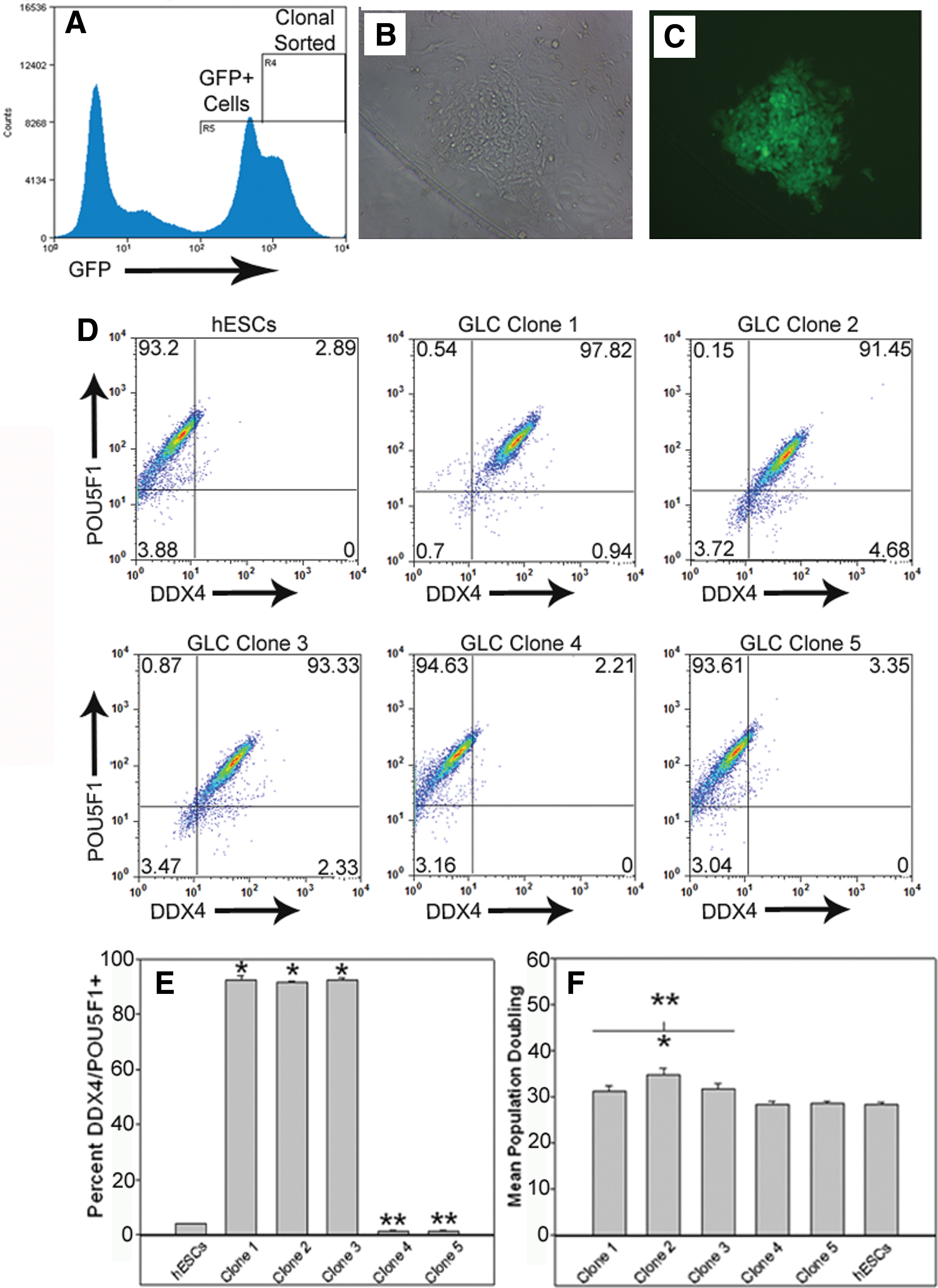
Homogenous clonal GLC and hESC populations can be isolated and expanded. GLC cultures expressing turboGFP at high levels were fluorescence-activated cell sorted
Clonal hESC lines differentiate into GLCs
Differentiation of hESCs into GLCs when hESCs frequently possessed a subpopulation of GLCs has previously raised the question of whether de novo GLCs are created from hESCs or whether conditions lead to enrichment of existing GLCs in the hESC cultures [3,6,7]. The isolation of 2 clonal hESC lines (4 and 5) allowed us to address this question. GFP+ clonal hESC lines were positive for the pluripotency markers POU5F1 (Fig. 5C), SOX2 (Fig. 5D), SSEA4 (Fig. 5H), and TRA 1-81 (Fig. 5L), whereas cocultured GFP− feeder cells (Fig. 5E, I, M; white arrows) were negative for all markers. These lines were differentiated for an additional 10 days as previously done without passaging and with every other day medium changes. Starting populations for both cell lines possessed <0.1% DDX4/POU5F1+ cells (Fig. 5N, O), whereas postdifferentiation 15.0% of hESC clonal line 4 and 22.8% of line 5 