
Abstract
The ability to control stem cell differentiation is the holy grail of regenerative medicine. Although significant progress toward this goal has been achieved, few efficient and straightforward methods have been developed, necessitating a better understanding of the mechanisms that influence differentiation. The extracellular microenvironment is emerging as a major player in controlling stem cell fate. Cell surface and secreted heparan sulfate glycosaminoglycans (HSGAGs) are one element of the extracellular matrix that regulates complex cell signaling networks. HSGAGs facilitate binding and availability of cytokines to cells as they progress through development. For example, growth factors such as fibroblast growth factor and vascular endothelial growth factor bind to specific HSGAG sequences during vasculogenesis. HSGAGs have been shown to be critical for stem cell vasculogenesis as well as other differentiation lineages. Understanding the role that the extracellular microenvironment plays in controlling cell fate can lead us closer to directing differentiation for developmental models and regenerative therapies. This review will focus on the role of extracellular microenvironment in regulating cell differentiation, with particular attention to the role of HSGAGs in vasculogenesis.
Introduction
T
Extracellular Microenvironment and Differentiation
The ECM supports cell function by surrounding the cell with a specific combination of structural and soluble proteins and proteoglycans [1]. The most abundant structural protein in the ECM is collagen, and its physical characteristics can influence cell spreading, morphology, and interaction with other cells [2,3]. Adhesive glycoproteins, such as laminin and fibronectin, induce cell–cell and cell–matrix interactions by binding integrins and cell adhesion molecules [4]. Proteoglycans such as heparan sulfate proteoglycans influence cell signaling by binding to soluble signaling molecules and influencing ligand–receptor interactions [5]. The extracellular microenvironment of stem cell and progenitor cell populations is referred to as a “niche” that provides a particular gradient of growth factors and cytokines, paracrine factors, and structural and physical cues that contribute to stem cell stability, proliferation, and differentiation [6 –8]. A niche can provide signals for stem cell recruitment to damaged tissues or for precise spatial differentiation in a developing embryo. Components of the extracellular niche vary from tissue to tissue and in developmental stage [9]. In addition to the molecules comprising the ECM, the molecules associated with remodeling the matrix play a critical role in the niche. External physical characteristics are constantly sensed and altered by internal feedback mechanisms. For example, tension-induced signaling through RhoA stimulates the unfolding of fibronectin fibrils to synthesize new ECM [10]. Remodeling of ECM facilitates cell motility and branching morphogenesis by removing restrictive barriers, providing cell adhesion sites, and gradients of growth factor exposure [11]. It is clear that the ECM plays an active role in determining cell behavior.
In addition to modifying protein signaling, changes in the ECM have been shown to influence epigenetic modifications responsible for lineage-specific expression profiles. Although little is known about epigenetic mechanisms, it is clear that changes in DNA methylation, histone acetylation, and chromatin packaging play a key role in lineage specification [12]. For example, sheer stress, shown to be regulated by ECM polysaccharides, induces epigenetic modifications in embryonic stem cells by increasing histone acetylase activity, resulting in cardiovascular lineage programming [13,14]. The combination of external cues provided by the extracellular niche in an inflammatory versus a regenerative state is directly linked to downstream chromatin remodeling and subsequent cell death or regeneration of muscle progenitor cells [15].
One of the early studies implicating the importance of ECM proteins in the stem cell niche discovered that specific structures of heparan sulfate proteoglycans were required for supporting hematopoietic lineage differentiation [16]. More highly sulfated HSGAGs bound hematopoietic signaling molecules including interleukin-2 and thrombospondin as well as hematopoietic progenitor cells more efficiently than unsulfated structures. The hematopoietic niche is the most broadly characterized to date; specific locations of bone marrow have been shown to support various stages of progenitor differentiation [17]. Hematopoietic stem cells (HSC) have been revealed to bind to osteoblast cells that secrete osteopontin, a proteoglycan that links HSCs with ECM and proteins that maintain quiescence and suppress expansion [18,19]. This highlights how a combination of cell–cell interactions, secreted proteins, and extracellular matrix interact to create a supportive stem cell niche.
Heparan Sulfate Glycosaminoglycans
HSGAGs are one element of the ECM that regulate multiple signaling cascades and have been shown to change composition in certain cancers, during development and cell death [20 –22]. Because of their complex compositions, the specific contributions of specific HSGAG structures are only beginning to be elucidated.
