
Abstract
Cell therapy represents the most promising alternative strategy for end-stage liver diseases and hepatic progenitors are the best candidates. We have identified a reservoir of immature hepatic precursors within human cord blood, which can derive engraftable bipotent progenitors. We isolated a stem cell subset CD133+/CD34+/OV6low expressing a surface-marker profile consistent with that of fetal liver cells. Upon induction of hepatic commitment by a medium containing cytokines and factors involved in vivo oval-cell activation, a heterogeneous cell population displaying characteristics of functional oval-cell-like bipotent hepatic progenitors was obtained. The cells expressed markers of hepatocytes and cholangiocytes and were highly enriched in OV6, c-Met, c-Kit, and Thy-1. They also displayed liver functional activity as glycogen storage, urea production, albumin secretion, and inducible CyP2B6 activity. When injected into liver-damaged severe-combined immunodeficient mice, induced bipotent hepatic progenitors appropriately engrafted livers of recipient animals, where they formed clusters of human-derived cells expressing human leucocyte antigen-class I, Hep-Par1, and OV6 antigens. Human-specific albumin, alpha-fetoprotein, and cytokeratin 19 were also expressed. In transplanted animals, AST serum levels showed a significative reduction with regard to controls. This human model for in vitro progenitor-cell activation may provide a powerful tool for elucidating the pathways and synergies that regulate this complex process and can represent a valuable source, exploitable for liver cell-based therapies and regenerative medicine.
Introduction
S
Significant ex vivo expansion and the possibility of subculturing hepatic stem cells have been found to only occur within oval cells (OC). These are a heterogeneous population of nonparenchymal cells present in biliary ductules of the normal adult liver, which emerges and proliferates when either massive damage is inflicted on the liver or when regeneration after injury is compromised. The human counterpart to rodent oval-cell activation has been identified in what has been termed “ductular reactions” [6]. The mechanisms underlying OC activation toward a fully hepatic lineage differentiation are yet not completely understood [7], and a reproducible and stable ex vivo culture system is still lacking.
OCs are bipotential: in vitro and in vivo they give rise to both liver and bile ductal epithelial cells and may constitute the liver stem-cell compartment [8 –11]. They express the hematopoietic stem markers, Thy-1, CD34, Flt-3-receptor, and c-Kit; the liver differentiation markers, alpha-fetoprotein (αFP), cytokeratins (CK) 7, 8, 18 and 19; and the oval cell marker OV6. High-level liver repopulation can be achieved with “oval cells,” but this requires substantial modification of the host's liver environment, similar to that required for liver repopulation by mature hepatocytes [12].
In the present study, we used UCB-derived CD133+/CD34+/OV6low cells and forced them to in vitro commitment toward the hepatic lineage. To recreate in vitro the microenvironment of oval-cell activation in vivo, we designed a differentiation medium supplemented with a selected pool of cytokines and growth factors that are acknowledged to be implicated in this process [13]. This modulation of the culture microenvironment allowed us to obtain a population of induced bipotent hepatic progenitors (iBHPs) that had an enhanced number of cells expressing oval markers and displayed hepatocytic functional activity. When transplanted into liver-injured mice, iBHPs showed good engraftment capacity and expressed human hepatic lineage and functional markers.
This human in vitro model, which mimics hepatic progenitor activation, may help elucidate the mechanisms controlling this multifactorial phenomenon and may provide a source of expandable and engraftable hepatic progenitor cells for liver regeneration.
Materials and Methods
Purification of UCB-derived CD133+ cells
Full-term delivery UCB specimens were obtained with informed consent. Mononuclear cells were isolated by Ficoll-Paque. CD133+ cells were then immunoselected using a midi-MACS selection kit (Miltenyi Biotec, Bologna, Italy), following the manufacturer's instructions.
Antigen immunophenotyping
The following monoclonal antibodies (mAbs) were used: fluorescein isothiocyanate (FITC)-conjugated against CD2, CD7, CD10, CD33, CD38, CD43, CD44, CD45, c-Kit, and Thy-1 (all from ImmunoTools GmbH, Friesoythe, Germany), allophycocyanine-conjugated anti-CD34, PE-conjugated anti-CD133 (both from Miltenyi Biotec), and anti-human/rat OV6 (R&D System Inc., Minneapolis, MN). Cells were incubated 30 min at 4°C in the dark with selected mAbs, in presence of FcR blocking reagent (Miltenyi Biotec). For OV6 mAb, a FITC-conjugated secondary antibody was used. After incubation, cells were washed twice with phosphate-buffered saline (PBS), resuspended in 200 μL PBS, and analyzed by flow cytometry using the FACSAria (Becton Dickinson, San Jose, CA) equipped with 3 air-cooled and solid-state lazers (488, 633, and 407-nm). To gate nonspecific fluorescence signals, cells were labeled with isotypic antibodies (ImmunoTools GmbH), whereas dead cells were excluded on the basis of propidium iodide (5 g/mL; Sigma-Aldrich S.r.l., Milan, Italy) fluorescence intensity.