cells were DDX4/POU5F1+, a significant (P < 0.05) increase. This suggests that the differentiation process does not just enrich for cells previously committed to the germ cell linage, but instead induced differentiation creating nascent GLCs.
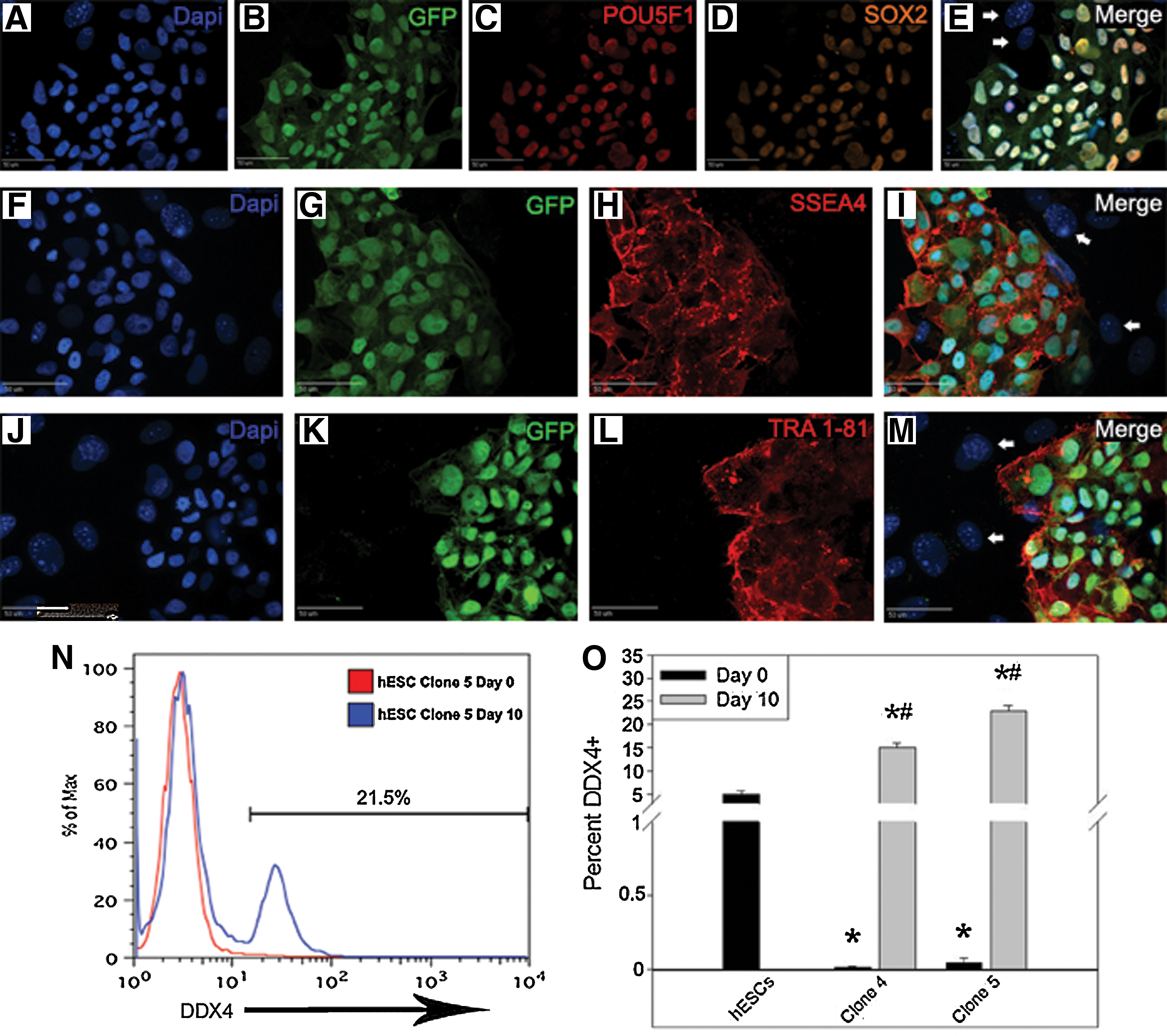
Clonal hESC cultures demonstrate de novo GLC formation and not simply enrichment. GFP+ clonal hESC lines 4 and 5 expressed the pluripotency markers POU5F1
Clonal GLCs produce haploid gametes in vitro
Clonal GLC lines were negative (<1% positive) for the early prophase meiotic markers SYCP3 and MLH1, suggesting that these cells were premeiotic. To determine if GLC lines could undergo differentiation into haploid cells in vitro, cells were cultured without passaging and with every other day medium changes for 10 days and then monitored by immunocytochemistry and flow cytometry for the early prophase meiotic proteins SYCP3 and MLH1. Both SYCP3 and MLH1 proteins were expressed at day 10 and these proteins were localized to the nucleus, where they may play a role in crossover events that occur during germ cell genetic recombination (Fig. 