
Abstract
The aryl hydrocarbon receptor (AhR) belongs to the basic helix-loop-helix family of DNA-binding proteins that play a role in the toxicity and carcinogenicity of certain chemicals. The most potent ligand of the AhR known is 2,3,7,8-tetracholorodibenzo-p-dioxin. We previously reported tetrachlorodibenzo-p-dioxin–induced alterations in numbers and function of hematopoietic stem cells (HSCs). To better understand a possible role of the AhR in hematopoiesis, studies were undertaken in young adult AhR null-allele (KO) mice. These mice have enlarged spleens with increased number of cells from different lineages. Altered expression of several chemokine, cytokine, and their receptor genes were observed in spleen. The KO mice have altered numbers of circulating red and white blood cells, as well as a circadian rhythm-associated 2-fold increase in the number of HSC-enriched Lin−Sca-1+c-Kit+ (LSK) cells in bone marrow. Primary cultures of KO HSCs and in vivo bromodeoxyuridine incorporation studies demonstrated an approximate 2-fold increased proliferative ability of these cells. More LSK cells from KO mice were in G1 and S phases of cell cycle. Competitive repopulation studies also indicated significant functional changes in KO HSCs. LSK cells showed increased expression of Cebpe and decreased expression of several hematopoiesis-associated genes. These data indicate that AhR has a physiological and functional role in hematopoiesis. The AhR appears to play a role in maintaining the normal quiescence of HSCs.
Introduction
T
Many of the molecular events leading to modulation of gene transcription following AhR activation by the potent xenobiotic ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) have been characterized. For example, induction of CYP1A1, a member of the cytochrome P450 drug-metabolizing enzyme family, and other genes including HES-1 has been ascribed to the recognition by the AhR•Arnt complex of Ah-responsive elements located in the upstream regions of responsive genes [5,6]. However, direct relationships between the modulation of known responsive genes and specific functional outcomes in cells/tissues have to be yet determined. Likewise, the normal function of the AhR is not known and true endogenous ligands have not been clearly identified, although recent data suggest likely candidates [7 –9].
Most of our understanding of AhR biology has come from studies using TCDD. This chemical causes, through its high affinity binding to and prolonged activation of the AhR, a wide range of biochemical and pathological changes in mammalian and nonmammalian species. TCDD has been shown to be a potent immunotoxicant, eliciting immune suppression in all animal species tested [10]. In humans, TCDD exposure has been associated with an increased incidence of lymphomas and leukemias [11,12]. Recently, AhR activity has been found to have a role in Th-17 cell development and autoimmune responses [13,14].
Given the profound effects of persistent AhR activation on the immune system, we have been investigating the cellular and molecular events underlying induced alterations in bone marrow (BM). The number of immature and mature B cells in mouse BM decreases following exposure to a single dose of TCDD [15]. Pre- or postnatal TCDD exposure results in a significant reduction in lymphoid-specific TdT and RAG-1 mRNA in BM, and thymic seeding by BM cells from TCDD-treated animals is substantially decreased [16]. However, TCDD treatment produces a significant increase in the number of hematopoietic stem cell (HSC)-enriched Lin−Sca-1+c-Kit+ (LSK) cells [17,18]. The use of radiation chimeras demonstrated that the presence of the AhR in hematopoietic cells, but not supporting stroma, was responsible for these effects [19]. It was also shown that marrow cells from TCDD-treated animals had lost the ability to fully reconstitute the immune system of irradiated mice [18,20]. Overall, these data indicate that both the numbers and functionality of HSCs and/or hematopoietic progenitor cells (HPCs) are directly modulated by AhR activation.
To better understand the physiological role of the AhR, Ahr-null allele (AhR−/− or KO) mice have been produced. These mice have abnormal vascular development and spontaneously develop other lesions in several organs [21,22]. Some lesions only become manifest with aging, and available data suggest that these mice have a decreased lifespan [23]. To date, only a limited characterization of the immune system in these animals has been performed. There is decreased mass and cellularity of the liver, spleen, and lymph nodes in 1–2-week-old KO mice. These alterations resolve by 3 weeks of age, but the spleen becomes enlarged in juvenile and mature animals. Young adult KO mice display shifts in the normal profile of developing B cells in the BM, with nearly 2-fold increases in the less mature phenotypes [24]. These mice are not susceptible to any of the immunotoxic endpoints from TCDD exposure that wild-type (WT) mice display [25,26], demonstrating a requirement for the AhR in these effects.
To further test our hypothesis that the AhR has a role in the function of hematopoietic precursor cells, and more specifically define what this function might be, more detailed studies were undertaken in KO mice. We report here that juvenile KO mice have altered numbers and characteristics of HSCs/HPCs. These and other data suggest that the AhR plays a significant role in maintaining the quiescence of hematopoietic precursors by acting as a negative regulator of proliferation.
Materials and Methods
Animals
A colony of AhR-knockout C57BL/6 mice (AhR
CD45.2+ (C57BL/6) and CD45.1+ (B6.SJL-Ptprc<a>/BoAiTac) mice used in the repopulation experiments were purchased from Taconic Farms.
BM and spleen cells isolation
BM of female mice, 5–6 weeks of age, was harvested from femurs and tibias as previously described [18]. Marrow was flushed from bone and single-cell suspensions were obtained by passing cells through a 22-gauge needle (0.7 mm × 40 mm). Cell suspensions from spleen were prepared by crushing the spleen between frosted microscope slides. The BM and spleen cell suspensions were passed through a Pasteur pipette lined with 80-gauge nylon mesh (Tetko) to eliminate large stromal aggregates. Cells were pelleted and resuspended in red blood cell (RBC) lysing buffer (Sigma) for 5 min. When necessary, lineage depletion of the BM cells was performed as previously described [18]. LSK cell population was obtained by laser-assisted sorting of lineage-depleted (Lin−) cells stained with fluorochrome-conjugated antibodies against Sca-1+ (E13-161.7) and c-kit+ (2B8).
Flow cytometric analyses
Lin− cell suspensions from BM with viability higher than 95% as measured by Trypan Blue dye exclusion were stained with fluorochrome-conjugated antibodies to detect the following populations of cells: Short-term repopulating HSCs (Lin−CD34+Sca-1highcKit+), common lymphoid progenitors (CLP) (Lin-IL7R+Sca-1lowcKit+), common myeloid progenitors (CMP) (Lin−CD34+FcRlowIL7−Sca-1−cKit+), granulocyte macrophage progenitors (Lin−CD34+FcRhighIL7−Sca-1−cKit+), and megakaryocyte erythroid progenitors (MEP) (Lin−CD34−FcRlowIL7−Sca-1−cKit+). The antibodies used to analyze BM cells were CD34 (RAM34), Sca-1 (E13-161.7), cKit (2B8), IL7R (A7R34), and FcR (2.4G2) (BD Pharmingen). Spleen cells were analyzed using fluorochrome-conjugated antibodies against CD4 (H129.19), CD8 (53-6.7), B220 (RA3-6B2), Gr-1(RB6-8C5), and Mac-1(M1/70) (BD Pharmingen).
Colony-forming assays
The high proliferative potential-colony forming cell (HPP-CFC) assays were performed using MethoCultTM M3231 medium (Stem Cell Technologies) according to the manufacturer's protocols and as previously described [18]. HPP-CFC colonies characterized by a diameter of >0.5 mm and a dense core of cells were scored at day 14.
Mouse colony forming unit-erythroid (CFU-E), burst forming unit-erythroid (BFU-E), CFU-granulocyte macrophage, CFU-granulocyte, CFU-macrophage, and CFU-Pre-B assays were performed by using the manufacturer's protocols (Stem Cell Technologies).
In vivo bromodeoxyuridine incorporation
Mice were given drinking water containing 1 mg/mL bromodeoxyuridine (BrdU; Sigma) + 4% sugar in light-protected bottles. Water was changed every 2 days and both the starting and ending volumes were recorded to ensure no water consumption bias between the groups of experimental mice. Mice were sacrificed after 5 days and incorporation of BrdU into Lin−cKit+Sca-1high and Lin−cKit+Sca-1− BM cells was assessed by flow cytometry.
In vivo 5-fluorouracil treatment
Mice were injected with 150 mg/kg body weight 5-fluorouracil (5-FU) (Sigma) via tail vein. After 24 h of treatment, mice were given BrdU in the drinking water as described above. Total BM cell counts and BrdU incorporation were measured at 6 days after treatment with 5-FU. Another group of mice were treated with 5-FU and sacrificed after 6 days. The BM cells were collected and Lin− cells were separated. Flow cytometry was used to analyze BrdU-positive LSK cells as described above.
Ex vivo proliferation assays
Ten thousand Lin−Sca-1+ cells were cultured in 200 μL of Iscove's modified Dulbecco media (IMDM) containing 25% fetal bovine serum, 2 mM
Cell cycle analysis
Lin− cells were stained for Sca-1 and cKit. Afterward, cells were stained with 7-aminoactinomycin (Molecular Probes) at room temperature for 20 min, followed by pyronin Y on ice in nucleic acid staining solution (0.1 M phosphate citrate buffer + 0.15 M NaCl + 5 mM sodium ethylenediaminetetraacetic acid + 0.5% bovine serum albumin + 0.02% saponin) for 10 min. Cells were analyzed using fluorescence-activated cell sorting (FACS) Calibur flow cytometer (BD Biosciences).
Hematology, histology, and immunohistochemistry assays
Peripheral blood was collected in an anticoagulant-coated tube from the retro-orbital plexus vein in the eye using a heparin-coated capillary tube. Blood samples were diluted in saline and RBCs were counted under the microscope using a Neuber's chamber. To obtain the white blood cell (WBC) numbers, blood was diluted in Turk's fluid, and blood smears prepared and stained with Wright's Giemsa stain. Differential WBC counts were determined on the basis of their morphology.