However, recent studies suggest that the chemical compositions of HSGAGs are a dynamic component of the cellular microenvironment by their ability to temporally and spatially bind important signaling molecules (Fig. 1). HSGAGs are linear polysaccharides linked to proteoglycans on the cell surface or in the ECM and are evolutionary conserved from Drosophila to vertebrates [23,24]. There are 4 core protein classes: syndecans and glypicans, which are anchored in the cell membrane by a glycosyl-phosphatidylinositol linker, and agrin and perlecan, which are excreted into ECM. Mutations in these proteins have been associated with developmental defects [25]. Once assembled, these core proteins are transported to the Golgi apparatus where HSGAG chains are attached and structurally modified by a series of enzymes. First, a common tetrasaccharide is attached to a serine residue on the protein core, followed by repeated units of glucuronic acid and N-acetyl glucosamine linked via 1-4alpha linkages to create a chain of 50–150 disaccharides [26]. The variable sulfation of the disaccharide chain creates the HSGAG diversity that is associated with assorted biological functions. The regulation of HSGAG structures is not completely understood, but it has been suggested that translation and assembly of HSGAG synthesis enzyme isoforms in a GAGosome complex in the Golgi is the means of controlling protein binding sulfation patterns [24,27]. The stoichiometry of enzymes in the complex may control the final sulfate composition. Isoforms of the various enzymes add an additional level of complexity. For example, 4 N-deacetylase/N-sulfotransferase (NDST) isoforms exist in vertebrates, which appear to have different substrate affinities. NDST1 and NDST2 are expressed in every cell type, whereas NDST3 and NDST4 are only expressed during development or in specific tissues [28,29].
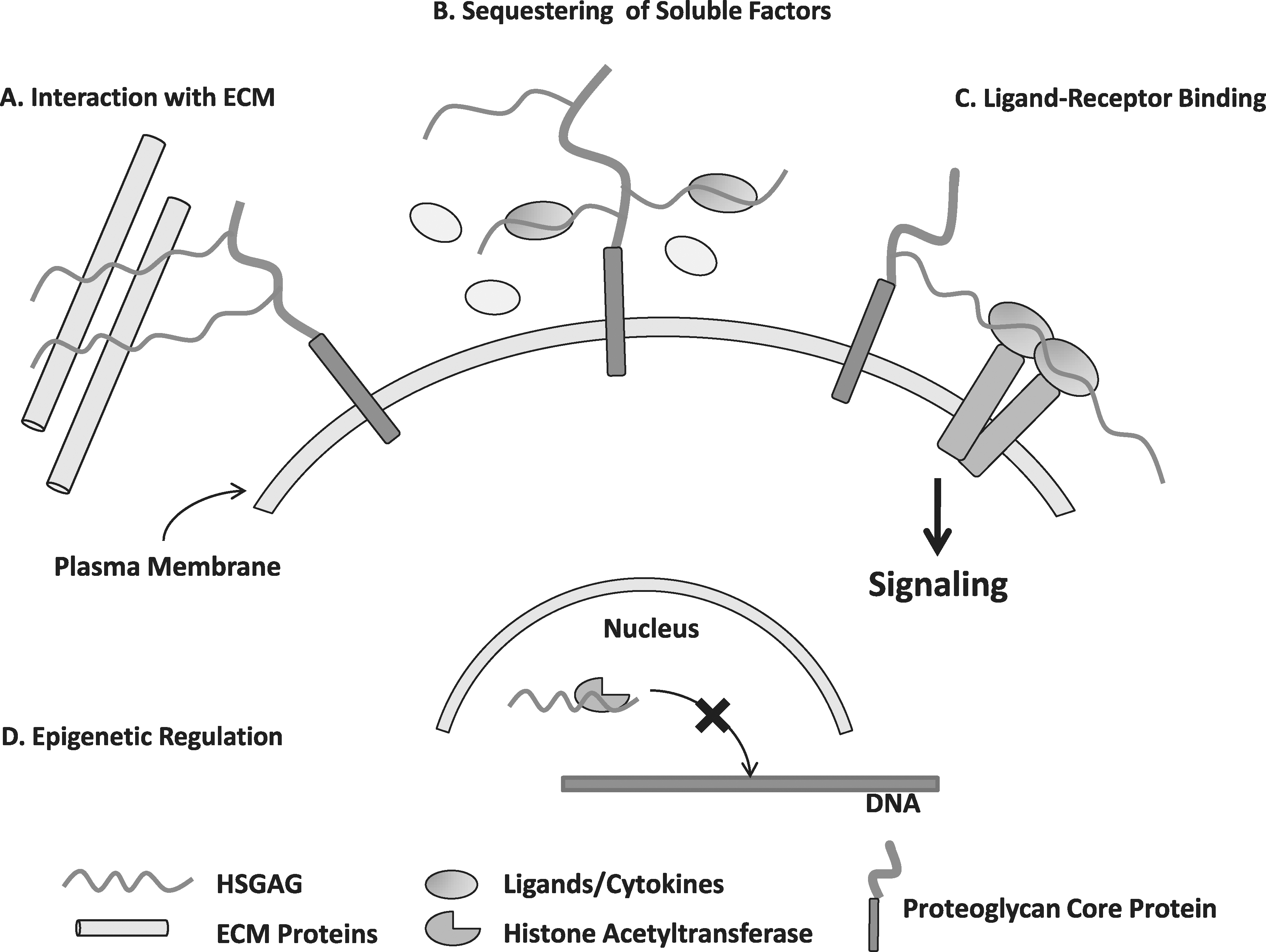
Proposed mechanisms of heparan sulfate glycosaminoglycan (HSGAG) function.