6A). Undifferentiated day 0 GLCs did not express SYCP3 or MLH1 proteins. On the basis of immunocytochemistry data, a high number of GLCs appeared to enter into meiosis. To quantify this progression, flow cytometry was then used to assess the number of cells expressing these meiotic proteins. To eliminate feeder cells, all cells were also stained with the human-specific human nuclear antibody. At 10 days, cells in all 3 GLC lines expressed SYCP3 and MLH1 proteins (Fig. 6B, C; P < 0.05) at significantly higher levels (>71%) than the starting population (<1%), whereas clonal hESC lines continued to be negative for both proteins (<0.5% positive).
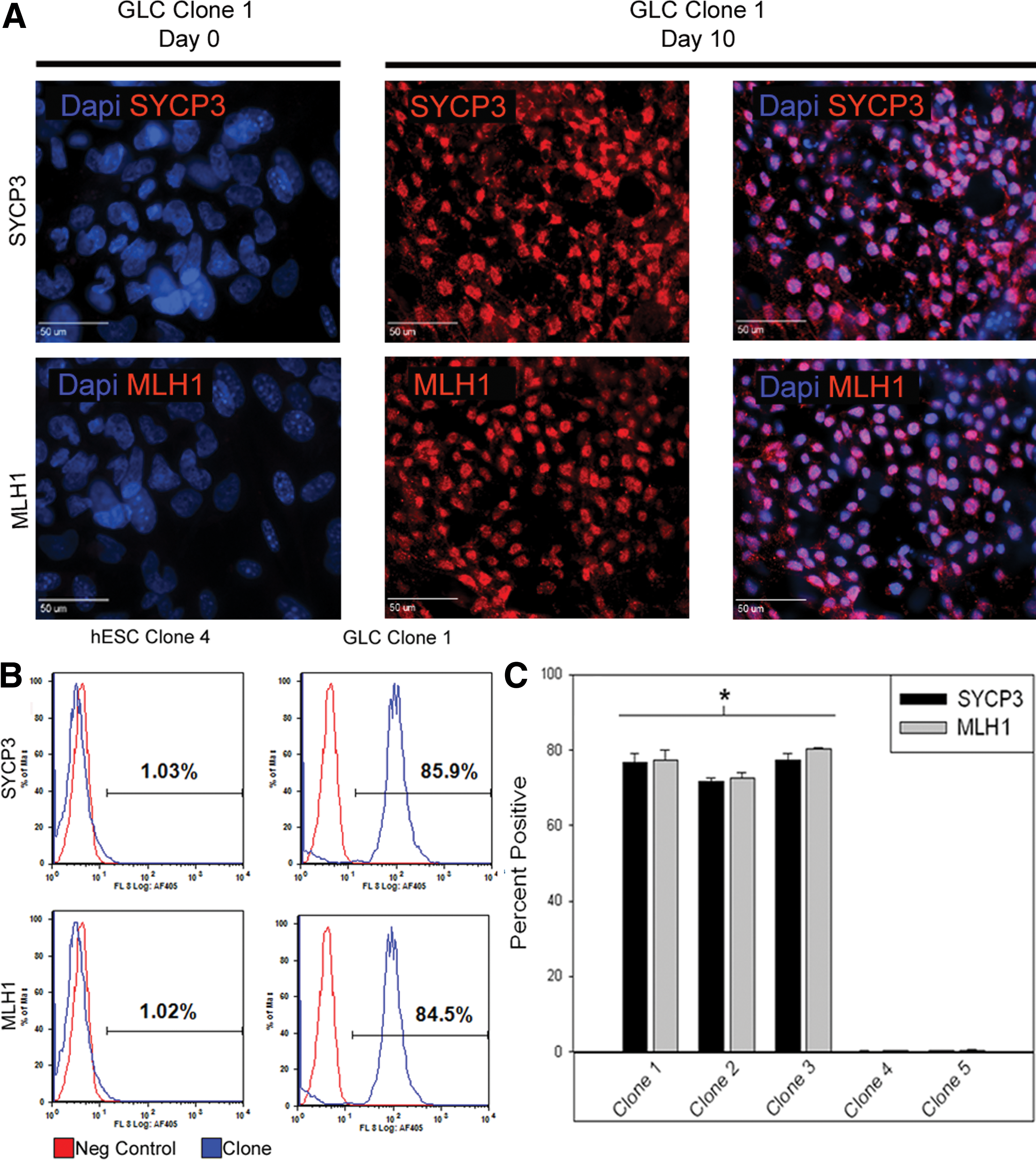
Differentiated clonal GLCs demonstrate meiotic entry. Clonal GLC cultures were SYCP3/MLH1−
Extended differentiation and additional conditions were necessary to reach a haploid state. GLCs were differentiated for 20 days and DNA content and postmeiotic germ cell markers were examined. The number of differentiated cells with decreased DNA content, indicative of a haploid state, was not above background levels after 20 days of differentiation. Despite this, ring finger protein 17 (RNF17) gene, normally expressed during meiosis and the postmeiotic genes acrosin, haprin, PRM1, PRM2, and TNP2 were expressed in clones 1 and 3 (as well as testis positive control), suggesting that a small subset of cells reached a haploid state. However, these genes were not expressed in clonal GLC line 2 (Fig. 7A), hESCs, or ovary negative control. To increase the number of growth factors and promote meiotic signaling, 20% KSR in the differentiation medium was replaced with 20% FBS, and GLCs were differentiated for 20 days and DNA content was re-examined. Under these conditions both clonal GLC lines 1 (2 of 4 replicates) and 3 (1 of 4 replicates) generated haploid cells ranging from 6.7% to 11.2% of total cells (Fig. 7B), a significantly (P < 0.05) higher percentage than negative treatments. Haploid cells were not observed at day 0 for any of the GLC lines (Fig. 7B), in GLC line clone 2, and were not detectable in the absence of FBS. Gene expression and DNA content analysis indicated that up to 11% haploid GLC population can be derived in vitro with the addition of FBS.