For histology and immunohistochemistry, spleens were fixed in 10% buffered formalin and embedded in paraffin. Tissues sections (6 μm) were prepared and stained with hematoxylin and eosin. Tissue sections used for immunohistochemistry were baked in an oven for 1 h, deparaffinized, hydrated, and quenched with 3% H2O2 in phosphate-buffered saline (PBS) for 5 min, followed by blocking with 20% normal horse serum in PBS for 20 min. Next, the slides were incubated with primary antibodies (anti-Pax5) overnight at room temperature, followed by incubation with secondary antibody conjugated with horseradish peroxidase. Sections were visualized by 3,3'-diaminobenzidine staining and counterstained with Meyers's hematoxylin. After each step of incubation and staining, sections were rinsed with several changes of PBS. The slides were dehydrated and mounted in nonaqueous mounting media.
BM transplantation assays
LSK cells were sorted using FACS Vantage SE sorter. The CD45.2+ donor (5 × 103 LSK) cells together with CD45.1+ competitive donors (2 × 105 BM) were transplanted into CD45.1+ irradiated recipients as previously described [18]. Six weeks after transplantation, mice were sacrificed and BM and spleen cells were collected. Cells were incubated with antibodies against CD45.1 (A20) and CD45.2 (104) and analyzed using a FACSCanto flow cytometer (BD Biosciences). CD45.2+ cells were phenotyped using the following markers: BM: Sca-1, c-Kit, Thy 1.2, B220, Gr-1, Mac-1; and spleen: CD4, CD8, B220, Gr-1 and Mac-1.
Quantification of long-term competitive repopulation units
The competitive repopulation units (CRUs) were analyzed by calculating the frequency of long-term HSCs per million of donor's BM cells injected into the irradiated recipients. A limiting-dilution approach was used for quantification [27]. Five concentrations (2, 1, 0.5, 0.1, and 0.05 × 106) of donor's (CD45.2+) BM cells isolated from WT and KO mice in 100 μL of PBS were mixed with 100 μL of PBS containing 2 × 105 competitive donor's (CD45.1+) BM cells. Recipient (CD45.1+) mice were irradiated with 2 doses of 5.5 Gy. Each concentration of donor's cells was injected intravenously in 8 recipients. After 20 weeks of BM transplantation, recipients were sacrificed and BM cells were isolated. BM cells were stained using fluorochrome-conjugated antibodies (BD Biosciences) against CD45.1 (A20) and CD45.2 (104) and analyzed using a FACSCanto (BD Biosciences) flow cytometer. The presence of more than 1% cells from donor's origin (CD45.2+) was considered as positive engraftment. The number of survivor mice negative for reconstitution in each concentration of donor's per group was recorded and frequency of HSCs was calculated using Poisson statistics [28] and the L-Calc software (Stem Cell Technologies).
Real-time reverse transcription–polymerase chain reaction analysis
Spleens from WT and KO were harvested and sections of around 20 mg each were snap-frozen in liquid nitrogen. Total RNA was isolated using RNeasy Mini Kit (Qiagen), treated with DNAse, and quantified using a spectrophotometer ND-1000 (NanoDrop Technologies). cDNA from 0.5 μg of RNA was prepared according to the manufacturer's protocol. Real-time reverse transcription–polymerase chain reaction [(RT)2-PCR] was done using the pathway-focused array for mouse inflammatory cytokines and their receptors (PAMM-011; SABiosciences Corporation) according to the manufacturer's protocols.
LSK cells from KO and WT (5 mice per group) were obtained using a FACSAria Sorter (BD Biosciences) as described earlier. RNA was isolated and cDNA was prepared from 100 ng of RNA as discussed above. The cDNA samples were analyzed by (RT)2-PCR of a pathway-focused array for HSCs and hematopoiesis (PAMM-054).
All (RT)2-PCR analyses were performed using an iCycler System (Biorad). Expression of mRNA for each gene was normalized using the expression of hprt, gapdh, and actin β as control endogenous genes. KO data were compared relatively with WT data using the 2
Statistics
When appropriate, the 2-tailed Student's t-test was used to analyze statistically significant (P value < 0.05) differences between genotypes.
Results
Juvenile AhR-KO mice have splenomegaly, changes in chemokine and cytokine genes in spleen, and altered blood cell populations
A previously published characterization of an AhR-KO mouse strain with a deletion of exon 1 within the Ahr locus revealed enlarged spleens with aging [23]. To confirm and extend that finding, spleens from juvenile WT, HZ, and KO mice (deletion in the Ahr locus at exon 2) [22] were weighed, and cells were counted and stained for a panel of leukocyte markers. Spleens were also processed for histology and immunohistochemistry. Figures 1A and B demonstrate that the spleens of juvenile KO mice are roughly 1.5-fold larger than those of their HZ littermates or WT mice in terms of weight and cellularity. Flow cytometry analysis of spleen cells revealed increases in the different lineages analyzed, with statistical significance for B220+ and Mac-1+ cells (Fig. 1C). Consistent with these data, spleen sections showed increased number and cellularity of splenic follicles (Fig. 1D, F). There was also an increased number of cells positive for Pax5 in KO mice, indicating the presence of higher numbers of B-lymphocytes (Fig. 1E, G).
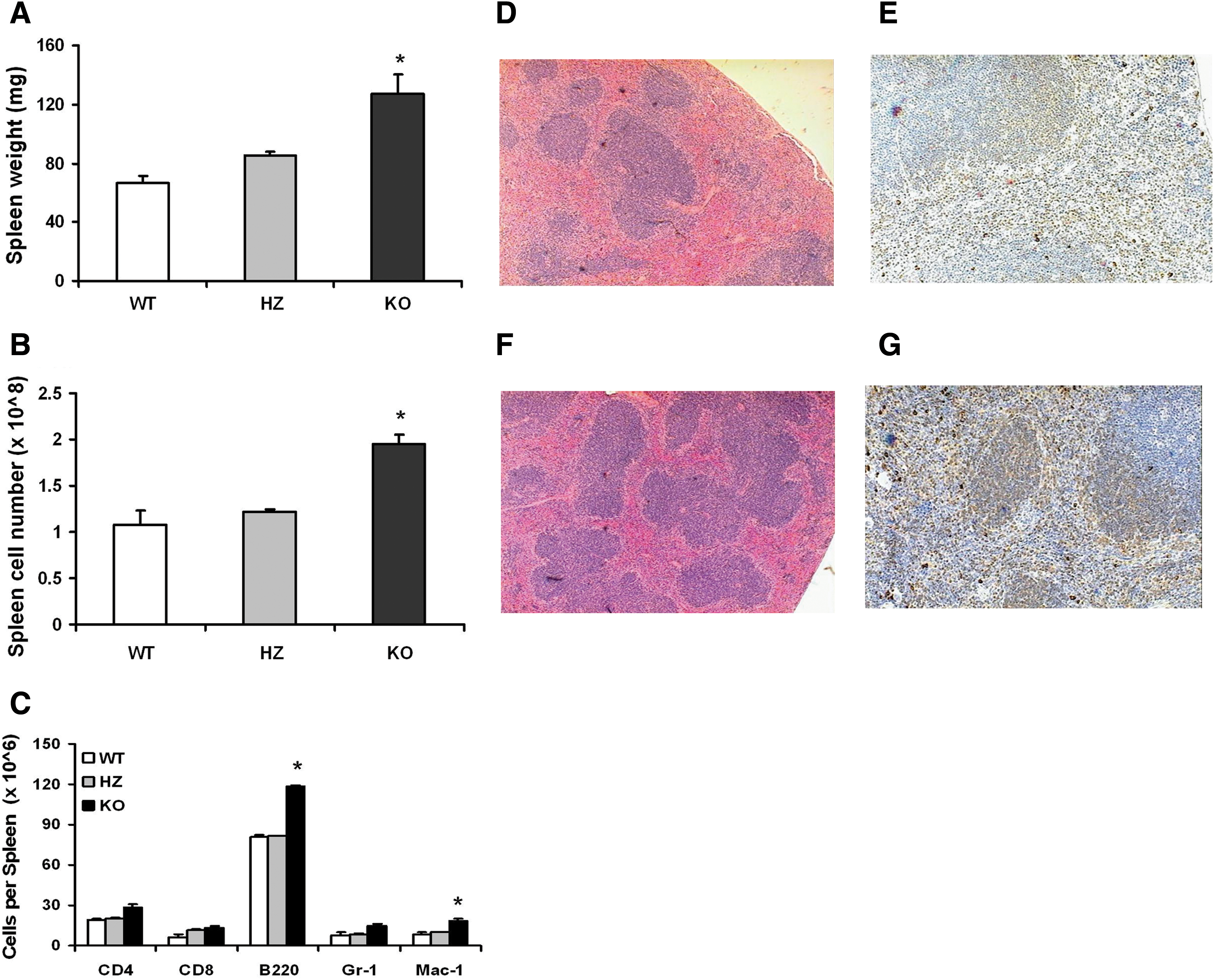
KO mice have enlarged spleens containing elevated numbers of myeloid and B-lineage leukocyte. Spleens were removed from WT, HZ, and KO mice (n = 5 each) and weighed
An analysis of peripheral blood showed a near doubling of the number of WBCs in the KO mice (Fig. 2B), which appeared to be mainly due to an increase in lymphocytes (Fig. 2C). However, the RBCs in peripheral blood decreased about 20% (Fig. 2A).