Once the disaccharide chain has been assembled, the enzymes modifying the HSGAG chains and sulfation patterns function sequentially (Fig. 2). The first alteration is by NDST, which forms a complex with exostosin enzymes EXT1 and EXT2 during the formation of the disaccharide chain. The expression of EXT enzymes was also found to affect NDST1 expression, which supports the notion of a precisely controlled GAGosome complex [30]. Because subsequent chain modifications occur at specific sites adjacent to N-sulfations, NDST may control final HSGAG sulfation patterns. After NDST functions, C5-epimerase converts glucuronic acid residues adjacent to N-sulfated groups to iduronic acid. Finally, sulfate groups are added to specific 2-O, 3-O, and 6-O positions of the sugars by their respective sulfotransferases using a 3′-phosphoadenosine 5′-phosphsulfate donor [23]. The iduronic acid unit has one possible sulfation site, the 2-O group, and the glucosamine unit can has 3 possible sulfation sites, the-3-O, 6-O, and free amine groups. Therefore, 32 possible disaccharide units linked linearly in an HSGAG chain generate an enormous array of chemical compositions that contributes to cell signaling and function [31,32]. Generally, sulfation occurs in clusters of disaccharides along the chain, creating regions of rigid negatively charged sites among flexible unmodified regions to allow for specific binding of proteins near the cell surface [33,34]. Soluble proteins interacting with HSGAG chains can be stored for future release, protected from degradation, or presented to a cell surface receptor to facilitate binding [35]. It is unknown whether sulfate sequences are specific for particular proteins or families of proteins. The activity of specific enzyme isoforms may be responsible for consistent differences in sulfation and spacing of sulfated regions in various cell types [36 –39]. For example, successive stages of developing neuroectodermal cells produce HSGAGs, which favor specific binding to fibroblast growth factor 1 (FGF-1) or FGF-2 [40]. The result of this complex, nontemplate-driven synthesis of sulfated HSGAG chains is a unique HSGAG “glycome” environment surrounding the cell. Changes in the cellular glycome contribute to the dynamic extracellular niche, which controls stem cell maintenance and differentiation.
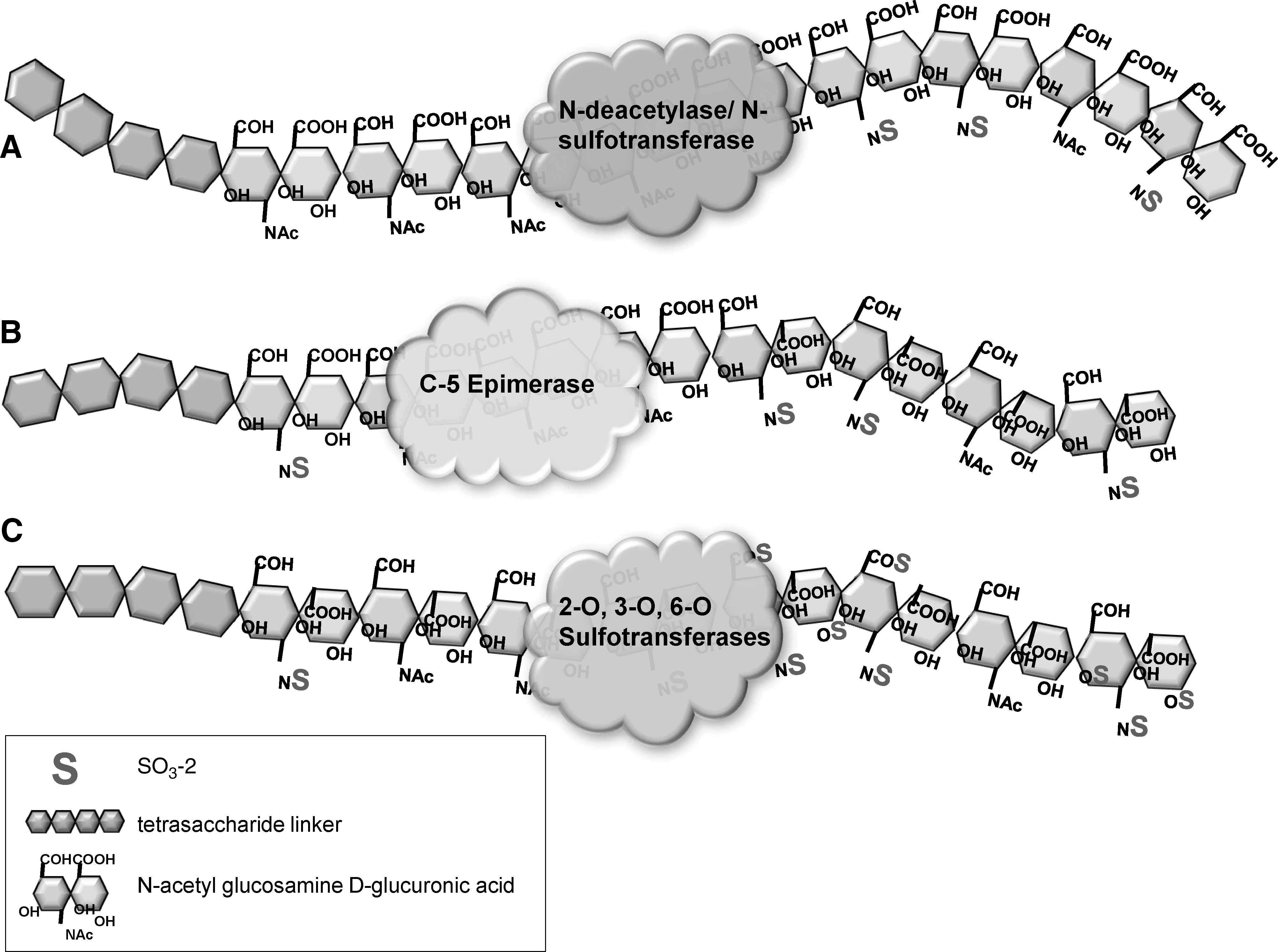
Sequential HSGAG sulfate modifications occur in the Golgi apparatus.
HSGAGs in Differentiation and Development
HSGAGs are required for natural embryonic development, and many studies have studied the effects of disruption of HSGAG chains on development by genetically manipulating HSGAG enzymes [41]. The most well-characterized pathways affected by HSGAG alterations are Wnt/Wg and FGF families. Drosophila mutants sugarless and sulfateless, expressing mutations in UDP-glucose dehydrogenase (responsible for glucuronic acid synthesis) and in NDST, respectively, display abnormalities in segment polarity and ventral patterning, which have been associated with disrupted Wg (Wnt-1) signaling [25,42]. These mutants were found to