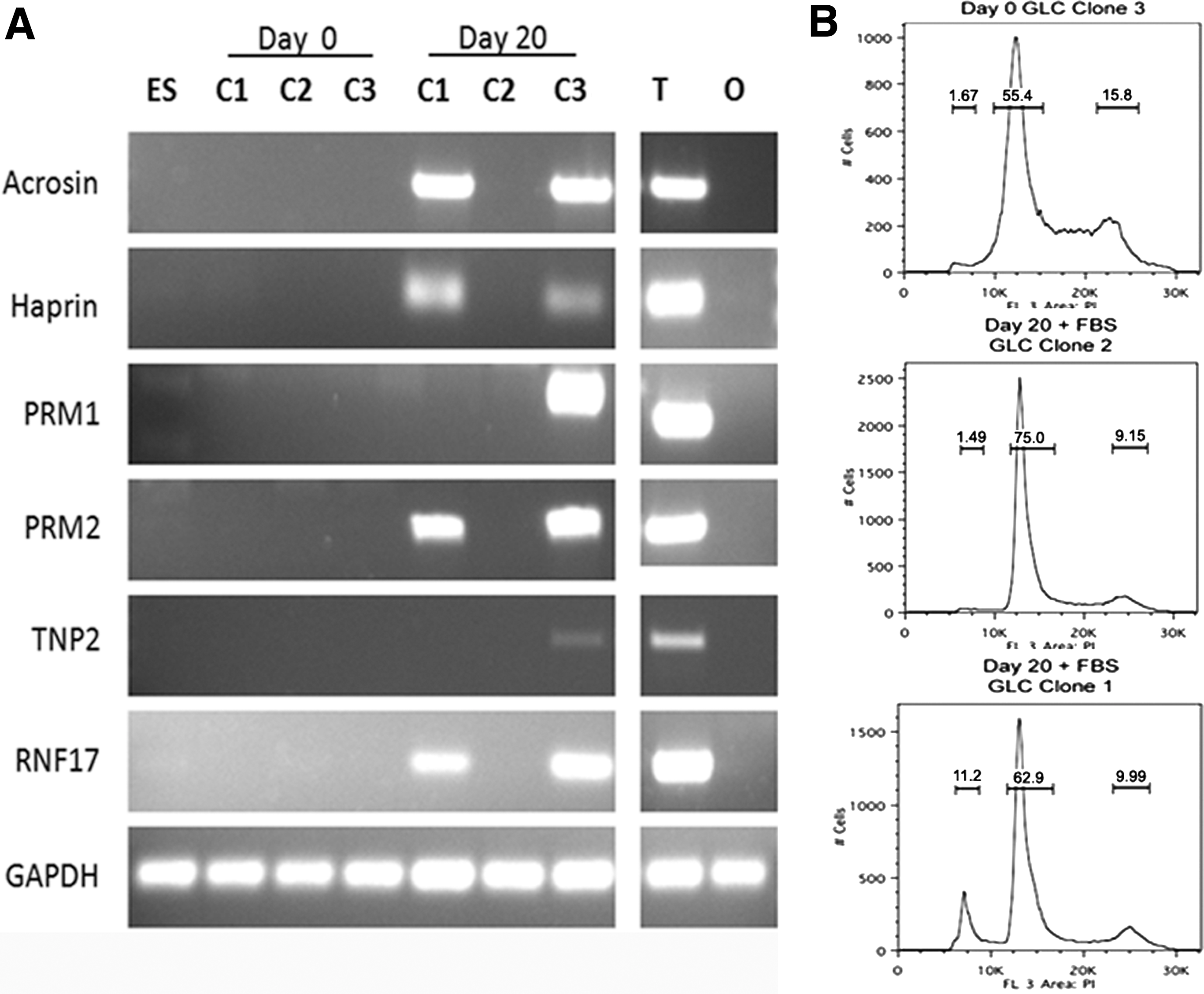
Differentiated clonal GLCs produce haploid cells that express postmeiotic markers. Differentiation of clonal GLC lines for 20 days in 20% KSR medium resulted in expression
Discussion
Despite attempts, little progress has been made toward understanding and developing the proper in vitro conditions that mimic in vivo-like mammalian germ cell development [15 –17] or deriving cells capable of completing meiosis [18]. In this study, we developed homogeneous (>90% DDX4/POU5F1+) clonal hESC-derived GLC lines able to be continually cultured with >71% of GLCs entering meiosis leading to a haploid population of up to 11% that expressed the postmeiotic genes acrosin, haprin, TNP2, PRM1, and PRM2. To our knowledge this is the first report that outlines the conditions necessary to generate homogeneous GLC lines that can be continually propagated and sustain germ cell competency. Utilizing these GLC lines, we found that FBS was responsible for potentiating meiosis and significantly enhanced the formation of haploid cells from undetectable levels to >11% of the differentiated population. Previously overexpression of the germ cell genes DAZL, DAZ, and BOULE also induced differentiation of GLCs into haploid gametes [8]. However, this strategy resulted in a relatively low percentage (∼2%) of haploid cells, similarly embryoid body differentiation in combination with retinoic acid produced very few haploid cells (∼0.5%) [10]. Serum has been a standard medium component utilized in short-term maintenance of gametes and has been show to improve germ cell survival, development, and meiotic competence in both spermatogonia [19,20] and oocytes [21,22]. However, the effect of serum on advanced differentiation and meiosis in hESC-derived GLCs have never before been determined. FBS in our culture conditions induced further differentiation of GLC lines to haploid cells (6.7%–11.2%). All of the reports that produced haploid cells have used the FBS-supplemented medium, but did not examine its effect nor would they likely have observed a difference given the low GLC starting population and numbers of haploid cells produced with FBS [2,8,10]. Findings of this study suggest that FBS contains essential components contributing to the successful formation of haploid cells. Serum contains multiple factors that increase meiotic competency, including epidermal growth factor [23,24], bovine serum albumin [21,24], follicle-stimulating hormone, and testosterone [25,26]. Determination of which serum containing factors increased meiotic potential remains to be reveled and will be intensely studied in the future.