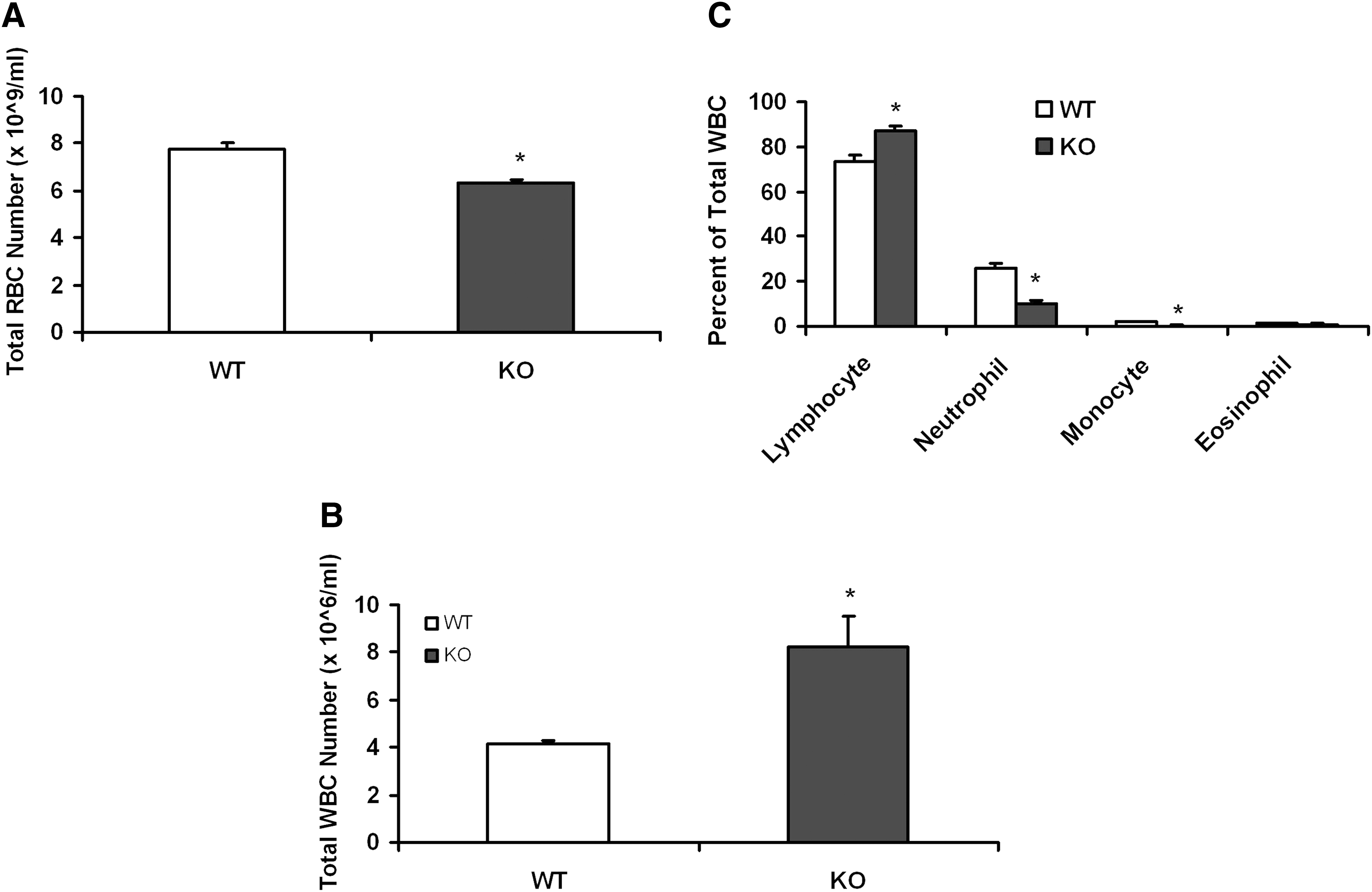
KO mice have changes in the peripheral blood cell number. Retro-orbital plexus blood samples were collected from the WT and KO mice (n = 8) eyes in heparinized vials for total RBC
To more closely examine the intrasplenic events that may contribute to splenomegaly, we used selective (RT)2-PCR arrays to analyze the mRNA expression of chemokines, cytokines, and their receptors (see Materials and Methods section). These analyses showed an increase (>2-fold) of 21 and a decrease (<2-fold) of 2 genes in splenic cells from KO mice (Fig. 3) that suggest alterations in hematopoietic cell kinetics. In particular, there was a significant decrease in the expression of genes for IL-8Rβ (CXCR2) and CCL2 involved in lymphocyte homing and trafficking [29,30]. Notably, CXCR2 KO mice, although reported to be healthy, also develop splenomegaly [29]. CCL2 (MCP-1) is a chemokine secreted by a variety of cells for the recruitment of monocytes, and in its absence, mice show increased susceptibility to a variety of infections including parasitic infections (eg, Toxoplasma gondii infection) [31]. In a recent study, AhR-KO mice were found to be more susceptible to T. gondii infection [32]. It is also of interest that the expression of the gene for CCR2 was increased over 15-fold in the KO spleens. CCR2 is the functional receptor for MCP-1, 2, and 3 cytokines and has been found to be expressed on the surface of HSCs/HPCs, and its role in the homing of these cells to other tissues has been suggested [33]. The relative percentages of B220+ and Mac-1+ and other subpopulations in the spleen (Fig. 1C) do not change to a large degree in KO mice in comparison to WT or HZ mice. For example, the relative percentage of B220+ cells present in the spleen of WT and KO mice was 74.6% and 60.8%, whereas for the Mac-1+ cells this was 8.1% and 9.5%, respectively. Thus, the changes observed in gene expression (Fig. 3) in KO mice cannot be totally explained by the increase in the total number of spleen cells or increase in any particular subpopulation.
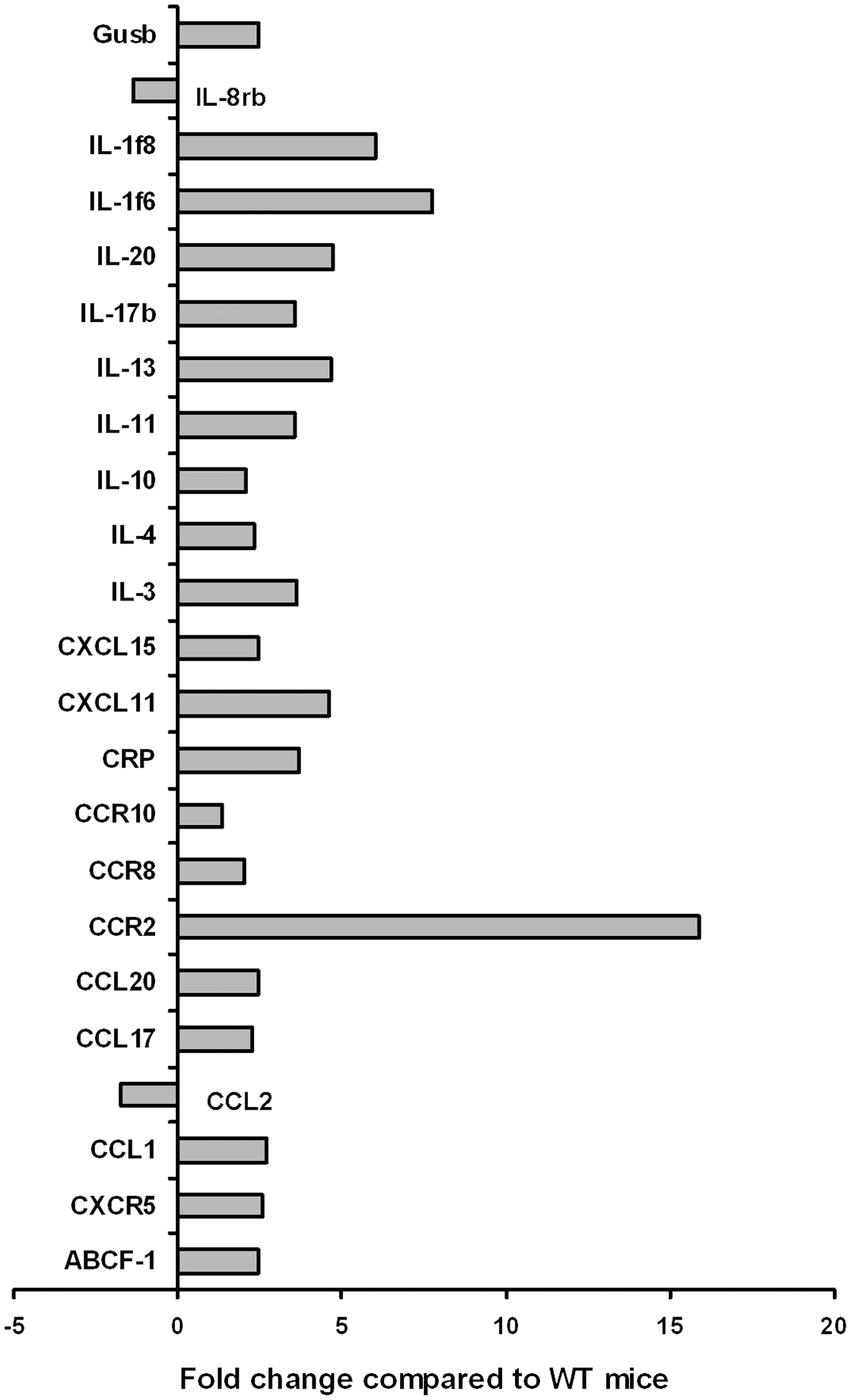
Spleens from KO mice have changes in the expression of chemokines, cytokines, and their receptor genes. Spleens were removed from the WT and KO mice (3 each) and processed for RNA isolation. The mRNA analysis of these genes was performed by using quantitative real-time polymerase chain reaction array analysis. The data are presented as mean of 2 experiments showing a fold change (≥2-fold) in KO mice mRNA expression level in comparison to control WT mice.
AhR-KO mice have a time-of-day–dependent increase of the number of specific hematopoietic populations
The above data suggest that the absence of a functional AhR may result in alterations in hematopoiesis within BM, resulting in changes to peripheral blood counts and contributing to splenomegaly. Consistent with this, we previously observed that BM from juvenile KO mice contains approximately double the numbers of B220+ cells, which were predominantly immature B lineage cells [15]. As such, we assessed the numbers and functions of HSCs/HPCs in BM.
