
Abstract
Neurons derived from human embryonic stem cells hold promise for the therapy of neurological diseases. Quality inspection of human embryonic stem cell-derived neurons has often been based on immunolabeling for neuronal markers. Here we put emphasis on their physiological properties. Electrophysiological measurements were carried out systematically at different stages of neuronal in vitro development, including the very early stage, neuroepithelial rosettes. Developing human neurons are able to generate action potentials (APs) as early as 10 days after the start of differentiation. Tyrosine hydroxylase (TH)-positive (putative dopaminergic, DA) neurons tend to aggregate into clumps, and their overall yield per coverslip is relatively low (8.3%) because of areas void of DA neurons. On the same in vitro day, neighboring neurons can be in very different stages of differentiation, including repetitive AP firing, single full-size AP, and abortive AP. Similarly, the basic electrophysiological parameters (resting membrane potential, input resistance, peak sodium, and peak potassium currents) are scattered in a wide range. Visual appearance of differentiating neurons, and number of primary and secondary dendrites cannot be used to predict the peak sodium current or AP firing properties of cultured neurons. Approximately 13% of neurons showed evidence of hyperpolarization-induced current (Ih), a characteristic of DA neurons; however, no neurons with repetitive APs showed Ih. The electrophysiological measurements thus indicate that a standard DA differentiation (dibutyryl cyclic AMP-based) protocol, applied for 2–5 weeks, produces a heterogeneous ensemble of mostly immature neurons. The overall quality of human neurons under present conditions (survival factors were not used) begins to deteriorate after 12 days of differentiation.
Introduction
T
The quality control of DA neurons derived from hESC has been previously assessed by functional methods, including neurotransmitter release [3 –8], or electrophysiological recordings of action potentials (APs) and synapse formation [5,6,9 –11], or calcium imaging [12]. However, in a greater majority of studies the stem cell differentiation protocols for generation of human DA neurons were evaluated based on the expression of proteins using standard immunostaining or polymerase chain reaction [3,7,13 –20]. Protein analysis is insufficient to determine the physiological properties of cells, since the functional aspects of proteins and the prevailing functions of the cells are not taken into account.
Here we use systematic electrophysiological recordings to evaluate the outcome of one previously established differentiation protocol for DA neurons based on dibutyryl cyclic AMP (dbcAMP) [14]. In the stem-cell field there are multiple designs for deriving DA neurons in vitro [21 –24]. For the purpose of the present study we chose the protocol initially developed in Wisconsin [23] and slightly modified in Philadelphia [14] for its simplicity and shortness. The design of the protocol rests essentially on the fundamental principle of neuroectodermal induction in mammalian embryos [25]. First, hESCs are turned into a population of neuroectodermal cells and later into a nearly pure population of neurons [23]. The protocol is chemically defined because it deliberately avoids unknown components, such as sera or stroma cells, in the system. The protocol is robust with reported efficiency of over 90% neurons [26]. In our hands the Iacovitti protocol has been highly reproducible, which allowed us to focus on electrophysiological properties of young human neurons.
In the past, the so-called neuronal markers (eg, TUJ1) have been used as a definitive proof of neuronal lineage, and as such, TUJ1-positive cells have been the ultimate goal of many studies [27]. Immunolabeled hESC-derived neurons are often assumed to constitute a synchronized population of functionally competent neurons. That is to say that TUJ1 positivity could be interpreted as neurons being in the same (postmitotic) phase, and therefore pure and homogeneous. Also, it could be interpreted that TUJ1-positive neurons are endowed with excitable membranes and ready to fire APs. To investigate the physiological status of hESC-derived neurons, we systematically patched dividing precursor cells or postmitotic neurons starting as early as 4 days from the beginning of the protocol (neuronal rosettes). Although started from synchronized neuroepithelial cells and treated under strictly controlled and identical conditions (incubator, same dish), the neighboring human neurons in the same culture dish exhibit an interesting variety of basic physiological parameters.
Materials and Methods
hESC culture
All cell culture reagents were from Invitrogen (Grand Island, NY) unless otherwise noted. H9 cells, passage 50–65, were cocultured with mouse embryonic fibroblasts (MEFs) in the ESC medium consisting of 80% Dulbecco's modified Eagle's medium (DMEM)/F12, 20% Knockout serum replacer, 1 mM glutamine, 1× nonessential amino acids, 4 ng/mL basic fibroblast growth factor (bFGF), and 7 nL/mL β-mercapto ethanol (Sigma, St. Louis, MO). Cells were grown on plastic dishes (Nunc, Rochester, NY) coated with 0.1% porcine gelatin (Sigma). The medium was changed daily, and cells were split using 1 mg/mL collagenase. MEFs derived from CF-1 mice were maintained in 90% DMEM, 10% fetal bovine serum, and 1× nonessential amino acid. MEFs were irradiated with 8,000 rads before seeding as feeder layers.
Neuronal differentiation of hESC
H9 cells, obtained from the institutional Stem Cell Core, were differentiated using the protocol of Iacovitti et al. [14]. Briefly, H9 colonies were disassociated by collagenase, and ESC aggregates incubated for 3–4 days in the ESC medium without bFGF on Ultralow adherence plates (Costar, Wilkes Barre, PA) (stage 2). ESC aggregates were then seeded on dishes coated with 1:100 Geltrex, and allowed to expand for 4 days in NEP-basal medium (stage 3). NEP-basal medium consisted of DMEM/F12, 1 mg/mL bovine serum albumin (Sigma), 1× N2, 1× B27 supplements, and 1× penicillin/streptomycin/anti-mycotic. Colonies with neuroepithelial morphology were removed by trituration, and seeded on glass coverslips coated with 1:100 Geltrex. For Rho-associated kinase (ROCK) inhibitor treatments, stage 3 cultures were treated with 3.3 μg/mL Y-27632 (Wako, Richmond, VA) 1 h before removal, and then for 1 day after seeding on coverslips. Cells were grown in NEP-basal medium with 20 ng/mL bFGF for 7 days (stage 4). Cells were then maintained in NEP-basal medium with 1 mM dbcAMP (Sigma) for 7 days, unless otherwise noted, with the treatments indicated in the figures.
Immunofluorescence
After differentiation, cells were fixed 30 min in 4% paraformaldehyde. Immunofluorescence was performed using 1:500 rabbit anti-TH (Pel-Freez, Rogers, AR) and 1:1,000 mouse anti-TUJ1 (Sigma T5076) antibodies. Cells were permeabilized in 0.2% Triton TM ×100 (Acros, Geel, Belgium). Cells were washed in phosphate-buffered saline (PBS), and then blocked for 1 h in 10% normal goat serum, 0.75% bovine serum albumin in PBS, and then incubated overnight at 4°C with primary antibodies diluted in blocking solution. Cells were washed in PBS and then secondary antibodies incubated for 1 h at room temperature. Goat anti-mouse Alexafluor488 and goat anti-rabbit Alexafluor594 (Invitrogen) were used in 10% goat serum. Cells were washed in PBS and then incubated 10 min in 1 μg/mL Hoechst 333258 (Sigma). Cells were mounted with FluoromountG (SouthernBiotech, Birmingham, AL). TH and TUJ1 signals were quantified from images selected using only the TUJ1 signal. ImageJ software was used for quantification [28]. Background was subtracted from each channel, integrated optical density calculated, and ratios of TH signal to TUJ1 signal determined for each image. Coefficient of variation (CV) was calculated as standard deviation/(mean ratio of TH/TUJ1). Each data group contained 22 or more images of differentiated cultures.