The inability to continually culture germ cells in vitro has impeded the study of mammalian germ cell development. In this study we showed that GLCs can be continually cultured without loss of germ cell phenotype, while retaining a normal karyotype. Previous attempts to derive GLC lines from PGCs resulted in developmentally restricted lines that could not form haploid cells [15,17,18]. Reports of germ cell culture systems that maintained the germ cell differentiation potential [27,28] could not be reproduced [18]. Here we demonstrate that serial passaging of GLCs resulted in not only a maintained germ cell phenotype, but further enrichment and that passaging was essential to preserving GLCs' germ cell potency. In the absence of passaging, germ cell phenotype associated gene and protein expression were lost. This is in agreement with our previous study where germ cell phenotype was diminished when GLCs were not passaged [3].
The lack of homogenous GLC cultures has often made it difficult to interpret GLC differentiation studies [1,2,5,7,8,10]. We isolated 5 clones from mixed GLC cultures: 2 hESC (DDX4−) and 3 GLC (DDX4+). Clonal hESC (DDX4−) lines presented a unique opportunity to decipher whether there was de novo GLC differentiation in these cultures or just enrichment of subpopulations of DDX4+ cells. The differentiation of clonal hESC (<0.1% DDX4+) lines into GLCs indicated that GLC formation is not exclusively an enrichment process, but does not rule out enrichment might have also occurred in addition to differentiation. In a clonal hESC population, presumably with identical differentiation potential, we might have expected most if not all cells to differentiate into GLCs. However, only a subset of cells expressed DDX4 upon differentiation in both clonal hESC lines and at a comparable level to nonclonal hESCs lines. Previously, we have shown that GLCs developed in clusters, suggesting that small groups of cells undergo germ cell differentiation in these cultures [3]. These clusters may receive various autocrine or paracrine factors, epigenetic changes or important feeder cell interactions [3,4,7] that only occur in these small pockets of cells; however, the defining germ cell signaling remains to be determined.