The lympho-hematopoietic system is temporally controlled by circadian rhythms, with the numbers of hematopoietic precursors fluctuating as much as 2–3-fold over a 24-h period. With this in mind, and because TCDD-induced AhR dysregulation alters the number of murine LSK BM cells [17,20], we evaluated the number of hematopoietic precursors in the BM of KO and their littermate HZ mice using a diurnal approach. Marrow was collected from HZ mice every 4 h for 24 h and the number of CD34+ LSK (LSK34+) cells was measured by flow cytometry. As HSCs from perinatal to 5-week-old mice predominantly express CD34 [34], and we used 5–6-week-old mice for this study, the LSK34+ markers were used to analyze HSCs in the BM. As seen in Fig. 4A, these mice were characterized by a near 2-fold greater number of LSK34+ cells at the acrophase (maximum value) when compared with a nadir (minimum or near minimum value), ZT3 and ZT15, respectively. These 2 time points were chosen to measure LSK34+ cells in HZ and KO mice. A 2-fold greater number of LSK34+ cells in KO mice were found to be time-of-day dependent, detectable at ZT3 but not 12 h later at ZT15 (Fig. 4B). This elevation coincided with the increase noted for HZ mice alone (Fig. 4A), suggesting that the lack of AhR enhances a daily increase in the number of LSK34+ that normally occurs. Notably, this experiment was performed weeks later using different groups of HZ and KO mice, and as such, the data in Fig. 4A cannot be directly compared with that in Fig. 4B.
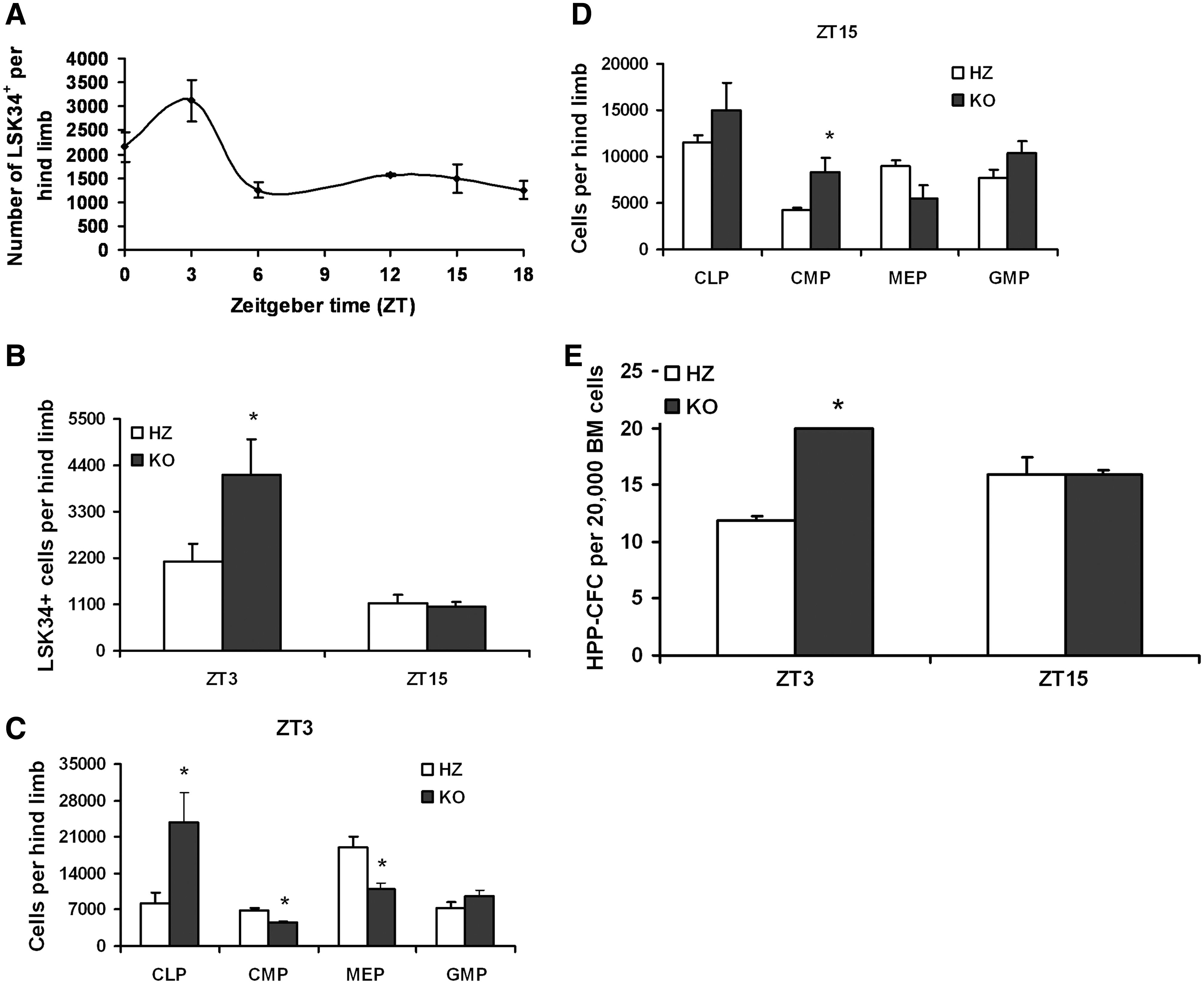
KO mice have time-of-day–dependent changes in the number of LSK34+ cells, hematopoietic progenitor cells, and colony-forming primitive hematopoietic progenitor cells. BM was isolated from HZ littermate control animals (n = 6) at ZT 0, 3, 6, 9, 12, 15, and 18
This diurnal KO phenotype was extended to phenotypically defined committed progenitors. The numbers of CLPs were increased by ∼2.5-fold at ZT3 but not at ZT15 (Fig. 4C, D). Numbers of CMP were abnormal at both tested times, although the nature of the change was diurnally dependent. MEPs were decreased in KO mice at ZT3 by nearly 36% (Fig. 4C, D). The 2 latter populations of cells also displayed a diurnal pattern in control animals alone. Decreasing numbers of CMP and MEP in HZ mice were associated with perceived night, much the same as LSKCD34+cells. No differences in the numbers of granulocyte macrophage progenitors were detected in any of the replicate experiments at these time points (Fig. 4C, D). Consistent with the increase in phenotypically defined HSCs/HPCs, HPP-CFCs were increased in KO mice in a time-of-day–dependent manner (Fig. 4E). Notably, the increases in phenotypically and functionally defined stem/progenitor cells, especially CLP, are consistent with the increase in immature B220+ BM populations that we previously observed [15]. Together, these data demonstrate that in the absence of a functional AhR the numbers of hematopoietic precursors in the BM of juvenile mice are disrupted in a time-of-day–specific manner, indicating a disruption to the temporal as well as lineage organization of hematopoiesis.
Hematopoietic precursors from AhR-KO mice incorporate more BrdU in vivo
The coincidence of the elevated numbers of HSCs and HPCs in KO mice BM with a circadian-regulated normal increase in control animals, and that there is low apoptotic activity in LSK cells [35], suggests that these cells are more likely to be dividing in KO animals. To investigate this possibility, BrdU-laced drinking water (1 mg/mL) was given to the mice over a 5-day period to track DNA synthesis. This period of BrdU exposure is necessary because the cycling time for HSCs and has been estimated to be progenitor 3 days [36]. Over the 5-day period, the percentage of LSK cells in the BM of KO mice incorporating BrdU was roughly 2-fold higher than in HZ mice (Fig. 5A). A similar effect was not observed in the Lin−Sca-1− fraction, a population representative of more mature progenitors (data not shown). These data suggest that in the absence of AhR signaling, the number of LSK cells undergoing division is significantly elevated.
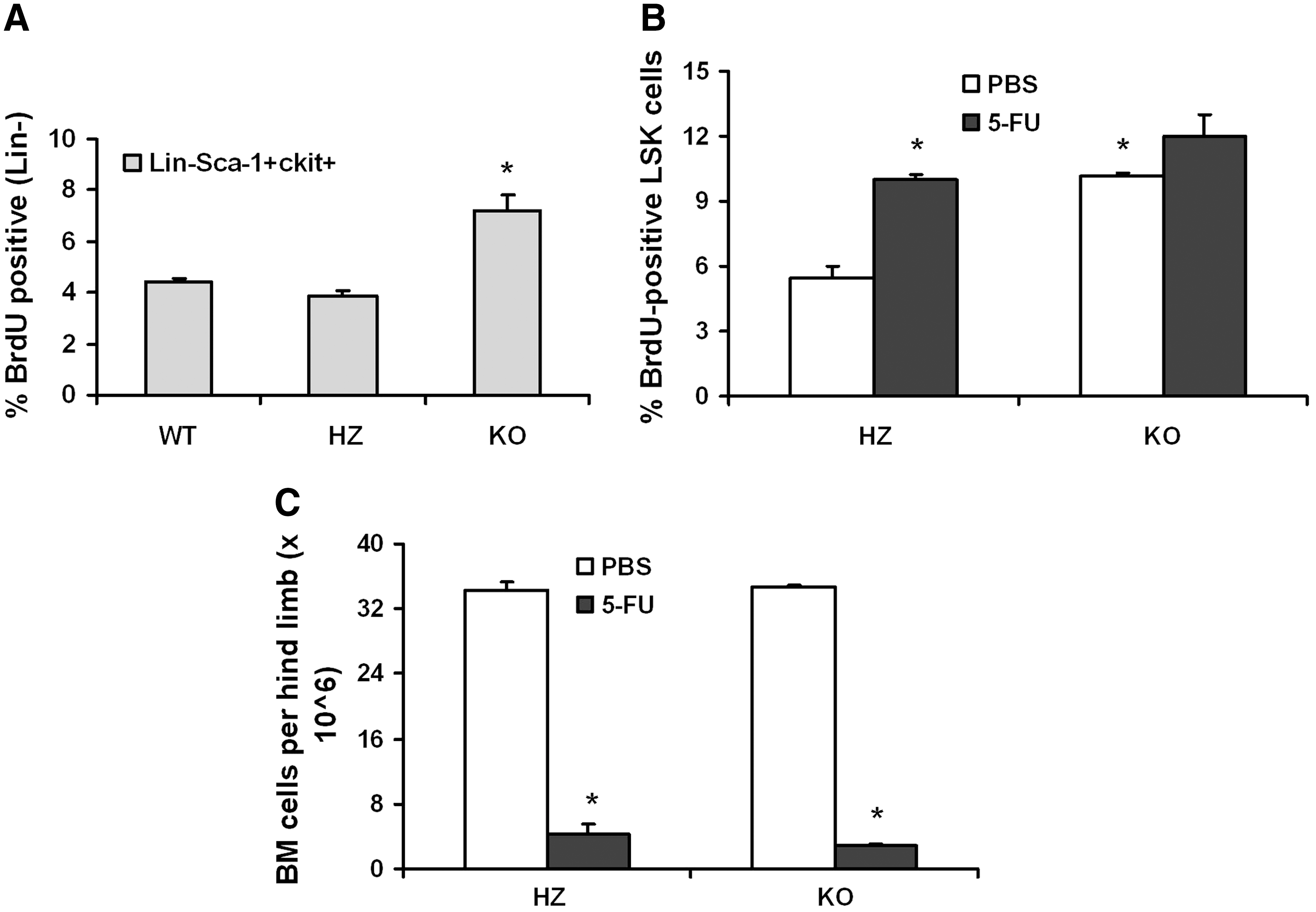
KO LSK cells incorporate more BrdU in vivo. Female HZ and KO mice (n = 6 each) received drinking water containing 1 mg/mL BrdU and 4% sugar for a period of 5 days, after which time they were euthanized. BM was harvested and stained with anti-BrdU-fluorescein isothiocyanate along with antibodies to Sca-1, lineage markers, and cKit
It was of further interest to test whether the observed increase in the proliferation of KO HSCs/HPCs would be different in response to hematopoietic stress. To test this, the chemotherapeutic agent 5-FU was administered to HZ and KO mice. This agent kills rapidly dividing cells in BM, partially ablating the hematopoietic compartment and stimulating the normally quiescent HSCs into division. One day after a single intravenous injection of 150 mg/kg 5-FU or saline vehicle to HZ and KO mice, BrdU-containing water was provided ad libitum. Five days later, BM cellularity and incorporation of BrdU into LSK cells were measured and compared with animals receiving an injection of PBS alone. Consistent with the data obtained above, LSK cells from saline-treated KO mice incorporated nearly double the amount of BrdU compared with cells from HZ mice (Fig. 5C). There was a modest but significantly greater loss of BM cellularity in the KO mice in response to 5-FU treatment, likely reflecting an increased number of dividing cells in these animals (Fig. 5B). We cannot conclude from this single time point (6 days after treatment) whether the KO BM may be significantly more sensitive to 5-FU. However, BrdU incorporation into 5-FU–treated KO LSK cells was similar to cells from 5-FU–treated HZ and PBS-treated KO mice (Fig. 5C). Together, these data indicate that the number of LSK cells in KO mice dividing in a given period of time is much greater than that in HZ mice, but identical to HZ animals responding to 5-FU-induced hematopoietic stress.