Electrophysiology
Whole-cell patch recordings were performed as previously described [29]. Briefly, before recordings, cells were transferred to an Olympus (Tokyo, Japan) BX51WI upright microscope (equipped with infrared video microscopy) and perfused in aerated (95% O2/5% CO2) artificial cerebrospinal fluid (ACSF) at 30°C. The ACSF contained 125 mM NaCl, 2.3 mM KCl, 23 mM NaHCO3, 1 mM MgSO4, 1.26 mM KH2PO4, 2 mM CaCl2, and 20 mM glucose, pH 7.3. Individual cells were selected for recordings based on a small round or ovoid cell body (diameters, 5–15 μm) and typically 2 or more extended processes. Pipettes (10 MΩ) were filled with an intracellular solution containing (in mM) 135 K-glutamate, 10 HEPES, 2 MgCl2, 3 ATP-Na2, 0.3 GTPNa2, and 10 P-creatine Na2, pH 7.3. Recordings were performed using Multiclamp 700B and Clampex 9.2 (Molecular Devices, Union City, CA). In voltage-clamp configuration, cells were given a series of voltage steps (duration, 50 ms) from −90 to +30 mV from a holding potential of −70 mV. In current clamp configuration we measured the resting membrane potential, and then a negative holding DC current (in the range: −2 to −15 pA) was applied to bring the membrane potential to approximately −60 mV. This was done to compare neurons under identical conditions during AP firing. To assess the AP firing pattern, in current-clamp configuration, we applied a series of current steps from −20 to +60 pA. Current intensities were modified depending on cell's input resistance. For fluorescent identification of cellular morphology, sulforhodamine 101 (50 μM, molecular weight 606.7; Molecular Probes, Eugene, OR) was added to the patch pipette. Electrical traces were analyzed using Clampfit 9.2 (Molecular Devices). Peak sodium current is defined as maximal transient inward current at any command voltage. Peak potassium current was measured at 40 ms from the onset of the command voltage pulse. Statistical analysis was performed using SigmaStat 2.03 (Systat, San Jose, CA). If data were normally distributed and variances equal, then we used Student's t-test or analysis of variance (ANOVA), followed by a Tukey test for pair-wise comparisons. If data points did not pass these criteria, we used a Mann–Whitney test or Kruskal–Wallis one-way ANOVA on ranks followed by Dunn's method for pair-wise comparisons.
Results
Clustering of DA neurons
The present study comprises 5 consecutive stages (Fig. 1A, stages 1–5). We used a batch of H9 hESCs between their 50th and 65th in vitro passage (Fig. 1B, stage 1). Suspended in the KSOR medium the hESC formed embryoid bodies (EBs, stage 2). Upon plating in the neuron differentiation media, these cells formed characteristic rosettes (stage 3). Stage 3 rosette cultures contained large clusters of cells displaying elongated cell morphology on the inside of colonies, with flattened morphology on the periphery of the colony (Fig. 1B, stage 3). Very little cell death was accounted by observation of live cultures in stage 3. Stage 3 colonies exhibiting elongated morphology were manually selected, lifted, and plated on glass coverslips, where they were allowed to expand first as a neuroepithelium (stage 4), and then as young neurons (stage 5). Rapid expansion during stage 4 indicated significant cell division, but also with many rounded dying cells. A rapid expansion was co-occurring with cellular migration, which resulted in the formation of dense cellular structures on some parts of the coverslip (Fig. 1B, stage 4). Neurites became visible during stage 4 (Fig. 1B). Although, patch-clamp recordings were made through all stages starting from stage 3 to stage 5 (Fig. 8), the majority of electrophysiological measurements were obtained after 21 days of differentiation (stage 5), for comparison with previous studies [5,6,10,11].

Differentiation of dopaminergic (DA) neurons from human embryonic stem cell (hESC).
In an attempt to reduce heterogeneity at the transition point from stage 3 to stage 4, the neuroepithelial rosettes were lifted off the bottom of the well as normal, treated with and without trypsin and trituration, and seeded evenly across the coverslip with and without ROCK inhibitor. ROCK inhibitor greatly reduced cell death caused by trypsin (Fig. 1C, left). Extensive trituration with ROCK inhibitor, but without trypsin, proved to be the optimal treatment. Variability was quantified by measuring the ratio of TH signal to TUJ1 signal after immunofluorescent staining, then calculating CV for each group. CV was calculated as standard deviation/(mean ratio of TH/TUJ1). Heterogeneity with ROCK inhibitor was reduced (CV=0.66) compared to control (CV=0.87).
Immunofluorescence of stage 5 cultures showed positive staining for both TUJ1 and TH (Fig. 2A, B), thus indicating the presence of DA neurons [30]. The differentiation protocol produced both single and clumped neurons, sometimes with tangled clusters of neurites, and clumping of cells in colonies or masses often occurred. Striking heterogeneity was found in the proportion of TH-positive neurons in different regions on the same coverslip (Fig. 2A–C). Neuronal clumps were usually round or oval in shape with diameters in the range between 400 and 800 μm. Neuronal clumps contained several strata of cells one upon another (usually 2–3 layers high), in contrast to space between clumps, which was filled with a monolayer of cells. TH staining revealed DA neurons in monolayer portions of the coverslip (Fig. 2C); however, the greatest density of TH-positive cells was detected inside the neuronal clumps (Fig. 2A). Occasionally, we found clumps void of TH-positive neurons (Fig. 2B), but these empty clumps definitively represent an exception to the rule. Identical differentiation protocols (Fig. 1A) were repeated 3 times and each time a major fraction of DA neurons was associated with medium and small size clumps (Fig. 2A).