Cell cultures
Ex vivo expansion
Cells (3÷4×104/cm2) were grown in ISCOVE medium supplemented with 25% FBS in the presence of 20 ng/mL of stem cell factor (SCF). Initial seeding: 0.5÷0.6×106 total cells. After expansion, aliquots of cells were kept in culture for hepatic differentiation or were frozen for further investigations.
Hepatic differentiation
To induce hepatic commitment, cells (5÷6×104/cm2) were seeded on gelatine-coated multi-well plates (8-well Lab-Tek II Chamber Slides RS treated) and grown in Eagle's Dulbecco Modified medium containing 10 ng/mL epidermal growth factor (EGF), 20 ng/mL hepatocyte growth factor (HGF), 10 ng/mL fibroblast growth factors (FGFs), 15 ng/mL stem cell factor (SCF), 10 ng/mL leukaemia inhibitory factor (LIF), 10 ng/mL oncostatin (OSH) (final concentration), 5% FBS, and 4% serum substitute (Bit 9500; StemCell Technologies Inc., Paris, France). Fresh medium was replaced weekly.
Reverse transcriptase polymerase chain reaction and quantitative polymerase chain reaction
Messenger RNA was extracted using AMBION Cell-to-DNA II (Applied Biosystems, Monza, Italy) following the manufacturer's instructions. Complementary DNA was amplified at 94°C for 40 s, 56°C for 50 s, and 72°C for 60 s for 35 cycles, after initial denaturation at 94°C for 10 min. Human hepatocytes (hH) (H1500.H15C; XenoTech, LLC, Lenexa, KS) were used as control. The RNA levels were normalized using human actin. Real-time polymerase chain reaction (PCR) was conducted using Sybr Green I Mastermix (Applied Biosystems) with an ABI PRISMTM 7000 Sequence Detection System. Each reaction was run in triplicate and contained 0,5 μl of cDNA template along with 250 nM primers in a final reaction volume of 25 μl. Cycling parameters were 50°C for 2 min, then 95°C for 10 min to activate DNA polymerase, 35 cycles of 95°C for 15 s, and 60°C for 1 min. Melting curves were performed using Dissociation Curves software (Applied Biosystems, Foster City, CA) to ensure only a single product was amplified. As negative controls, tubes were always prepared, in which reverse transcriptase was omitted during the Reverse Transcriptase (RT) reaction. Primers used are listed in Table 1.
PCR, polymerase chain reaction.
RT, reverse transcriptase.
Immunoblotting
Cell lysates (20 μg/lane) were separated in 7%–10% sodium dodecyl sulfate polacrylamide gel, transferred on nitrocellulose membrane (Bio-Rad, Milan, Italy), and incubated with anti-human AFP (FP FP-C3, Santa Cruz Biotechnology Inc., Heidelberg, Germany), anti-human Albumin (ALB) (clone HAS-11), anti-human CK18 (clone Cy 90); anti-human CK19 (clone A 53-B/A2), all from Sigma-Aldrich (Sigma-Aldrich S.r.l.); anti-human HGF receptor/c-Met (clone 95309; R&D Systems, Wiesbaden, Germany); oval cell marker OV6 (R&D System Inc.). Protein concentration was determined with Bradford-assay (BioRad, Richmond, CA), and sodium dodecyl sulfate polyacrylamide gel electrophoresis was carried out according to Laemmli [14]. For immunoblotting analysis of cellular proteins, human hepatocytes (H1500.H15C, XenoTech, LLC) were used as control.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde (PFA), permeabilized with 0.1% Triton X-100 (Sigma-Aldrich S.r.l.) for 10 min at room temperature, and incubated with mAbs in a wet chamber for 30 min in the dark at +4°C. When required, a secondary antibody was used. Each step was followed by 3-fold washing with PBS + 1% bovine serum albumin. mAbs against human αFP (clone C3), CK19 (clone A 53-B/A2), human ALB (clone HAS-11), CK18 (clone Cy 90); all from Sigma (Saint Louis, MO); oval cell marker OV-6 (MAB2020; R&D System), Thy1 (HIS51, Santa Cruz Biotechnology Inc.), human HGF receptor c-Met (clone 95309; R&D System); stem cell receptor c-Kit (clone 104 D2, Santa Cruz Biotechnology Inc.) were used. For double immunofluorescence (IF) staining, the following additional mAbs were used: goat antihuman CK18 (H-80 rabbit polyclonal) and rabbit anti-human CK19 (C-17 goat polyclonal) both from Santa Cruz (Santa Cruz Biotechnology Inc.). Cells were quantified by counting positive cells in 5 sequential fields of view per sample, at 20× magnification. Average cell numbers were then pooled for each experimental group, and the overall mean and standard error were determined. To avoid any possible fluorescent bleed through or spectral overlap between fluorochromes, appropriate filters were used, namely U-MWIG, BP520-550/BA580IF for rhodamine and U-MNIBA, BP470-490/BA515-550 for FITC. Controls on uninduced cells were always performed. To gate nonspecific fluorescence staining, cells were also incubated with isotypic or secondary antibodies alone.