AhR-KO mice have more LSK BM cells in the S phase of cell cycle than their HZ littermates
To further test the premise that the increase in BrdU incorporation into LSK cells of KO mice indicates a decrease in quiescence, Lin− BM cells were stained with antibodies to cKit and Sca-1, followed by dual staining for DNA and RNA using 7-aminoactinomycin and pyronin Y, respectively (Fig. 6A). KO mice have roughly 40% fewer LSK cells in G0 when compared with HZ mice, with a concomitant increase in the number of G1 and S phase cells (Fig. 6B). These data demonstrate that LSK BM cells in KO mice are less likely to remain in quiescence when compared with mice with functional AhR protein.
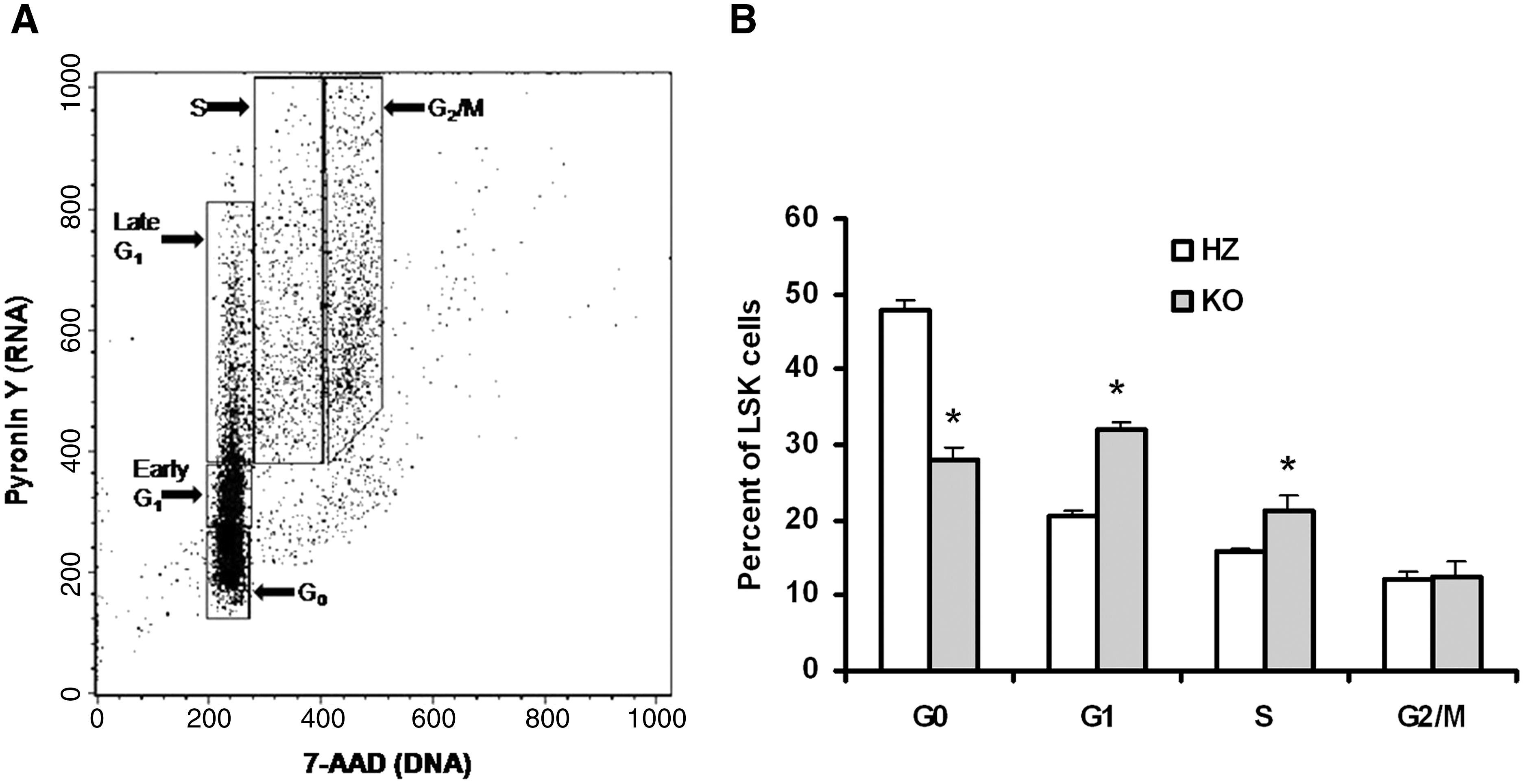
KO mice have more LSK BM cells in the S phase of cell cycle than HZ mice. BM cells were isolated from HZ and KO (4 each) mice. Lin− cells were separated from the BM cells. Lin− cells were stained with Sca-1 and cKit antibody. Lin− cells were then stained with 7-AAD and pyronin Y. The cells were analyzed by fluorescence-activated cell sorting Caliber flow cytometer:
Hematopoietic precursors from KO mice proliferate more rapidly ex vivo
The knockout strain used in these studies has the Ahr gene nullified in all tissues at all times, preventing a conclusive interpretation of whether increased proliferation of LSK cells is due to alterations inherent to those cells or to enhanced stimulatory signals from stromal cells in the BM niche. To address this, we measured the expansion potential of these cells under different ex vivo stimulatory conditions in isolation of any confounding effect of stromal cells from KO mice. Lin−Sca-1+ cells were placed into culture under basal and maximal growth conditions [37], and cell proliferation was assessed over 2 weeks. Under basal stimulatory, but not maximal stimulatory conditions, primary KO cells expanded more rapidly than their HZ counterparts. After 14 days of culture in SCF alone, Lin−Sca-1+ cells from HZ mice increased 6-fold, whereas the same cells from KO mice expanded 13-fold (Fig. 7A). Analysis of these cultures demonstrated 10-fold increased number of LSK FcR− cells in the KO culture (data not shown). The addition of thrombopoietin and Flt3L to the same culture system stimulated HZ cells to expand 13-fold over the 12-day culture period, whereas KO cells expanded 21-fold (Fig. 7C). Lin−Sca-1+ BM cells from HZ and KO mice expanded at identical rates under the most maximal stimulatory conditions, SCF + Flt3-L + IL-6 and SCF + FLT3-L + IL-11 (28-fold and 22.5-fold, respectively). These data demonstrate that the absence of the AhR in Lin−Sca-1+ cells, a population highly enriched for HSCs/HPCs, is sufficient to drive an approximate 2-fold elevation of their ex vivo proliferation potential. The finding that this happens only under submaximal stimulating conditions suggests that the AhR may serve as a negative regulator under normal homeostatic conditions. Further, these data suggest that this is a property inherent to the KO stem cell populations, that is, dependent on the loss of functional AhR expression within those cells.