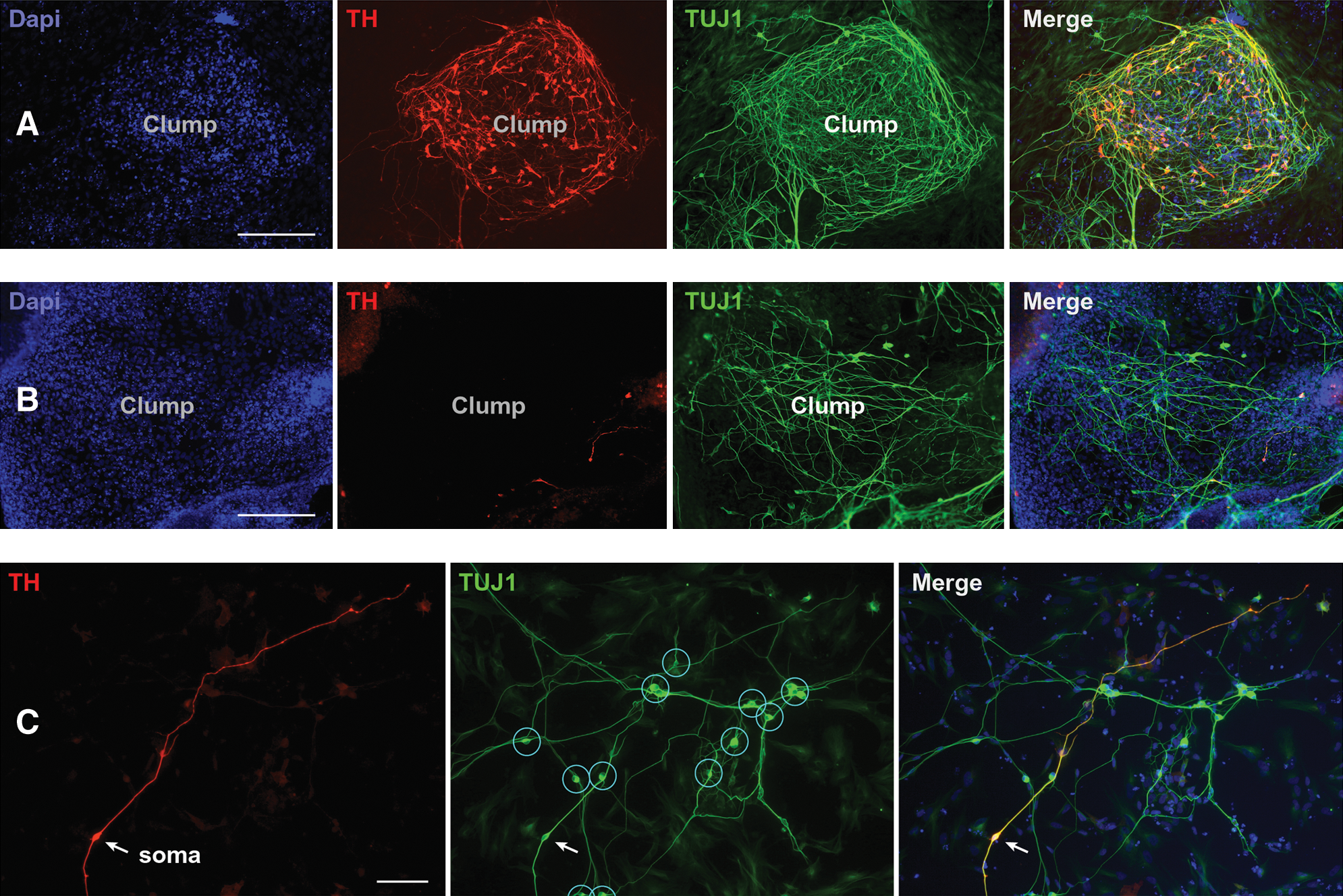
Clustering of hESC-derived DA neurons.
AP firing in hESC-derived neurons
Robust expression of voltage-gated sodium channels that endow the plasmalemma with transient inward current, and the ability of the membrane to generate an AP upon adequate depolarization are 2 major hallmarks of postmitotic human neurons [29]. In human fetus, the insertion of voltage-gated sodium channels into the plasmalemma of developing human neurons increases gradually following the time course of differentiation [29]. At one critical point in human development the density of voltage-gated sodium channels inserted and accumulated in the plasmalemma provides enough sodium conductance for the generation of self-perpetuating (regenerative) sodium current upon adequate stimulus, which is manifested as a stimulus-evoked voltage spike in intracellular recordings. Here we sought to determine the earliest time point when putative stem cell-derived neuron progenitors adopt an irreversible path toward the neuronal lineage. In our view this point is manifested by the first instance of a regenerative sodium spike, dubbed as an abortive AP (Fig. 4B).
There are no previous reports of electrophysiological recordings carried out in cells that constitute neuroepithelial rosettes. In mature rosettes (day 7 of the differentiation protocol, 4 days after the EBs were seeded on a glass coverslip; Fig. 3A1) we patched individual cells characterized with a pear-shaped cell body and a radial orientation, where the apical pole points toward the center of the rosette (Fig. 3A2). Although their voltage-gated potassium currents were quite prominent (Fig. 3E), none of the neuroepithelial cells exhibited any signs of voltage-gated sodium current in voltage clamp measurements (n=11; Fig. 3D), or any signs of regenerative spikes upon strong current stimulus in current clamp recording configuration (not shown). The same batch of neuroepithelial rosettes used for recordings was lifted, triturated, and re-seeded on Geltrex-coated glass coverslips, which allowed us to continue the electrophysiological monitoring of differentiating cells. On the fourth day after re-seeding (protocol day 10 in Fig. 1A), we found plenty of attached cells (Fig. 3B1), and we patched cells that appeared to have neuronal morphology under the infrared video-microscopy (Fig. 3B2). On the differentiation day 10, as many as 9 out of 15 cells had detectable transient inward sodium currents. The average peak sodium current increased significantly from day 10 to 12 (Student's t-test, P=0.013), suggesting rapid dynamics of differentiation in this particular segment of the Iacovitti protocol (Fig. 3D). Interestingly, the outward potassium currents that occur in response to depolarizing voltage steps did not correlate to the increasing age of differentiation. Between the 3 age groups (Fig. 3E), one-way ANOVA failed to detect any significant differences. APs were absent at day 7 (stage 3), but were occasionally found in neuron-like cells during stage 4; on day 10 (Fig. 3F), as well as on day 12. Protoplasmatic processes from putative neurons also first appear in stage 4. The Iacovitti differentiation protocol produced electronically passive cells, neurons with abortive AP, and neurons with single and repetitive APs (Fig. 4). We systematically performed both voltage-clamp and current-clamp measurements from each cell in the data set. This allowed us to compare the amplitudes of transmembrane currents with the voltage waveforms of evoked spikes. In Fig. 4, the sequential voltage clamp and current clamp traces obtained in the same cell are aligned with microphotographs of the actual human cells. The current and voltage traces in the present study were similar to those obtained in human fetal brain slices [29], and the AP waveforms and AP firing patterns are similar to the published data from hESCs used in other differentiation protocols [5,11]. One of the most surprising discoveries of the present study was that the cellular morphology of differentiating stem cell-derived neurons was not predictive of AP firing patterns (Figs. 4 and 5). In spite of the intensive experimental effort to search and locate mature neurons based on their visual appearance (infrared video microscopy), we failed to identify or establish any morphological features that would indicate a postmitotic neuron capable of firing a full-size AP. The shapes of cell bodies and the number of primary processes emerging out of the cell body were not predictive of the AP firing waveform or pattern. In many instances the neuronal processes were intertwined and difficult to follow under infrared microscopy (Fig. 4D). To provide a definite account of whether the number of processes and their branching pattern may correlate with the AP firing pattern, we introduced a fluorescent dye into the patch micropipette. Rhodamine injections eliminated any uncertainty about the neuronal processes (Fig. 5); however, once again the number of primary branches and the number of secondary ramifications did not provide any clues about the physiological maturity of neurons understudy. For example, abortive APs were documented in bipolar, tripolar, and multipolar neurons in the present data set (Fig. 5A–C). In summary, we conclude that heterogeneity in neuronal morphological and physiological properties is developing as early as day 10 and that significant neuronal maturation occurs even before dbcAMP is added to initiate the differentiation stage 5.