The isolation of clonal GLC lines (>90% of cells are DDX4/POU5F1+) allowed for easy identification of haploid GLCs and may enable the elucidation of factors and biochemical pathways in serum responsible for haploid cell formation. These types of studies are not possible using mixed cell population cultures. Previous studies successfully producing haploid GLCs showed that ∼0.5%–2% of cells had reached a 1 N state [8,10], which is often considered near or below background/noise levels for even the most sensitive of flow cytometry assays. Without the ability to more efficiently produce haploid cells, studies with the aim of examining the meiotic process will be challenging. The low percentage of haploid GLCs may be a direct result of a small GLC starting populations in some studies, often <6% DDX4+, suggesting that a larger and clonal starting population may produce more robust results [8,10]. Here differentiation of homogenous clonal GLC lines resulted in a greater percentage of haploid cells than previously described, supporting this working hypothesis. Additionally, >71% of cells showed synchronized entry into meiosis as indicated by protein expression of SYCP3 and MLH1, which potentially would have been masked in cultures with low numbers of GLCs and even fewer numbers of meiotic cells. Previous studies that demonstrated low levels of haploid differentiation in GLC cultures had shown mis-localization of SYCP3 protein outside of the nucleus, which also may account for lower haploid cell numbers [2]. Here we show appropriate localization of SYCP3, a synaptonemal complex element, and MLH1, a meiotic recombination protein, proteins to the nucleus, suggesting that they will take part in their normal function in crossover events. However, SYCP3 did not appear to form the typical synaptonemal complex nor did MLH1 proteins associate with DNA as they would when performing their normal DNA repair function after crossover events. This may account for the low and, in some cases, absence of cells converting from a diploid to haploid state. Since a small proportion of cells did appear to reach a haploid state, the complexes may have formed at some undetected point. The absence of verified complexes and recombinant proteins associated with DNA is a common error in hESC-derived germ cells and remains an important topic of study [2,6]. Homogeneous GLC lines also show a significant reduction in nongerm cell types that are a confounding variable that potentially introducing nongerm cell signaling [1,2,5,7,8,10]. Eliminating nongerm cells is of particular interest when studying ubiquitous germ cell pathways in germ cell differentiation cultures such as bone morphogenetic proteins (BMPs), where contaminating cell types may respond to these factors but are expected to respond differently than germ cells. In this study we produced 3 clonal GLC lines all showing differences in differentiation potential. The development of GLC lines with differences in germ cell competency is an important screening tool to identify novel germ cell proteins.
The ability to derive homogenous clonal GLC lines that can be continually cultured presents a powerful system to study the major factors that orchestrate, enhance, and inhibit human germ cell development. Our studies demonstrated that clonal GLC lines could be derived from mixed GLC cultures and possess increased germ cell phenotype due to serial passaging. These cells not only demonstrated the ability to enter early meiosis, but meiotic completion as indicated by postmeiotic gene expression and haploid DNA content, without having randomly inserting overexpression gene constructs containing germ cell-related transgenes. Utilizing clonal GLC lines, we showed that FBS was a meiotic enhancer and, based on clonal hESC lines, that hESC to GLC differentiation systems did not only enrich pre-existing GLC subpopulations but participate in de novo GLC formation. The derivation of 3 clonal GLC lines with differences in differentiation potential also provides future opportunities to conduct comparative studies that can be used to dissect abnormal germ cell phenotypes. Ultimately, these homogeneous cell populations may lead to high-throughput toxicology and drug discovery screens that generate robust, reliable, and reproducible data and a deeper understanding of infertility and the many conditions caused by abnormal germ cell development.
Footnotes
Acknowledgments
We thank R. Nilsen and K. Huff for their quantitative RT-PCR assistance, J. Chilton at ArunA Biomedical Inc. for her aid in transduction and J. Nelson at the Center for Tropical and Emerging Global Diseases Flow Cytometry Facility, University of Georgia. We are also grateful for the funding support from Georgia Research Alliance and University of Georgia Graduate Recruitment Opportunities Program.
Author Disclosure Statement
No potential conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.