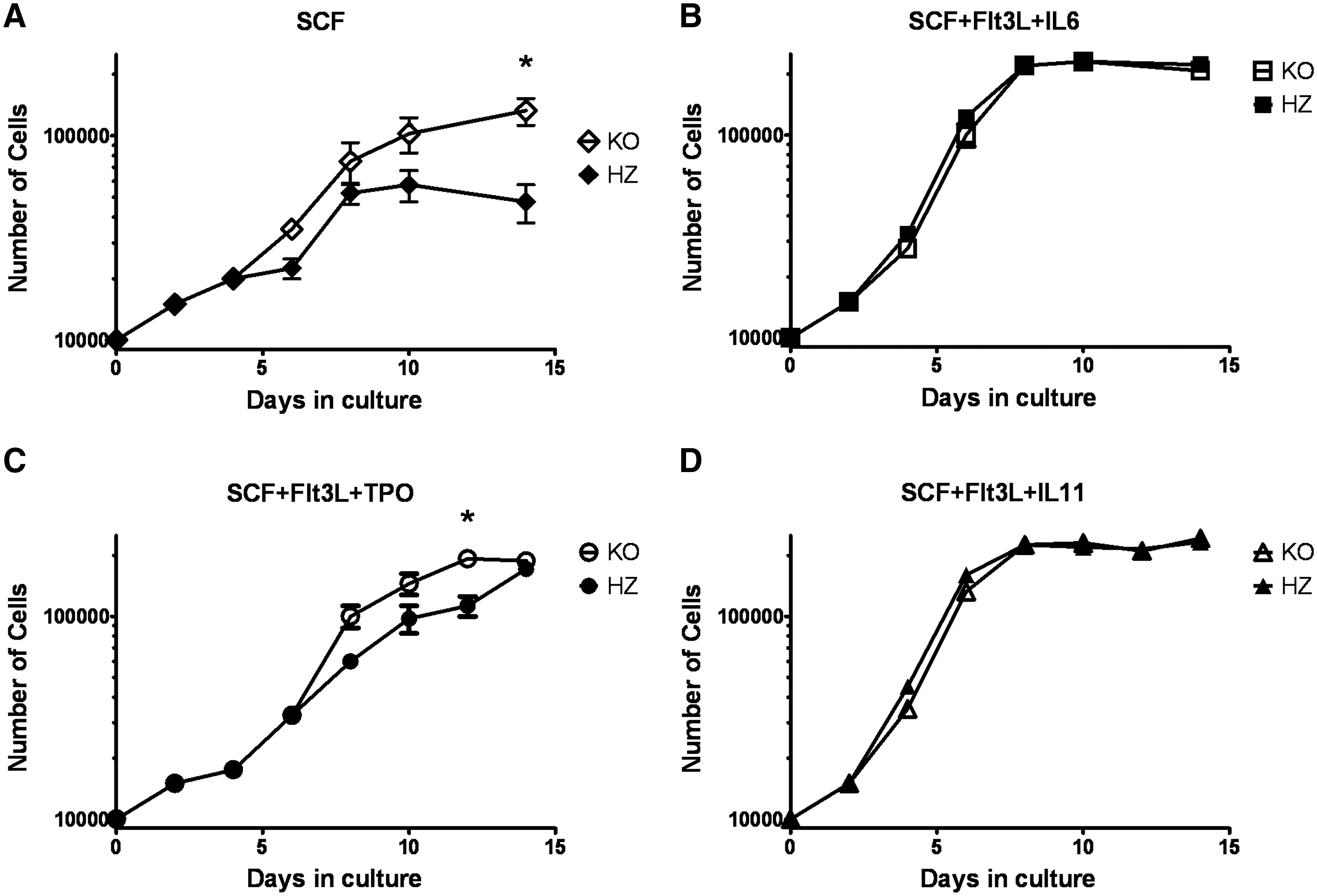
KO Lin−Sca-1+ BM cells proliferate more rapidly to low stimulatory ex vivo conditions than cells from HZ littermates. BM cells were isolated from HZ and KO mice (n = 6 each) and incubated with antilineage biotin, followed by streptavidin microbeads (Miltenyi Biotec). An autoMACS magnetic cell sorter was used to isolate the Lin− fraction, which was subsequently incubated with anti-Sca-1-microbeads and sorted. Ten thousand Lin−Sca-1+ BM cells were placed into culture in Iscove's modified Dulbecco media (IMDM) with following combination of cytokines:
There are functional changes in HSCs/HPCs from KO mice
Although phenotypically defined HSCs/HPCs are increased in KO mice (Fig. 4), this may or may not reflect the functional capacity of these cells. To determine whether the numbers of functional HSCs are increased in KO BM, long-term CRU were quantified using a limiting-dilution approach (see Materials and Methods section and Fig. 8A). Consistent with the phenotypic data, this analysis indicated that KO mice have increased numbers of HSCs with long-term repopulation potential (Fig. 8B).
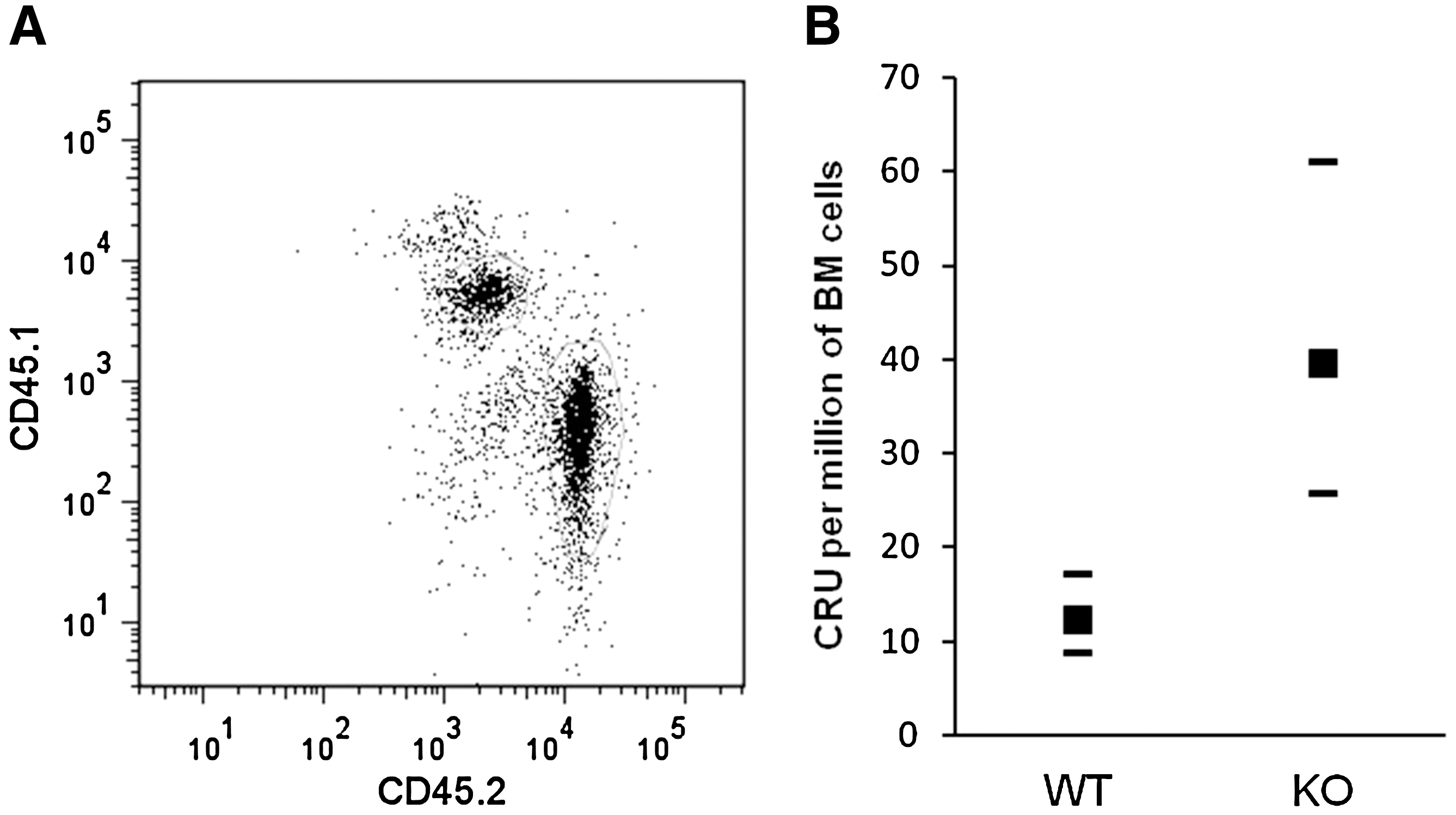
AhR KO mice have increased numbers of HSCs with long-term functional ability in BM. Limiting dilutions of donor's (CD45.2+) BM cells isolated from WT and KO mice were co-injected with competitive donor's (CD45.1+) BM cells into irradiated recipients (CD45.1+, n = 8 per group). After 20 weeks of BM transplantation, recipients were sacrificed and the percentages of donor (CD45.2+) or competitor donor (CD45.1+) cells in total BM were analyzed using flow cytometry. The presence of >1% cells from donor's origin (CD45.2+) was considered as positive engraftment.
To further examine engraftment and differentiation potential of LSK cells from KO mice, we performed a competitive repopulation study in which 2 × 105 recipient competitor mononuclear cells (CD45.1+) were transplanted into irradiated WT (CD45.1+) mice in the presence of 5,000 LSK cells (CD45.2+) from either KO or WT mice. BM was collected from recipient mice at 8 months after transplantation and presence of donor and recipient cells in BM and within different lineages were determined by flow cytometry. There was a slight but nonsignificant increase in total number of BM cells in recipient mice, which were transplanted with the cells of KO mice in comparison to WT donor mice (Fig. 9A). The analysis of total donor (CD45.2+) cells and other lineage-positive cells (B220+, Thy1.2+, Mac-1+, Gr-1+, and Ter-119+) of donor origin present in BM of recipient mice (CD45.1+) also showed a slight but nonsignificant increase in KO LSK cells in recipient mice (Fig. 9B). However, the total spleen cell number was increased dramatically in recipient mice receiving the KO cells (Fig. 9C), and this increase was due, in part, to an increase in CD45.2+ KO cells within all lineage-positive cells, that is, B220, CD8, CD4, and Mac-1 (Fig. 9D). However, the increase in spleen cell number in the KO-repopulated group does not appear to be accounted for by only an increase in KO cells; there may be an increase in competitor population also. The KO cells may have stimulated proliferation and/or differentiation kinetics in competitor cells.