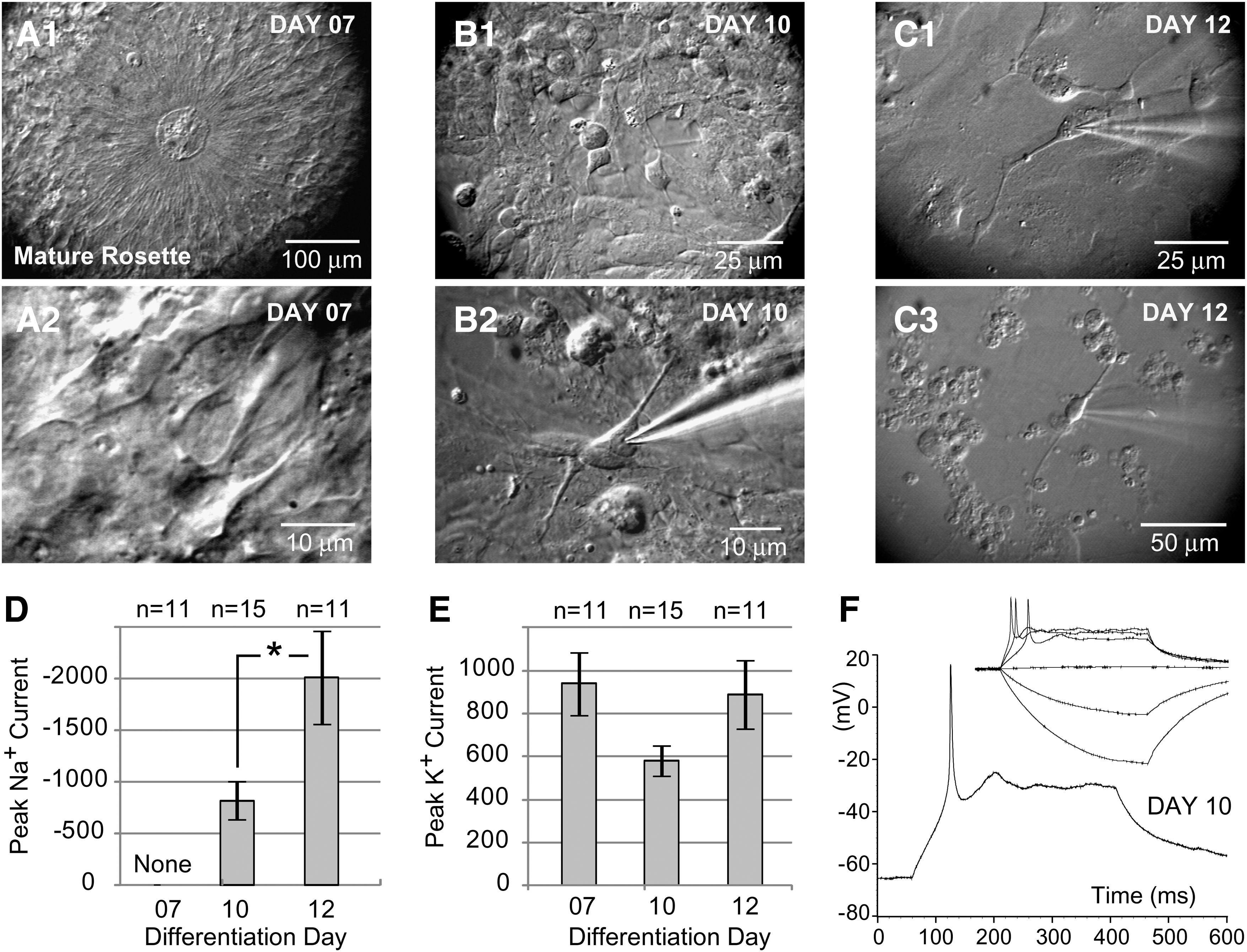
Physiology of the neuroepithelial rosettes.
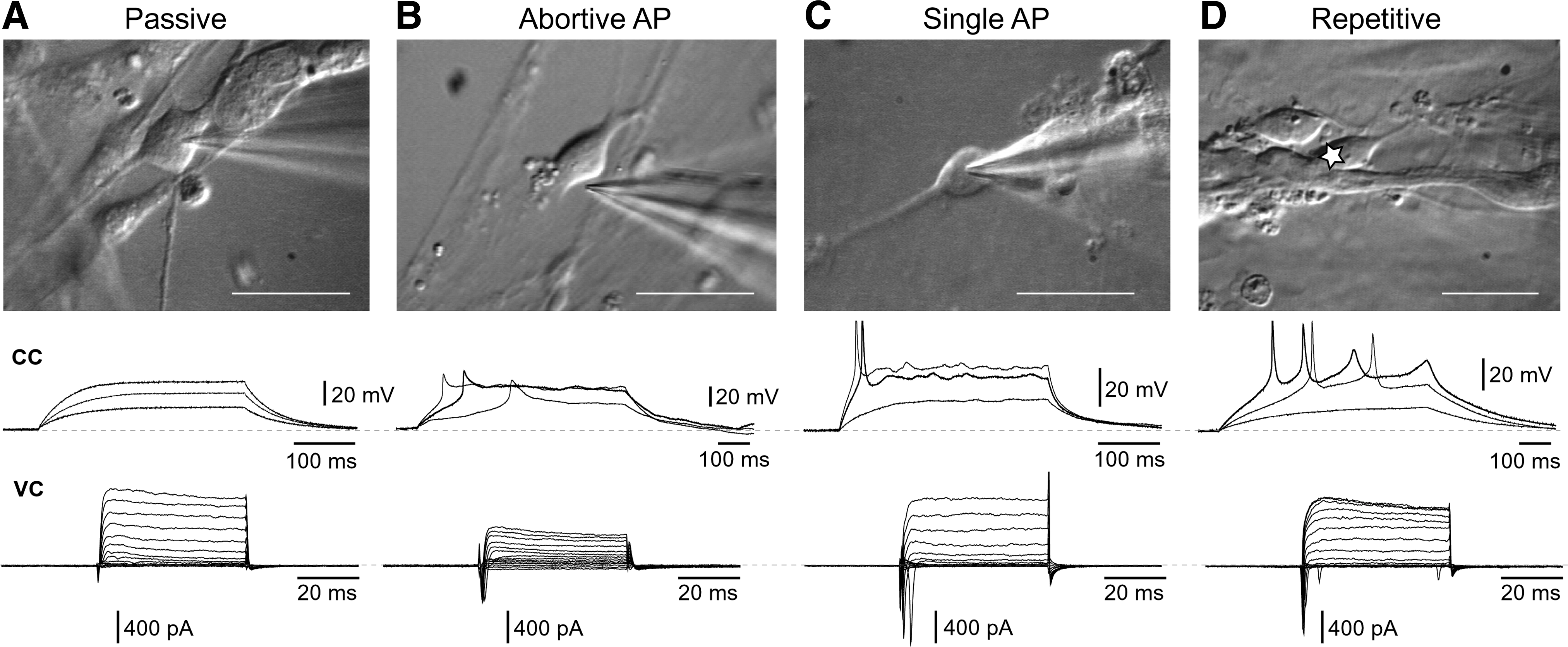
hESC-derived neurons and their AP firing patterns. Each panel
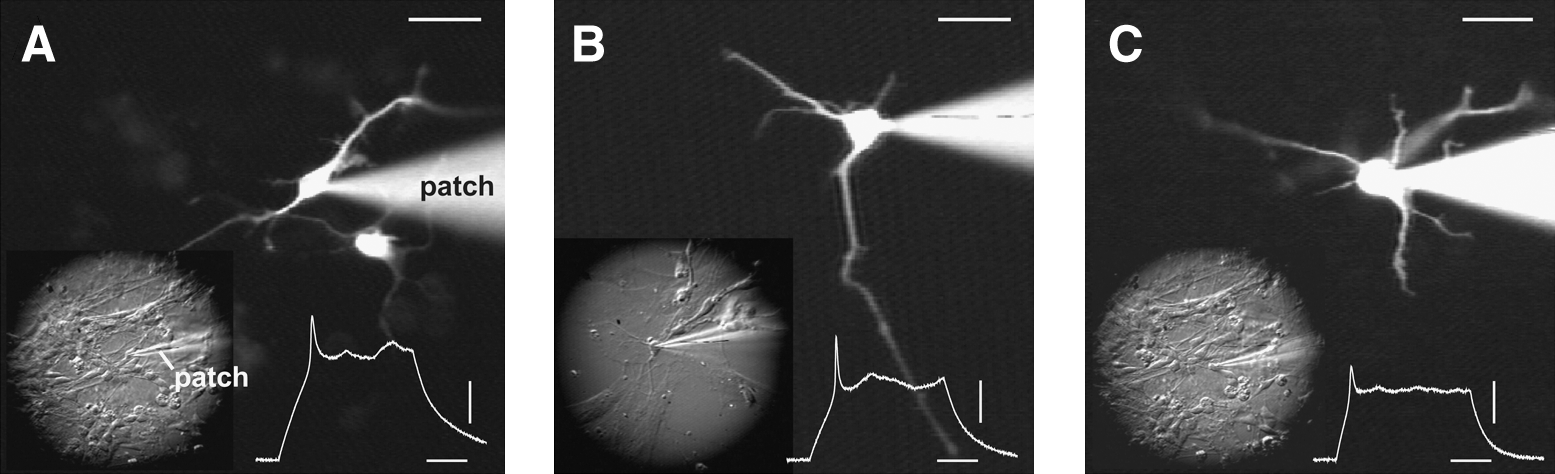
Morphology of hESC-derived neurons.