be similar to Drosophila mutants with diminished FGF signaling functionality, which leads to disrupted tracheal development, mesodermal cell migration, and determination of particular cell fates. It is now well established that HSGAG sulfation is required for proper FGF ligand/receptor complex formation, which plays a crucial role in developmental cell migration and differentiation [43 –45]. Specific 6-O, N, and 2-O sulfate patterns bind different FGF isoforms to receptors [46 –48]. In the mouse, mutation of the 3-OST using a gene trap method was demonstrated to be lethal to the development of the kidney [49], whereas NDST1 mutation produced embryonic lethal embryos with immature lung formation [50]. And, in the zebrafish, Chen et al. demonstrated that 6-OST1 but not 6-OST2 exhibited abnormalities in the branching morphogenesis of the caudal vein during embryonic development [51]. Because of the critical role that HSGAGs play in development by controlling critical signals, it will be interesting to uncover their functions in stem cell differentiation and progenitor cell fate.
Although this review focuses on vasculogenesis, emerging studies have begun to associate HSGAG compositions as important for stem cell differentiation potential or differentiation into many specific lineages [31,52]. Embryonic Stem Cells (ES) lacking HSGAGs are unable to differentiate because FGF signaling is disrupted, causing pluripotency transcription factors to remain active. ES primed for differentiation but deficient in HSGAG chains maintained markers of pluripotency significantly longer than controls [53]. Knockdown of phosphoadenosine phosphosulfate, the donor of sulfate moieties to HSGAG chains, results in a stunting of mesodermal differentiation and an increase in ectodermal differentiation [54]. Signaling molecules important for neural progenitor development and axonal guidance, including netrins, slits, and their receptors, are modulated by HSGAGs [55 –57]. Irie et al. demonstrated that the addition of exogenous HSGAGs disrupts axonal targeting and discovered that specific sulfation patterns caused more significant changes [58]. This study confirmed that specific sulfate compositions of the HSGAG backbone are critical for controlling signaling and not simply the degree of sulfation. Another study using HSGAG epitope-specific antibodies reported that the sulfation motifs in pluripotent embryonic stem cells are different from those of neural progenitors [59]. HS-specific antibodies have also been used to identify a unique and transient sulfate epitope expressed in stem cells differentiating along a mesodermal lineage. Separated cells possessing this epitope had a dramatically increased potential to form endothelial colonies [60]. Thus, HSGAG-specific epitopes can be used as a novel means to identify and sort cell types as well as determine signaling molecules that bind to transient epitopes during developmental pathways. These observations raise the possibility that unique HSGAG epitopes could be harnessed to control the differentiation of stem cells into specific lineages.