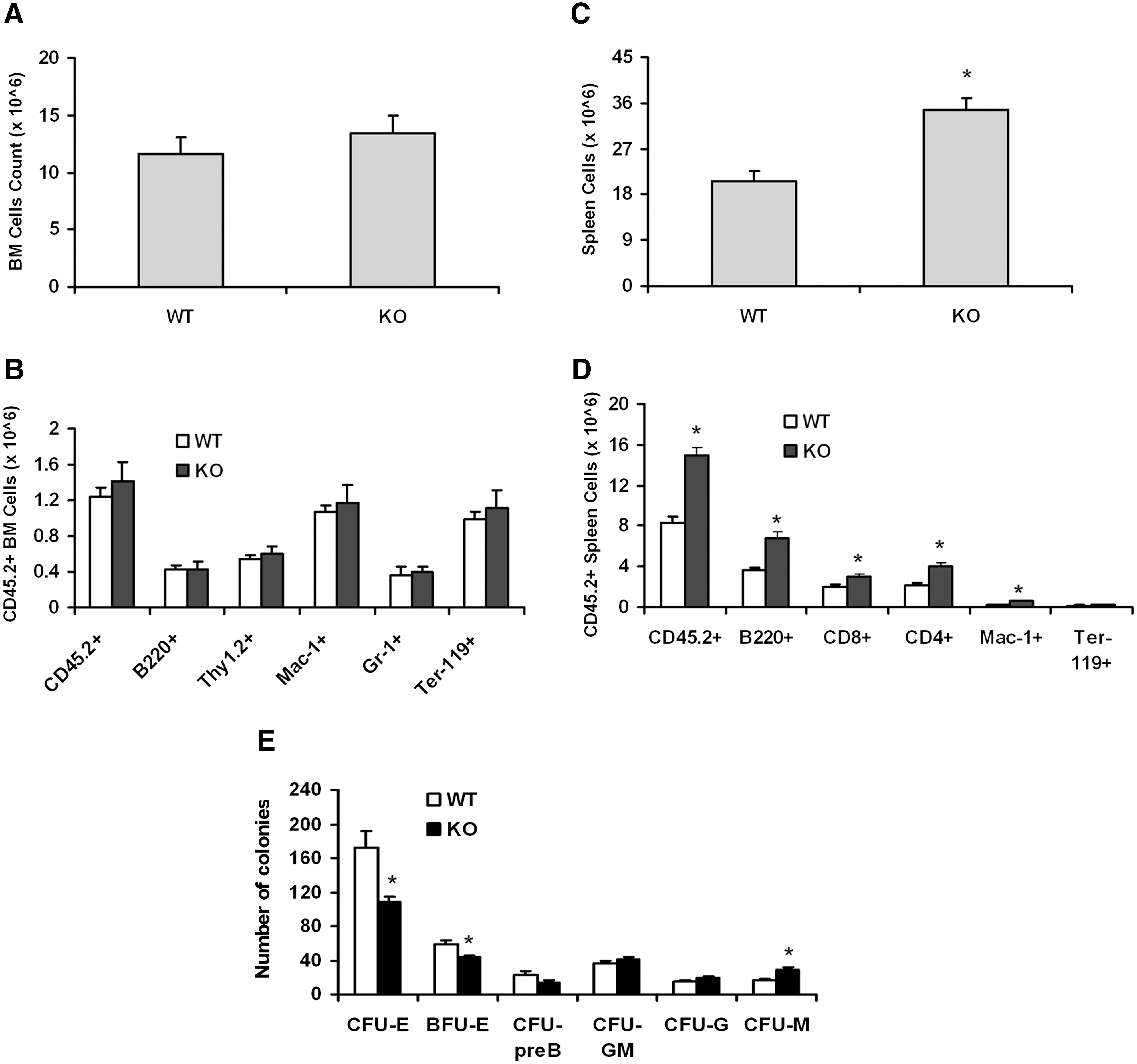
LSK cells from KO mice have changes in the competitive repopulation ability and hematopoietic progenitor cells colony-forming ability. BM cells from WT and KO donor (45.2+) mice were isolated and LSK cells were sorted. The recipient mice (CD45.1+) were irradiated, and after 3 h, mice were injected intravenously with 5,000 LSK and 2 × 105 CD45.1+ recipient competitor mononuclear BM cells. Recipient mice were euthanized at 8 months following transplantation and the presence of CD45.2+ cells of donor origin was analyzed by flow cytometry.
The BM cells from normal naive WT and KO mice were also used for functional evaluation of progenitor cells by using CFU assays. There was a significant decrease in the BFU-E and CFU-E numbers in KO mice compared with cells from WT animals. There was a significant increase in CFU-macrophage, but no differences in CFU-preB, CFU-granulocyte macrophage, or CFU-granulocyte (Fig. 9E).
LSK cells from KO mice have a differential expression of hematopoiesis-associated genes
The expression of mRNA for HSCs and hematopoiesis-related genes were analyzed in LSK cells sorted from BM of KO and WT mice (Fig. 10A). This fraction is enriched for HSCs but contains some early progenitor cells also. Of the 84 genes analyzed by a pathway-focused array (see Materials and Methods section), a >2-fold decreased expression of 22 genes and increased expression of 1 gene was observed (Fig. 10B). Notably, the upregulated gene, Cebpe, is known to be involved in the reprogramming of myeloid lineage commitment and differentiation as well as help in chemotactic function [38]. There was also a nearly 6-fold depression in the expression of Lef-1 mRNA. Lef-1 acts as a central transcription mediator of Wnt signaling, regulating cell cycle and growth through relevant genes such as cyclin D1 and c-myc [39], and plays major role in T-cell development, B-cell proliferation, and apoptosis in pro-B cells [40]. Importantly, there are several putative binding sites for Lef in the promoter regions of the Ahr gene in humans, rats, and mice [41]. Consistent with altered cycling and number of LSK cells in KO animals, we also found the downregulation of many genes (eg, Jag-1, KitL, Tal-1 (Scl), Tlr-3, and Tlr-4) associated with HSC maintenance, growth, differentiation, and trafficking within the BM niche. Of those genes found to be significantly downregulated, a number of these encode proteins that have various roles in the development, signaling, and trafficking of T- and B-lymphocytes, for example, CD2, CD3δ, CD3γ, CD4, CD8α, CD80, IL-1α, IL-10, IL-2, and Pax5. Notably, the conditional loss of Pax5 expression has been shown to result in progenitor cell lymphomas in the mouse [42], and aging KO mice show a greater increase in the development of lymphomas [43]. Several of the downregulated genes express proteins that are also important in normal erythropoiesis, for example, ERAF, TAL-1 (SCL), INHBA, and TRIM-10. The decreased expression of these genes may be associated with the noted decrease in RBC numbers in peripheral blood (Fig. 2A) as well as decreases in BFU-E and CFU-E in BM of KO mice (Fig. 9E). Notably, the long-term deletion of TAL-1/SCL has been also found to produce anemia, splenomegaly, and maturation defects in erythrocyte cells [44].
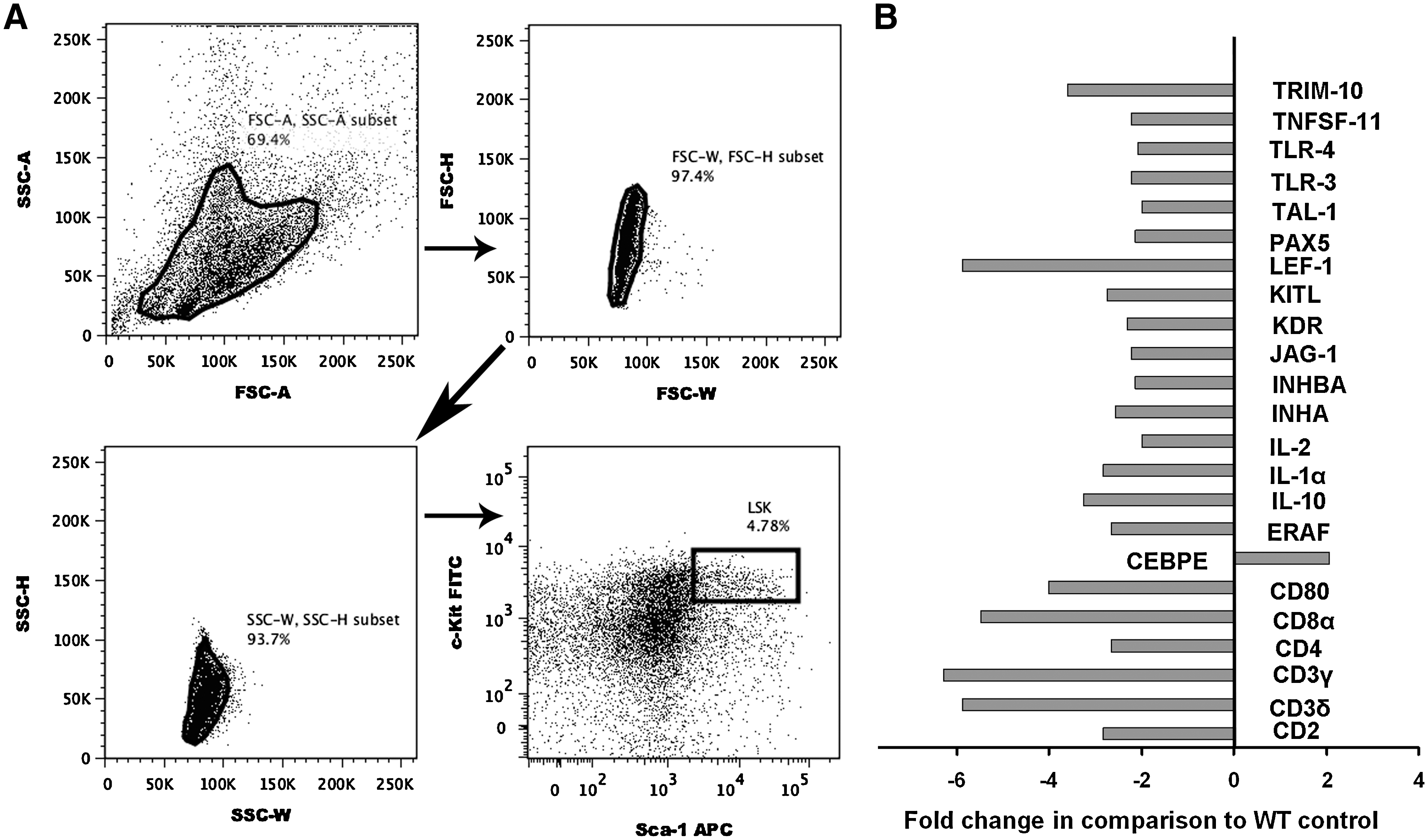
KO mice have altered expression of hematopoietic stem cell and hematopoiesis genes.