Late phase of the differentiation protocol
We allowed human stem cells to undergo 21 days of the Iacovitti protocol before engaging in the second wave of systematic electrophysiological measurements. Coverslips were taken out of the incubator between protocol day 21 and 32. Individual cells were patched in a semirandom fashion, with an experimental aim to collect as many samples as possible, before deterioration of the preparation. Typically, a coverslip would last 2–3 h inside the heated (30°C) ACSF-perfused recording chamber before the cultured human cells began to change their appearance. The signs of deterioration included shrinkage or swelling of cell bodies, loss of membrane flexibility, a decrease in transparency, poor light reflection, and detachment from the glass surface. These visual signs were invariably paralleled by poor electrophysiological features. Unhealthy cells were difficult to patch (fragile). They had bad resting membrane potentials (more positive than −20 mV) and did not fire APs upon stimulation. At this point the old coverslips were removed from the recording chamber and replaced with fresh coverslips directly from the incubator.
Summary statistics of basic electrophysiological parameters of 46 cells in this data set are presented in Fig. 6. The mean peak sodium current, peak potassium current, input resistance, and resting membrane potential were −510 pA, 432 pA, 3.2 GΩ, and −43 mV, respectively (Fig. 6A–D, circle inside box). The peak sodium current plot indicates maximal transient inward current amplitude at any command step (see Materials and Methods). The measurements of resting membrane potential (Fig. 6D) refer to values obtained immediately after the whole-cell breakthrough, and before application of negative holding current (see Materials and Methods). To illustrate the high variability seen in all 4 basic parameters, next to the box plot we display a scatter plot of data points, where each data point represents one cell (n=42). Two main conclusions emerged from electrophysiological measurements performed at the end of the differentiation protocol. First, many human neurons were found in immature states (low peak amplitude of sodium and potassium current, high input resistance, and depolarized resting membrane potential). Second, the seemingly identical human neurons exhibited a large variety of physiological parameters. More than often a cell with large sodium current and full-size AP (Fig. 4C) resided next to a cell with zero sodium current and passive membrane response to current injection (Fig. 4A).

Postmitotic hESC-derived neurons in stage 5—basic electrophysiological parameters.
Hyperpolarization activated current
For areas of dispersed cells adherent to the glass (where counting of cells was possible), the percentage of TH-positive neurons (Fig. 7A) ranged from 0% to 15%. The fraction of TH-positive neurons in these areas was 8.3% (Fig. 7B, inset, immunoreactivity), similar to the 6.5% reported by Cai et al. using the same protocol [31]. We searched for electrophysiological parameters that may identify a DA lineage among patched neurons. In current clamp recordings performed in animal preparations, the mature DA neurons display a characteristic “sag” upon injection of a negative current pulse (Fig. 7B, sag). This sag is associated with hyperpolarization activated cationic current Ih [32], which represents an electrophysiological hallmark of DA neurons [33,34]. In the present study, ∼13% of semirandomly patched neurons displayed the hyperpolarization-induced sag (Fig. 7B, inset, patch). Although DA neurons are not the only cell type to exhibit hyperpolarization sags, the electrophysiological account (13%) is very close to the TH-positive cell count (8.3%) obtained using immunoreactions (Fig. 7A). When combined, these 2 results (immunoreactivity and patch clamp) suggest that the majority of Ih-positive cells in the data set were also positive for TH, an obligatory enzyme in the catecholaminergic pathway, and are likely to belong to a DA lineage.
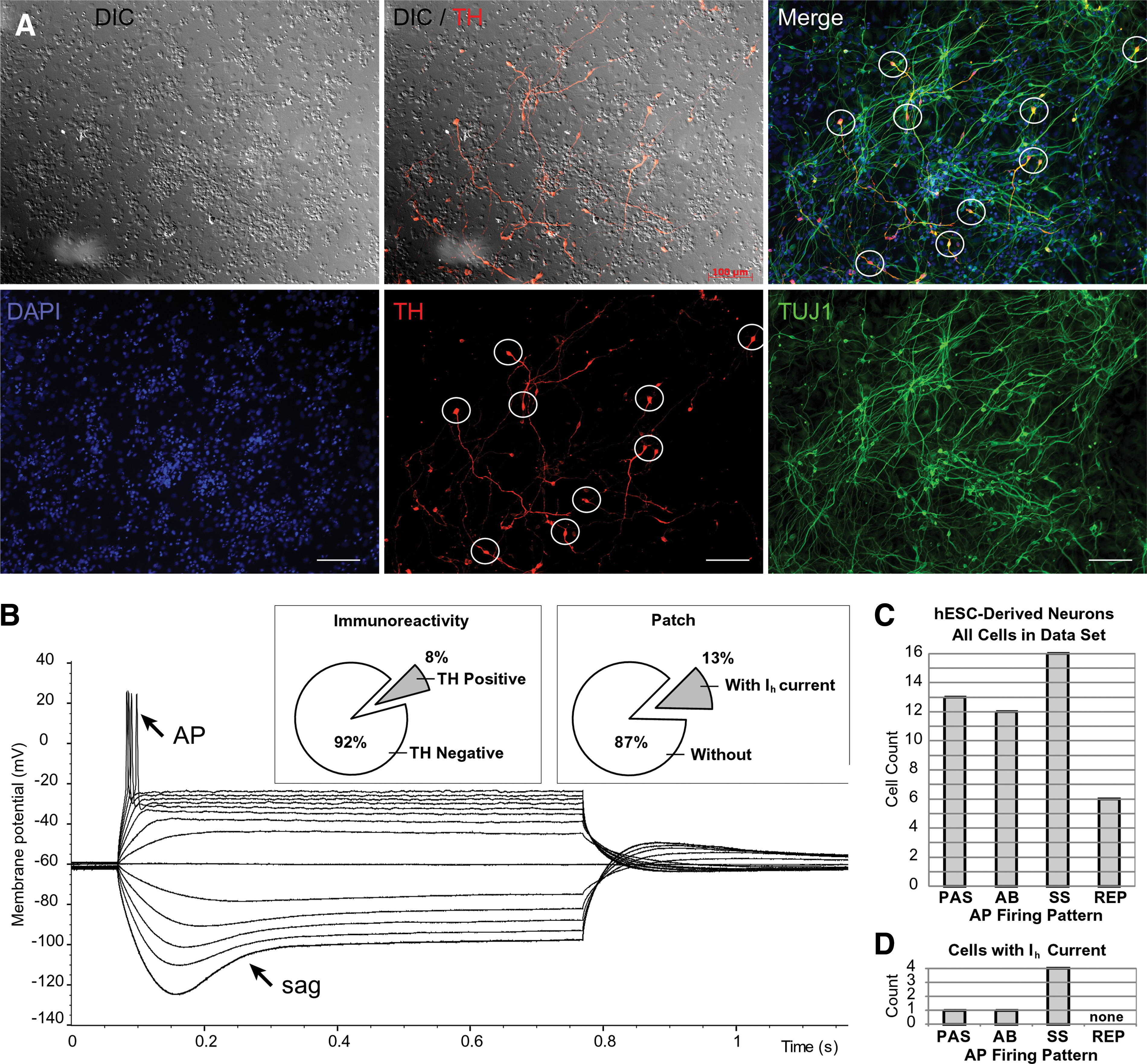
Human ESC-derived neurons in culture.