ECM and HSGAG Modulation of Vasculogenesis
Stem cell differentiation into endothelial cells is the first step of vasculogenesis [61,62]. Differentiation into endothelium occurs from ventral mesoderm following embryonic gastrulation. Gain- and loss-of-function studies have revealed many signaling molecules critical for progressive steps of endothelial differentiation. FGF, transforming growth factor-β, and bone morphogenic protein 4 are critical inducers of mesoderm formation and differentiation [63,64]. Following FGF stimulation, mesodermal cells expressing vascular endothelial growth factor receptor 2 (VEGFR-2) are triggered by threshold amounts of secreted VEGF from adjacent endoderm toward an angioblast lineage. VEGF is the most important regulator of vasculogenesis and angiogenesis; mice deficient in VEGFR-2 die in utero with a nearly complete lack of vasculature, which defines a necessity of the growth factor for endothelial differentiation [65].
Many studies have focused on methods of directing endothelial differentiation in vitro that result in higher efficiency yields than a spontaneous embryoid body (EB) model and subsequent endothelial isolation. The most common methods to induce differentiation include exposing cells to combinations of growth factors or controlling the culture substrate, with or without first inducing EB formation [66 –71]. Collagen IV appears to be an efficient ECM coating for promoting mesodermal differentiation and generating increased expression of Flk1 [72,73]. In contrast, fibronectin is more efficient at inducing later stages of endothelial development from isolated progenitors [74,75]. 3D substrates are advantageous because they mimic the physiological ECM context and can encourage the formation of larger tissue networks [76,77]. 3D scaffolds used for vascular differentiation include agarose, dextran, hyaluronic acid, poly-L-lactide/poly (lactic-co-glycolic acid) (PLLA/PLGA) and poly (glycerol-co-sebacate-acrylate) (PSGA) [78,79]. A dramatically efficient differentiation method was achieved by McCloskey et al. by growing Poly(glycerol-co-sebacate-acrylate) Embryonic stem cells (ES) on collagen IV coatings, isolating Flk+ cells by fluorescence activated cell sorting (FACS), treating with VEGF, and finally isolating and expanding cells based on endothelial-like morphology for up to 25 doublings. Although this method is clearly labor intensive, the study reported a final differentiation efficiency of >96% endothelium [80].
Our laboratory has investigated the necessity of sulfated HSGAGs for vasculogenesis [81]. We used an analytical capillary electrophoresis method preceded by sequential enyzmatic digestion of cell-surface HSGAGs to detect an increase in HSGAG sulfation as ES differentiated to endothelium. Sodium chlorate can be used to prevent backbone sulfation by inhibiting phosphoadenosine phosphosulfate sulfate moiety donors. Sodium chlorate and heparinase treatment as well as siRNA silencing of NDST1 effectively suppressed the sulfation of cell-surface HSGAGs and prevented vasculogenesis. The deleterious effect of NDST1 inhibition on vasculogenesis was translated to a zebrafish model, in which inhibition resulted in aberrant formation of dorsal aorta, caudal vein, and intersegmental vessels. In the absence of a patent caudal vein plexus, stagnant blood cells moved synchronously with the heartbeat, which points to a specific disruption of vasculogenic signaling and not blood cell formation. RNA microarray comparison of control and NDST1-suppressed zebrafish embryos uncovered possible signaling pathways disrupted by HSGAG modulation that affect vasculogenesis, including the insulin-like growth factor (IGF) axis and downstream transcription factor Foxo5 (Foxo3A in human/mice). IGF is a heparan sulfate binding ligand whose role in vasculogenesis has not been fully elucidated (Fig. 3). This demonstrates the capacity for using HSGAG to identify novel pathways that control stem cell differentiation.