Discussion
Previous studies examining the consequences of persistent AhR activation in BM cells suggest a normal role for the AhR in controlling HSCs and HPCs that constitute the murine hematopoietic compartment. Treatment of mice with TCDD results in an increased number of HSC-enriched LSK cells, concomitant with a decrease in B-cell progenitors and increase in myeloid progenitors consistent with a skewing of lineage development from lymphoid to myeloid populations [18,19]. HSCs from TCDD-treated mice exhibit diminished capacity to reconstitute BM of irradiated recipients [18,20], suggesting a role of the AhR in regulating the functional capacity of these cells. AhR mRNA is expressed in stem/progenitor subsets but is downregulated during proliferation [45,46]. Activation of the AhR by TCDD also alters circadian rhythms, quiescence, and expression of clock genes in hematopoietic precursor populations [47]. The processes that control the proliferation of HSCs/HPCs in and release from the BM niche are, in part, regulated by circadian-related mechanisms [48,49]. Our flow cytometric analyses confirmed that phenotypically defined hematopoietic precursors increased or decreased in a time-of-day–specific manner (Fig. 4A). Importantly, LSK34+ cells were elevated in AhR-KO mice by 2-fold at 3 h after the onset of perceived day but not after the onset of perceived night (Fig. 4B), suggesting that the elevation in KO animals is an exaggerated response to a normal process. This could be the manifestation of disruption in several specific processes including proliferation, death, and differentiation into downstream phenotypes. Because of the coincidence of this elevation with what appears to be a normal period of proliferation in HZ mice, we hypothesized that the number of dividing cells was increased. After 5 days of exposure to BrdU, we observed that an approximate 2-fold higher number of LSK cells had incorporated BrdU in KO mice when compared with HZ mice (Fig. 5A). Further, the largest percentage of LSK cells from KO mice are in the G1/S phase of the cell cycle (Fig. 6B). We further hypothesized that the elevated proliferation observed in KO mice would be equated with an elevated response to 5-FU. The most intriguing aspect of these data is that BrdU incorporation into LSK cells was identical between 5-FU- and PBS-treated KO mice (Fig. 5C). This is similar to what was observed in the ex vivo studies where the expansion of KO Lin−Sca-1+ cultures was increased over HZ cultures under minimal but not under maximal growth conditions (Fig. 7), suggesting that KO LSK cells are dividing rapidly, possibly due to a hypersensitivity to proliferative signals. Notably, it has been recently shown that a human specific AhR antagonist promotes ex vivo expansion of human HSCs [50].
All of the data presented here, that is, increased numbers of LSK cells in KO mice BM, increased HPP-CFC and CRU, increased numbers of LSK cells incorporating BrdU and no increase in this parameter following 5-FU treatment, increased numbers of LSK cells in G1/S phases of cell cycle, as well as the increased expansion of hematopoietic precursors under ex vivo conditions, are consistent with a hypothesis that the AhR functions directly in HSCs/HPCs to regulate the proliferation of these cells. In KO BM, this could occur through a loss of a proliferation block and/or increased sensitivity to proliferation signals. Notably, this hypothesis is also consistent with a previous postulate that, based on the finding that the Ahr promoter is silenced by hypermethylation in human acute lymphoblastic leukemia, the AhR is a cell-specific negative regulator of cell proliferation [51]. Further, the finding that the Ahr gene is turned off during the proliferation and self-renewal of hematopoietic precursor cells, but is expressed during periods of quiescence [46], also supports this role of the AhR.
Although together these data suggest that the AhR is a negative regulator of HSC/HPC proliferation, it is not clear whether this regulation may be relevant to asymmetric cell division as well as symmetric division of these cells. If asymmetric division is also influenced, one would also expect to see increased numbers of more mature progenitors in the absence of the AhR. Our findings that there are circadian rhythm-dependent increases in both phenotypically defined CLPs as well as CMPs (Fig. 4) suggest this might be the case. In addition, we previously reported a nearly 2-fold increase in immature B-cells in BM from young adult KO mice [24]. On the other hand, in the competitive reconstitution study we observed only a slight, but nonsignificant, increase in the total number of donor KO cells in BM and lineage cell populations (Fig. 9). Although this suggests that the loss of AhR has little or no influence on the ability of HSCs to engraft and differentiate into lineage immune cells, it is difficult to conclusively determine this from the present study using a single dilution of KO cells with competitive WT cells and examining only a single time (8 months) after cell infusion. It is possible that, for example, increased engraftment and differentiation into lineage cells may occur early (ie, prior to 8 months), but be diminished at later times because of the senescence of rapidly cycling stem and progenitor cell populations. Notably, others have observed a skewing of lineages toward myeloid at the expense of lymphoid populations in conditions of aging and BM senescence [52]. In preliminary studies, we observed a significant depression of B-cell progenitors in 6- and 12-month-old KO when compared with WT or HZ mice (unpublished observations). Although this observation, along with data we report here, are consistent with the reported shortened lifespan in KO mice [43] and the inverse correlation of lifespan with HSC cycling in mice [53], additional work is needed to determine whether in fact the increased cycling of HSCs/HPCs in KO mice will eventually lead to premature BM senescence. Notably, an association between the AhR and aging was made years earlier when it was found that there was a correlation between AhR responsiveness in various mouse strains with lifespan of those mice [54,55].
A clearer indication that HSCs/HPCs from KO mice are dysfunctional comes from the finding that the total number of cells and donor cells in the spleen from the transplanted animals was profoundly increased (Fig. 9), indicating that the splenomegaly observed in KO mice (Fig. 1) is transplantable by KO HSCs/HPCs. These data, taken together with the increased cycling of KO HSCs/HPCs, are consistent with an increased throughput of lineage cells from BM and/or increased trafficking and residence in the spleen. Other data indicating an increase in the number of early B cells in KO BM [15], as well as increased numbers of WBCs in the blood of KO mice (Fig. 2B), are also consistent with this interpretation. The finding that splenic cells from KO mice have the altered expression of many genes involved in lymphocyte homing and trafficking further support this interpretation. Together, these data also suggest that the splenomegaly in KO mice is not due to increased hematopoiesis in the spleen, but is a function of the lack of AhR in hematopoietic cells.
The data presented here also indicate that lack of a functional AhR significantly modifies several important signaling pathways that ultimately influence the balance between quiescence and proliferation and further functioning of HSCs/HPCs.
However, clearly more work is needed to define these relationships. Although our data suggest several “likely suspects,” we do not yet know the precise signaling pathways controlled by the AhR that regulate the altered cell biology observed for HSCs/HPCs. (1) In addition to the genes (especially Lef-1) that we have shown to be altered in LSK cells, AhR is also known to regulate the expression of c-Myc, Hes-1, p21cip1, and p27Kip1 [56], all of which are known to play a central role in the regulation of HSCs. (2) Transforming growth factor-β levels are increased in AhR-KO mouse cells [57], and a role of transforming growth factor-β signaling has been found in normal and malignant hematopoiesis [58]. (3) AhR-KO mice have an approximate 3-fold increase in retinoids related to a decrease in retinoic acid metabolism [59], and retinoids and the retinoic acid receptor play significant role in development and hematopoiesis [60]. Further, the retinoic acid receptor-γ is highly expressed in HSCs and is critical for maintaining balance between HSC self-renewal and differentiation. (4) KO mice have significantly increased oxidative stress in BM cells and primitive HPCs [43]. Recent evidence suggests that oxidative stress plays a significant role in the regulation of normal and neoplastic hematopoiesis. Accumulation of reactive oxygen species in HSCs results in damage to DNA, leading to loss of quiescence and change in cell cycle pattern [61].
The depression of the gene expressing KDR (vascular endothelial growth factor receptor-2) in LSK cells from KO mice is of particular interest, given the roles of this protein in hematopoietic and vascular development [62], the reported defects in vascular development in KO mice [63], and the association of these vascular defects with vascular endothelial growth factor-A depletion [64]. It is tempting to speculate that, given the similarities in phenotype and origin between hemangioblasts and HSCs [65], the origin of defects in angiogenesis in KO mice may be closely related to the altered properties of HSCs that we report here.
In conclusion, we have presented data indicating that the loss of AhR signaling has a dramatic impact on BM hematopoietic precursors. These data support our hypothesis that the AhR is a physiological regulator of HSCs. Further, the data support the contention that the AhR is a negative regulator of HSC proliferation by promoting HSCs to remain in quiescence. Following AhR downregulation, resulting from niche signaling to proliferate, or its nullification as in the KO mice, HSCs may become more susceptible to proliferative signals, allowing these cells to escape from quiescence. Ultimately, it appears that the AhR regulates critical genes within HSCs and/or other progenitors that allow them to differentially respond to signals in their microenvironment. This is consistent with a proposed function of Per-Arnt-Sim superfamily proteins as “sensors of environmental and developmental signals” [1]. In this case, the presence and functional activity of the AhR may provide an important advantage to organisms by preventing the premature exhaustion of HSCs and sensitivity to genetic alterations, thus preserving HSC function and long-term multilineage generation. Additional work is needed to more clearly define these relationships and the signaling pathways responsible for them.
Footnotes
Acknowledgments
The authors are thankful for getting financial support from the National Institutes of Health (Center Grant ES01247, Training Grant ES07026, Grant 04862, and Grant ES016606) for carrying out this research work. The authors thank Jason Walrath, for managing the mouse colonies, and Dr. Ellen Henry, for critical reading of the manuscript.
Author Disclosure Statement
No competing financial interests exist.