In animal preparations mature DA neurons fire repetitive APs [33,34]. With this piece of information in mind we proceeded to assess the maturity of hESC-derived Ih-positive cells based on their ability to fire APs. One of the Ih-positive cells was purely passive (Fig. 7D, Passive), one cell showed an abortive AP (Abortive) and 4 Ih-positive cells were able to fire a full-size AP (Fig. 7D, single spike, SS). However, none of the neurons endowed with Ih current were able to generate repetitive AP firing upon depolarizing current injection (Fig. 7D, REP). The distribution of 4 AP-firing patterns (Passive, Abortive AP, SS, and Repetitive) among hESC neurons is essential for putting the Ih data into an accurate context. For this reason we display data from 47 cells (day 12 to 32), and we assign each cell to one of the aforementioned 4 categories (Fig. 7C, PAS, AB, SS, REP). We conclude that at the end of the Iacovitti differentiation protocol (nominal days 21–32) the DA neurons were too young to show repetitive APs, and, interestingly, the ability to sustain Ih seems to precede the ability to fire a full-size AP.
Time profile of physiological maturation
To study the temporal aspect of physiological maturation of hESC-derived neurons, we performed electrophysiological measurements at multiple time points during the course of the protocol (Fig. 8). We focused our attention on 4 basic electrophysiological parameters: the peak sodium and potassium currents (INa+ and IK+ ), effective input resistance (Rin), and resting membrane potential obtained immediately upon whole-cell breakthrough (Vr). These measurements were grouped based on the precise age of cultured cells (particular differentiation day; Fig. 8A). In such orderly temporal arrangement, our data provide a unique description of cell physiological properties in 3 phases of one differentiation protocol: (1) early phase (day 7, 10, and 12); (2) mid-phase (day 21); and (3) late phase (day 25, 26, and 32). A trend of reduced peak potassium current was found during stage 4 (compare day 12 to days 25 and 26), suggesting reduced health of the cultures with increasing time spent in the incubator (under present experimental conditions; see Materials and Methods). One-way ANOVA test performed on peak potassium currents produced P<0.001, so at least one group is significantly different from others. Tukey test (P<0.05) determined that the following pairs were significantly different: day 7 versus 21; day 7 versus 25; day 7 versus 26; day 7 versus 32 (asterisks not shown). More interestingly Tukey test also showed that 2 pairs, day 12 versus 25 and day 12 versus 26, were significantly different (Fig. 8B, Peak K+ Current, asterisks). The resting membrane potentials of animal neurons show gradual hyperpolarization during the course of development [35]. The developing human neurons in the present study showed the opposite trend. At older ages (days 21–32) the mean Vr was generally more positive than in the beginning of differentiation protocol (days 7–12). One-way ANOVA test performed on Vr values produced a P value of 0.002, so at least one group is significantly different from others. Tukey test (P<0.05) determined that the following pairs were significantly different: day 7 versus 10; day 7 versus 21; day 7 versus 26 (Fig. 8B, Resting Potential, asterisks). What could be the reason for time-dependent decay in outward current (IK+), and gradual depolarization of resting membrane potential (Vr), other than the decreasing viability of cultured human neurons? Following initial differentiation (day 12), the cultures of postmitotic human neurons begin to deteriorate. These unexpected findings pose 2 important questions. First, what should be done in the future to sustain the physiological quality of differentiating human neurons beyond 3 weeks (eg, addition of survival factors)? Second, should experimental and clinical applications of hESC neurons be limited to neurons produced in the earliest stages of the current differentiation protocols (around day 12)?
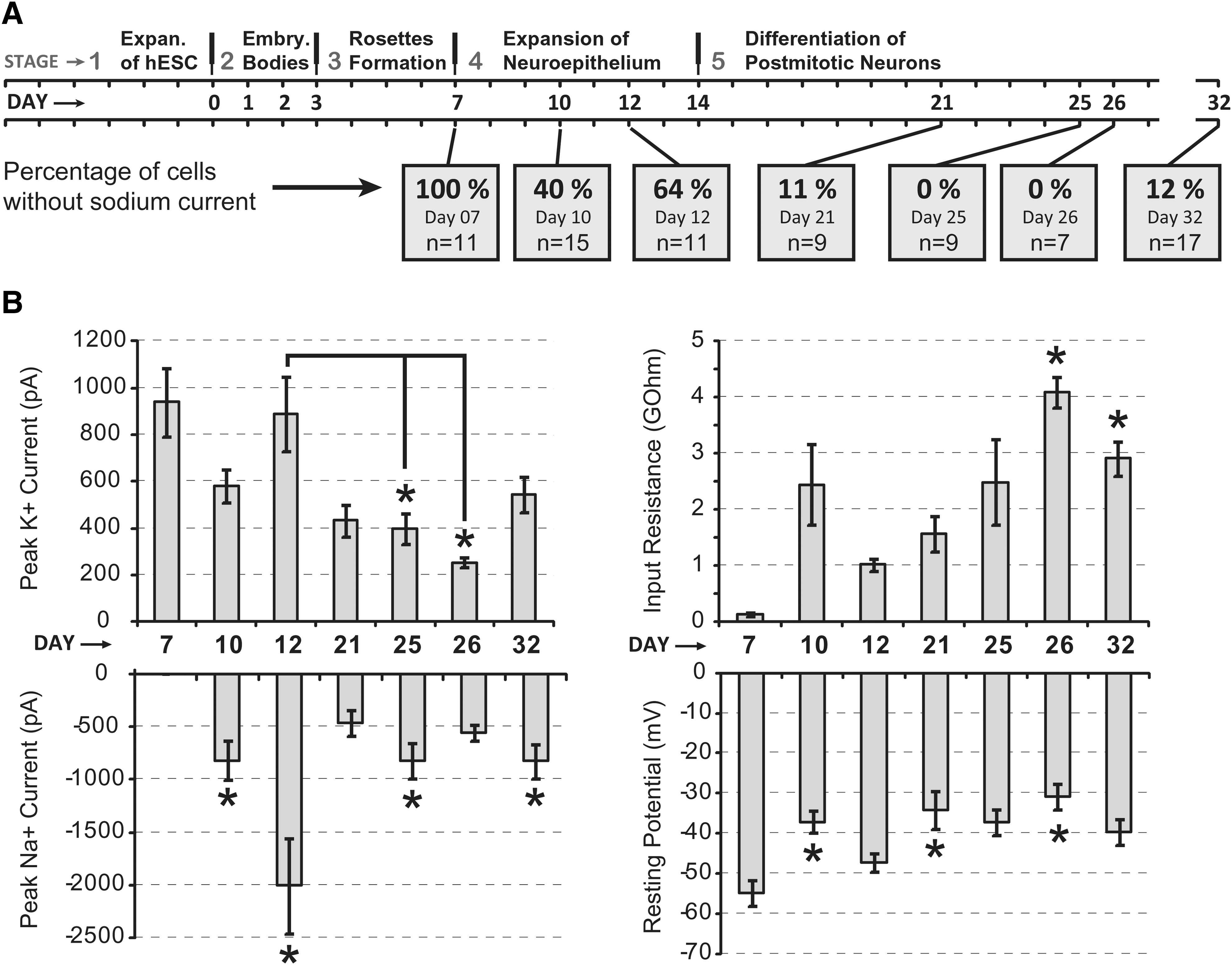
Time course of physiological changes.