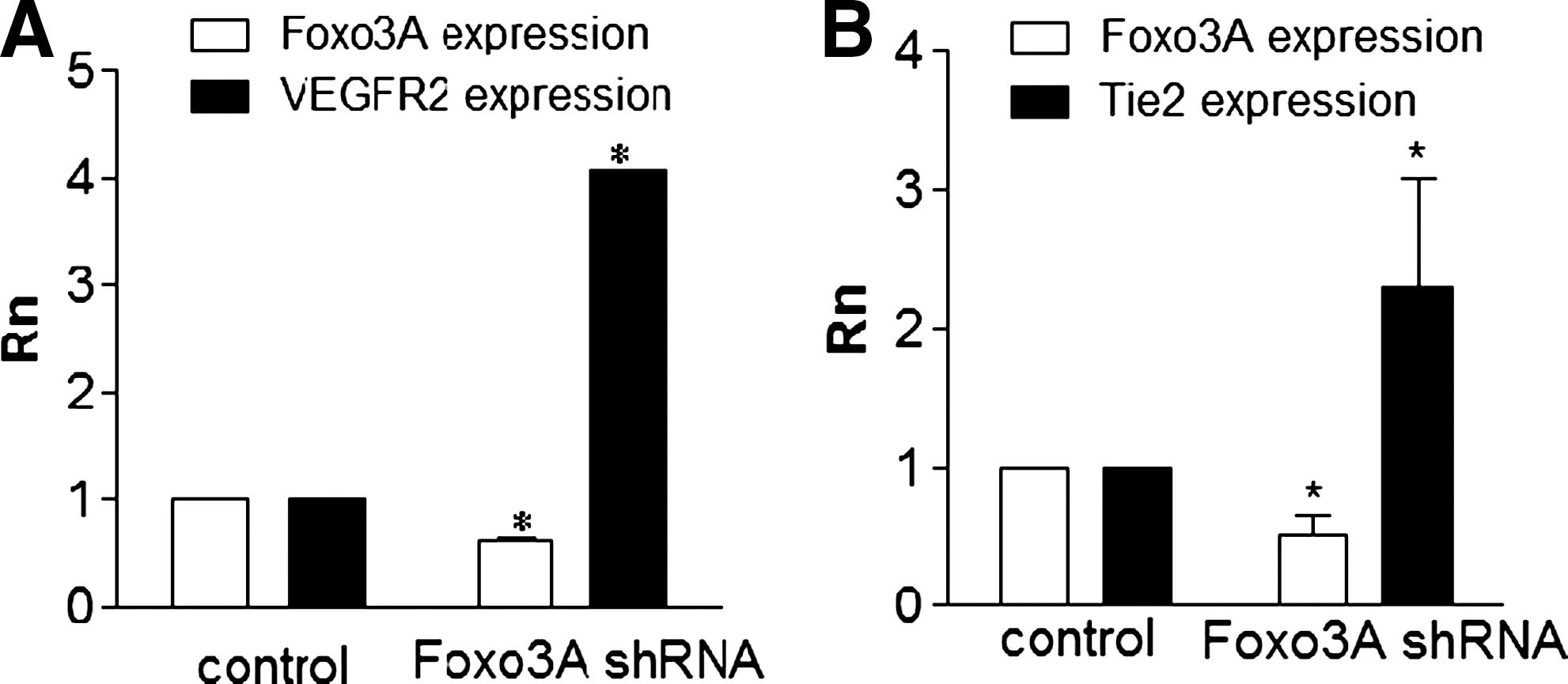
Perturbing the glycome can uncover new pathways implicated in developmental pathways. In this example, modifying HSGAGs and monitoring the resulting effects on vasculogenesis enabled us to discover a regulatory network governed by Foxo3A transcriptions factor. Silencing Foxo3A using shRNA increases the vasculogenic potential of embryonic stem cells, as characterized by an increase in the mRNA levels of endothelial-specific genes vascular endothelial growth factor receptor 2 (VEGFR-2)
Interestingly, in the above study, we found that inhibition of vasculogenesis could be rescued in both mouse ES and zebrafish with the administration of exogenous HSGAGs. This underscores the fact that compositions of HSGAGs on neighboring cells or in the surrounding matrix can play critical roles in controlling a cell's fate and opens the door for engineering specific microenvironments to do so. This concept of heparan sulfate crosstalk in trans was corroborated by Jakobsson et al., who elegantly demonstrated that suppression of vasculogenesis in ES devoid of surface HSGAGs could undergo endothelial differentiation when cocultured with ES possessing typical HSGAGs but lacking vasculogenic potential [82]. HSGAG modulation disrupted VEGF gradient formation, which is critical for vascular development. Disruption of FGF-2 and VEGF binding via endothelial-specific NDST1 suppression was found to selectively lessen tumor neovascularization, a process increasingly believed to be generated by angiogenesis and vasculogenesis [83]. Reduced N-sulfation was also found to affect pericyte recruitment during vascular development in a similar model—NDST1−/− mice displayed vessels with defective pericyte attachment due to inhibited platelet-derived growth factor signaling [84]. Lanner et al. found that NDST1−/− EBs failed to differentiate at all [85]. Specifically, disruption of FGF-4 signaling maintained elevated expression of pluripotency factor Nanog, which could be modulated by exogenous heparin. Knockout of NDST1 disrupted FGF-4 but not FGF-2 signaling, pointing to specific sulfation epitopes for the FGF isoforms. The difference between this and our study may be the complete abrogation of N-sulfated HSGAGs compared with partial knockdown.
HSGAG Modulation of Vasculogenic Factors
HSGAGs are capable of modulating the vasculogenic niche by providing specific binding motifs along the sulfated backbone that affect growth factor gradients and ligand–receptor binding (Fig. 4). They may bind to receptors and maintain threshold levels required for downstream signaling, or they may bind to soluble growth factors and store them near the cell surface [86].
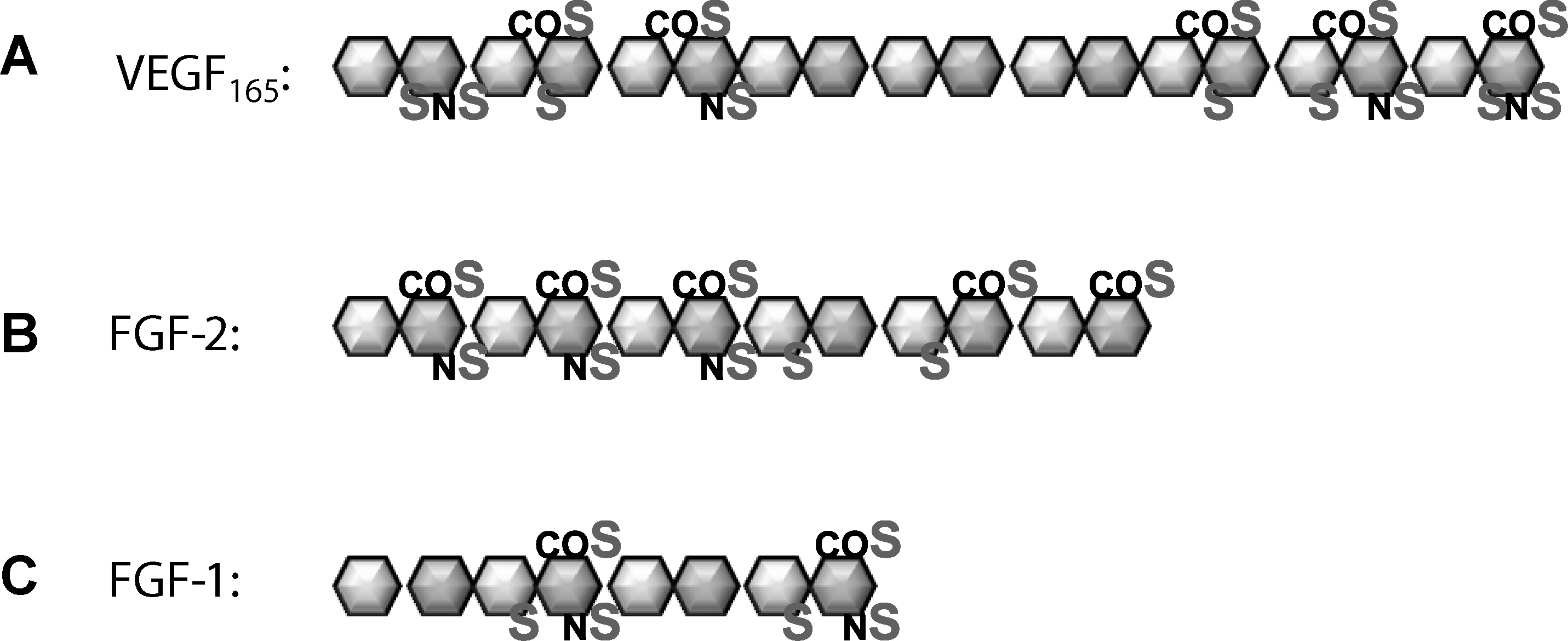
Specific heparan sulfate sequences are required for binding of provasculogenic growth factors.
In many cases, the disruption of HSGAG sulfation patterns can be linked to specific vasculogenic signaling pathways that are inhibited. For example, morpholino inhibition of HSGAG core proteins syndecan-2 or perlecan causes abnormal development of vessels in zebrafish embryos that could be partially rescued by overexpressing VEGF [87]. A small molecule antagonist of HSGAG, surfen, interrupts FGF-2, VEGF165, and fibronectin interactions with endothelial cells and prevents sprouting [88]. Specific HSGAG patterns can be exploited to control the activity of vasculogenesis signaling.
VEGF165 is the most potent mitogen for vessel formation and binds to specific HSGAG sequences. Ono et al. used chemically modified HSGAGs and determined that a high content of 2-O sulfation is not required for VEGF binding and that 2-O-desulfated HSGAGs could sequester VEGF from proliferating endothelial cells in culture, whereas 6-O and N-S desulfated fragments could not [89]. The requirement of 6-O sulfation for VEGF binding was confirmed by Chen et al. when morpholino-induced knockdown of 6-O sulfation enzymes in zebrafish impeded caudal vein morphogenesis and had a synergistic effect with VEGF knockdown [51]. This effect was linked to defects in late endothelial differentiation events because markers of late-stage endothelial development, tie-1 and tie-2, were decreased. Using a library of HSGAGs isolated from different cell types and selectively desulfated, Robinson and colleagues reinforced that all sulfate positions played a role in VEGF binding affinity, but 2-O sulfation was less important than N-S or 6-S [90]. Varying disaccharide lengths were also explored, which led to a proposed model wherein 2 separate highly sulfated domains along the HSGAG chain that is at least 6 disaccharides in length and contains a central 6-O sulfated glucosamine residue results in extremely stable binding to VEGF165. Heparan sulfate interacts with the exon-7 coding region of VEGF165 and interacts with VEGFR-2 to increase endothelial proliferation and tube formation [91].