The peak sodium current of cultured human neurons showed an expected tendency in the first 3 points (day 7, 10, and 12), but its trend over the entire course of the protocol was not indicative of linearly progressive maturation (Fig. 8B, Peak Na+ Current). It must be noted that the first 3 time points (day 7, 10, and 12) were obtained on the same differentiation trial, which may have been accidentally more successful than other differentiation trials in the present study (ie, day 21, 25, 26, and 32), in spite of the fact that all differentiation trials were performed under identical experimental conditions (same protocol). Kruskal–Wallis one-way ANOVA on Ranks produced P<0.001. However, the only significant differences were detected between day-7 group and the groups marked by asterisks (Fig. 8B, Peak Na+ Current, asterisks). After many days spent in stage 5, the hESC-derived cells showed no improvement in neurophysiological parameters associated with healthy mature neurons: the percentage of cells with sodium current began to decrease (Fig. 8A, bottom), resting membrane potentials declined slightly (Fig. 8B, Resting Potential), and peak potassium currents showed a decreasing trend (Fig. 8B, Peak K+ Current). These data suggest that as far as 32 days into the Iacovitti differentiation protocol (day 32), most of the hESC-derived neurons are not completely mature (Figs. 4 –8), and extending the differentiation protocol after day 12 does not appear to produce more mature neurons.
Discussion
Electrophysiological measurements as a practical tool
Electrophysiological recordings of hESCs differentiated into neurons have been employed in the past; however, previous authors typically show data from 1 or 2 cells [5,11]. Here, we attempt to present data points from all the cells characterized over the course of a differentiation protocol to give a more complete description of some practical problems in hESC research; mainly, the physiological immaturity and heterogeneity of human neuron cultures (Figs. 6 –8). We found excitable neurons at a very early time point, 10 days after formation of EBs (Fig. 1A, day 10), an earlier age than any other report. This may be due to spontaneous differentiation in the maintenance culture; however, great care was taken to remove all differentiated colonies before making EBs. The presence of neurons at an early stage may be due to stochastic variability in the culture. Functional neurons at day 10 may provide a rationale for optimization of shorter differentiation protocols, which will save time and expense. After completion of the differentiation stage of this protocol (day 21), both immature and mature neurons were present in the cultures. Cellular morphology was not predictive of maturity (Fig. 5). We found no benefit for extending the protocol for >5 days into stage 5, as average potassium currents, peak sodium currents, and resting membrane potential did not improve with extended time (Fig. 8).
At any given differentiation day during stages 4 and 5, we found a striking heterogeneity in AP firing pattern among neighboring cells; from purely passive membrane response (Fig. 4A) to repetitive firing of APs (Fig. 4D). The heterogeneity in the AP firing pattern is a result of diverse expression of voltage-gated sodium channels. Unlike immunostaining, or single-cell polymerase chain reaction, the whole-cell patch-clamp recordings provide quantitative estimates of the number of functional voltage-gated sodium channels inserted into the plasmalemma [36]. We observed a strict correlation between the peak amplitude of sodium current and the quality of the AP waveform, as previously shown in human fetus in vitro [29]. For example, cells with zero sodium current have purely passive response (Fig. 4A). Cells with moderate amount of sodium current generate abortive APs (Fig. 4B). Cells with large sodium currents produce full-size APs (Fig. 4C, D). The variability in sodium channel expression levels among cultured human neurons (Figs. 4 and 6A) reflects different differentiations stages. Each member of the neuronal lineage passes through these differentiation stages on an individual basis [29]. The variability in sodium channel expression is unlikely to be caused by long-range extracellular modulatory influences secreted into the medium, since neighboring cells are exposed to the same extracellular environment. Short-range neuromodulation based on a physical proximity or physical contact with glial cells and/or transmitter-secreting neurons (Figs. 3B1–C1 and 4, top panels) cannot be ruled out at this point. Some differentiation protocols are reported to achieve higher fractions of TH-positive cells than the protocol chosen here [5]. The advantages of the Iacovitti protocol used in this study are its shortness and simplicity, in that it is 21 days long, does not require a specific type of stromal feeder cell to induce differentiation, nor does it require a cocktail of added ligands. We also note that the media used contains 50 μM glutamate, which might be predicted to cause excitotoxicity in some neurons, but, on the contrary, glutamate may actually be beneficial for DA neurons [37,38]. The major weaknesses of the current differentiation protocol are (1) the heterogeneity that develops early on (stage 4), (2) the relatively low yield of DA neurons (Figs. 6 and 7), and (3) the decline of physiological parameters in late stages (Fig. 8).
Heterogeneity of hESC-derived neurons
Several factors may influence heterogeneity. The EB (stage 2) comprises several cell lineages [39]; thus, stage 3 includes a selection step where colonies of the correct morphology are chosen. This selection of neuroepithelial colonies at the end of stage 3 is imperfect as morphologically distinct clusters of cells grow in contact with each other, making the physical separation by microdissection difficult. The selection step is hindered by the occasional layering of non-neuroepithelial cells derived from the margins of the spreading EB over the top of neuroepithelia, which form in the center of the colony. During stages 3–5, other ligands such as Noggin may be needed to inhibit the production of nonectodermal and non-DA lineages [24]. However, in the present study we deliberately avoided any amendments to the protocol (including neuron survival factors such as glial cell line-derived neurotrophic factor, GDNF, and brain-derived neurotrophic factor, BDNF) to preserve its simplicity. The lack of survival factors (GDNF and BDNF) may explain a slow decline of physiological parameters (resting membrane potential and peak potassium current) in the late stages of the current protocol (Fig. 8).
In a given well the cellular milieu (Figs. 3B1–C1 and 4, top panels) may contribute to profile of cells developing on the coverslip via the production of soluble factors, matrix, and cell–cell contacts. Therefore, colonies need to be disassociated into cells before seeding to reduce well-to-well heterogeneity. Disassociation by trituration and addition of ROCK inhibitor modestly reduced heterogeneity (Fig. 1C, right). In addition, cells in a monolayer early in stage 4 migrate to form large masses, and this process appears to create the most heterogeneity visible in the live cultures. Further research is needed on the signals inducing migration and migration mechanism during stages 4 and 5. At the end of stage 5, monolayer and large clusters of cells are present, with the highest density of TH-positive found in some clusters (Fig. 2).