The addition of HSGAG chains to neuropilin-1, a coreceptor for VEGFR-2, had opposing effects on VEGF signaling in endothelial and smooth muscle cells, although the particular sulfation sequences were not explored [92]. Sulfate binding motifs have different effects on various cell types and cell stages, and in addition to facilitating growth factor-receptor binding, they may act as decoys or sinks for signaling molecules. As mentioned earlier, differentiating EBs derived from NDST1/2 null mice are defective in HSGAG sulfation and unable to spontaneously generate endothelium because of lack of VEGF165-VEGFR-2 response. Soluble heparan sulfate was unable to rescue this phenotype, but chimeric EBs containing cells capable of producing sulfated HSGAGs could do, suggesting the possibility of intercellular crosstalk in stabilizing receptor interactions [82]. However, our data demonstrate that soluble heparan sulfate is able to rescue endothelial differentiation potential from EB with siRNA suppression of NDST1 [81]. Neovascularization in tumors derived from mice with an endothelial-specific deletion in NDST1 was dramatically suppressed despite intact pericytes surrounding vessels [83]. Collectively, the results indicate that regulation may be context dependent or that cells can draw on a variety of HSGAG sources to proceed through developmental stages.
FGFs are the most well-studied protein with respect to their dependence on HSGAG for signaling. Isoforms of FGF, along with bone morphogenic proteins, play a major role in maintaining stem cell pluripotency [93]. They are also crucial inducers for mesoderm differentiation and the generation of angioblasts among other developmental roles [94]. HSGAG serves as a reservoir of FGF for its receptor by binding a large quantity of FGF but with a lower affinity than that of the receptor. It also increases the half-life of receptor binding [43]. HSGAG composition changes during development or in different tissues to accommodate the binding of various FGFs—FGF-2 preferentially binds to HSGAG harvested from neuronal tissue at embryonic day 9, whereas FGF-1 favors HSGAG from day 11. Both isoforms are produced during this time span, which means that HSGAG composition is critical for regulating specific developmental signals [22]. Like VEGF, 6-O glucosamine sulfation is required for interaction with FGF. 6-O endosulfatase strikingly reduces FGF-2 signaling in ovarian cancer cells, chick, and Xenopus embryos [95 –97]. The first studies to associate intact HSGAG for proper FGF signaling occurred in Drosophila melanogastar, when mutants of UDP-GlcA-dehydrogenase, HS polymerase, and NDST disrupted mesoderm development in a manner similar to FGF knockdown [98]. These mutations lead to complete disruption of FGF, Wnt/Wg, and hedgehog signaling cascades, all of which generate gradients to control cell polarity and pattern formation during development. Based on the above studies, it is easy to imagine a model wherein HSGAG compositions are generated at particular cell development stages to attract specific molecular isoforms.
Besides soluble growth factors, HSGAGs interact with ECM proteins, which can exert control over cell differentiation through mechanical stimulation and integrin signaling [99]. The unfolding and polymerization of fibronectin is a requisite for neovascularization [100]. HSGAGs catalyze this process by binding to and changing the conformation of fibronectin. Specific HSGAGs with 6-O and N-sulfated glucosamine expose a fibronectin binding motif for VEGF165 [101]. This indicates another level of HSGAG control over growth factor presentation in the context of vasculogenesis.
Synopsis and Future Directions
The tremendous array of cell lineages that are generated simultaneously in a developing embryo is testimony of the remarkable control that the extracellular niche has over stem cell maintenance and differentiation. HSGAGs play a central role in controlling intra- and extracellular crosstalk by providing variable binding motifs for differentiation signals. Many of the mechanisms responsible for vasculogenesis have been shown to be modulated by specific HSGAG sequences. Most studies have focused on HSGAG regulation of the critical vasculogenic factors VEGF and FGF, but new vasculogenic growth factors and HSGAG binding proteins are constantly being discovered. We found that inhibition of sulfated HSGAGs inhibited vasculogenesis in mouse embryonic stem cells and correlated with upregulation of Foxo transcription factor and upstream IGF family members, which we are currently exploring. It is clear that HSGAG regulate the extracellular niche, and intriguingly, recent studies have identified a possible role for HSGAG in directing epigenetic changes in the nucleus. HSGAG with specific sulfation patterns were shown to effectively inhibit histone acetyltransferases and thus regulate chromatin structure [102]. This would mean that HSGAGs have a critical role in mediating both extracellular and intracellular regulation of cell lineage commitment. Overall, identifying new levels of cellular control over differentiation allows us to understand the mechanism of vasculogenesis and harness these processes for potential cell therapies.
Footnotes
Author Disclosure Statement
No competing financial interests exist.