Cell clusters
TH-rich cell clusters were observed starting from day 10 (stage 4). It is not clear what constitutes the preference of DA neurons for neuronal clumps. One possible explanation for the formation of such clusters is based on cell migration mechanisms [40]. Upon re-seeding of neuronal rosettes on day 8 the cultures are nominally monolayered (Fig. 1B). After the initial settlement on the surface of the coverslip the re-seeded cells migrate during stages 4–5 and aggregate based on chemotactic and/or contact clues [40,41]. Alternative explanation for the origin of cell clusters is based on an imperfect dissociation of neuroepithelial cells upon lifting of the neuronal rosettes. In spite of agile trituration some large aggregates of neuroepithelial cells remain. They attach to the glass coverslip and provide basis (nucleus) for the growing clusters. Regardless of their exact nature of origin (migratory or nuclear), the presence of clusters among hESC-derived neurons poses one important question. Given the highest density of DA neurons inside clusters (Fig. 2), would it be prudent to develop strategies for increasing the number of cell aggregates in cultures of differentiating neurons, and to select only TH-rich clusters for grafting? One can envision 2 straight forward benefits of such strategies: (1) a greater initial density of grafted DA neurons would results in greater number of surviving DA neurons; (2) clusters may provide a protective environment for grafted DA neurons, again resulting in their better survival inside the host. Note that current grafting protocols implement agile dissociation of DA neurons before grafting [9]. Agile dissociation of grafting materials may be counterproductive.
Hyperpolarization activated current
During current clamp recordings, only 13% of patched cells exhibited a characteristic sag indicative of Ih (Fig. 7B). Data are unavailable from adult human tissue concerning the percentage of TH-positive neurons showing Ih, but in rodent slices, Ih is found in all DA neurons [42,43]. In rodents, a population of non-DA midbrain neurons may also exhibit Ih [44], although it has been reported that false-negative anti-TH immunofluorescence may account for Ih +/TH-negative neurons [45]. Our data (Fig. 7) from differentiated hESC are consistent with the finding in rodents that TH-positive DA neurons are a subset of neurons that show Ih. Several reports describe DA neuron differentiation protocols and show electrophysiology of hESC-derived neurons without any indication of the presence of Ih [5,9,10,46], which might be interpreted as evidence that some cells shown in those reports are not DA. Because all DA neurons are thought to display Ih, this current may be a useful marker for identification of non-DA neurons in differentiated hESC cultures, and used to assess the efficiency of the differentiation protocol along with other parameters such as percentage of TH-negative cells. Therefore, we believe that an adequate number of cells should be assayed using standard electrophysiology methods to determine the percentage of Ih-positive neurons in the population, as done in this report. Although the presence of Ih is not proof that a particular neuron is DA, we expect all dedicated DA neurons to show Ih. Techniques such as immunofluorescence also yield unknown proportions of false-positive and -negative results, so multiple methods to determine the efficiency of a particular differentiation protocol are useful.
Stimulation-induced dopamine release detected by high-pressure liquid chromatography of the culture media is a powerful evidence of DA neurons, but does not yield information on the status of individual neurons. Single-cell fast-scanning cyclic voltammetry or amperometry [47] would be an appropriate addition to detailed evaluation of DA-neuron yields. Ideally, one would like to combine immunocytochemistry (Figs. 2 and 7A), electrophysiology (Figs. 3 –5), and microelectrode-based electrochemistry to assess the quantity and quality of hESC-derived DA neurons. The major deficiency of our present study is lack of a cell-fate reporter to identify DA neurons before recordings [48], or postrecording immunostaining using back-labeling techniques for post-hoc analysis of recorded neurons that expressed markers of DA neurons [49]. We have attempted the latter experimental approach. We were able to inject cells with fixable fluorescent dyes (Fig. 5), but we were not able to keep the cell bodies of patched neurons attached to the coverslip for subsequent immunolabeling reactions. The electrode pull-out maneuver invariably compromised the adhesions between the recorded neuron and Geltrex-coated coverslip. Recorded neurons were either ruptured or detached from the coverslip during the pipette pull-out (outside-out configuration), or these loose cells were irretrievably lost during lifting of coverslips out of the recording chamber.
Neuromelanin pigment is missing
Our data indicate that most of the cells patch-clamped in the present study were immature (Fig. 7C, D). It is unclear whether this is due to an inhibition of differentiation, the death of mature differentiated neurons, or a combination of the 2. It is also unclear, using the criteria of presence of both repetitive AP and Ih, if completely mature DA neurons have ever been created in vitro in this report. Pigmentation is another important characteristic of substantia nigra DA neurons, and a subpopulation of DA neurons containing black pigment are preferentially lost in Parkinson's disease [50]. However, at present, no protocols for producing hESC-derived TH-positive neurons containing neuromelanin have been described. We also did not find TUJ1-positive/TH-positive cells containing melanosomes. One possible explanation for the lack of concurrent repetitive APs, Ih, and neuromelanin is that the present differentiation protocol is inadequate to create mature DA neurons with physiological characteristics of those found in the substantia nigra. Thus, a need to develop a differentiation protocol to produce pigmented and physiologically mature DA neurons remains. Such a protocol could, in theory, be produced by a slight modification of the Iacovitti procedure and used in an in vitro toxicological model for Parkinson's disease. In contrast to stem cell-based research strategies for analyzing mechanisms of disease, the therapeutic use of stem cells, on the other hand, requires immature, yet lineage committed neurons. Immature neurons are preferable material for grafting, because mature neurons experience greater physical damage during removal and injection. Here we have shown that the majority of neurons created in this protocol are physiologically immature, therefore this protocol may be better suited for therapeutic endpoint (grafting) rather than a toxicity model.
Concluding Remarks
• The standard DA differentiation protocol, applied for 2–5 weeks, produces a heterogeneous ensemble of mostly immature neurons (Fig. 8).
• Inside the so-called neuroepithelial rosettes (4 days in differentiation medium), there are no signs of cells dedicated to neuronal lineage (zero sodium current, zero excitability, zero TUJ1 expression; Fig. 3).
• The earliest full-size AP firing (signature of a nerve cell) was observed 4 days after re-seeding the neuroepithelial rosettes (10th day from the beginning of the differentiation protocol, day 10; Fig. 3).
• On the same in vitro day, neighboring neurons can be in very different stages of differentiation, where one cell is only capable of producing an abortive AP (immature), whereas another cell in its immediate vicinity is mature, firing APs, repetitively (Fig. 4).
• DA neurons tend to aggregate into clumps (Fig. 2), which may offer 2 potential benefits. First, those laboratory strategies that promote clustering may also improve the yield of hESC-derived DA neurons. Second, isolation of TH-rich clusters may improve the number of surviving neurons upon grafting into a living organ, because clusters may have encapsulating and protective influences.
• Visual appearance of differentiating neurons, and the number of primary and secondary dendrites, cannot be used to predict the peak sodium current or AP firing properties of cultured neurons.
• Approximately 13% of neurons are endowed with hyperpolarization induced current (Ih), a characteristic of DA neurons; however, no neurons with repetitive APs showed Ih, which is evidence of their physiological immaturity.
• Extending the differentiation protocol after day 12 (in the absence of survival factors such as GDNF and BDNF) does not appear to produce more mature neurons. On the contrary, the quality of samples (neuronal physiological properties) deteriorates in later stages of the protocol.
• The present study demonstrated that electrophysiological measurements yield information concerning the identity and maturity of derived neurons, inaccessible by other methods, and therefore electrophysiological measurements may be useful in optimizing differentiation protocols.
Footnotes
Acknowledgments
Supported by Connecticut Stem Cell Initiative/Connecticut Innovations grant to S.D.A. We are grateful to Jing Cai for introducing the Iacovitti Lab differentiation protocol to us at the SFN meeting. We are grateful to Xue-Jun Li and Zhibo Wang for comments on the article.
Author Disclosure Statement
Glenn S. Belinsky, Anna R. Moore, Shaina M. Short, Matthew T. Rich, and Srdjan D. Antic have no competing financial interests.