All antibodies used for immunoblotting and IF are listed in Table 2.
IHC, immunohistochemistry; IF, immunofluorescence.
Functional activity
Low density lipoprotein uptake
The Dil-Ac-LDL assay (Biomedical Technologies, Stoughton, MA) was performed following the manufacturer's instructions. Briefly, complete growth medium containing Dil-Ac-LDL 10 μg/mL final concentration was added to live cells and incubated for 4 h at 37°C. Medium was removed, cells were washed several times and then visualized under fluorescence microscopy.
Pentoxyresorufin O-dealkylase assay
Cytochrome P450 2B fluorescence Detection Kit (Sigma-Aldrich S.r.l.) was used. At week 2, 3, and 5 postinduction, cells were maintained under the same conditions in the presence or absence of 1 mmol/Phenobarbital (PB) for 72 h. Cytochrome P450 activity was assessed by treatment with 10 mol/L pentoxyresorufin and detected under fluorescence microscope.
Periodic acid-schiff stain for glycogen
Cells were fixed in 4% PFA, permeabilized with 0.1% Triton X-100 for 10 min at room temperature, and then oxidized with 1% periodic acid for 5 min. After 3 successive washings with dH2O, cells were treated for 15 min with Schiff reagent, rinsed in dH2O for 10–15 min, stained with Mayer's hematoxilyn for 1 min, and assessed under a light microscope. All reagents were from Sigma-Aldrich (Sigma-Aldrich S.r.l.).
Urea assay
Urea concentration in the medium was determined by Urea/Bun Kinetic UV Assay Kit (Roche Diagnostic GmbH, Mannheim, Germany) following the manufacturer's instructions.
ALB assay
ALB secreted into the medium was determined by the Albumin Bromocresol Green colorimetric assay (ALB Plus; Roche Diagnostic GmbH), based on the ability of serum ALB to produce a color change from yellow-green to green-blue in the presence of Bromocresol Green at pH 4.1.
Transplantation into severe-combined immunodeficient mice
All animals received human care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” (NIH publication 86–23 revised 1985). Male severe-combined immunodeficient (SCID) mice 6–8 weeks old were used. All animals were anesthetized with ketamine (Ratiopharm, Ulm, Germany)—Domitor (Bayer, Leverkusen, Germany) mixture. To induce liver damage, mice were intraperitoneally treated with a sub-lethal dose of N-acetylparaminophen (150 mg/kg) 6 h before cells transplantation. Liver-injured mice were randomly divided in 2 groups, one group (4 mice) was used for transplantation and the another group (4 mice) served as control. One-week-induced cells (2.0×105) [15,16] were intravenously tail injected in the first group of mice. The cell suspension was kept on ice and heated to 37°C immediately before transplantation. After 21 days, mice were sacrificed, and the peritoneal cavity was immediately opened. Each animal's left liver lobe was removed and longitudinally separated into 2 pieces, one piece was snap-frozen and stored in liquid nitrogen until using and the other one was formalin fixed and paraffin embedded: the specimens were fixed in 4% PFA overnight and stored in 70% ethanol. Tissues were embedded in paraffin. Specimen no. 1 was used for IF staining on fresh frozen tissue and for RT-PCR analysis, specimen no. 2 was used for classical immunostaining.
Tissue immunohistochemistry and IF
Immunohistochemistry
Formalin–fixed, paraffin-embedded tissues were cut into 5-μm sections, deparaffinized with xylene, and rehydrated with decreasing alcohol series. For antigen retrieval, the sections were microwave treated in 1 mM EDTA at pH 8 for 10 min and allowed to cool for 20 min. The sections were then added with PBS containing 0.5 g/L Triton×100 and incubated at room temperature for 5 min. Endogenous peroxidase activity was blocked with 3% H2O2 for 3 min at room temperature. Endogenous biotin was saturated using a biotin blocking kit (Vector Laboratories, Burlingame, CA). For detection of human cells in mouse parenchyma, mouse anti-human human leucocyte antigen (HLA) mAb specific for all HLA class I molecules (Sigma, Saint Quentin Fallavier, France) was applied to slides overnight at 4°C. Horseradish peroxidase-coupled (HRP) goat anti-mouse IgG (Amersham, Les Ulis, France) was incubated for 30 min and visualized with diaminobenzidine. Finally, slides were counterstained with hematoxylin. Omission of the primary antibody or substitution with irrelevant mouse or goat IgG served as negative control. Mayer's hemalun solution (Merck, Darmstadt, Germany) was used for nuclear counterstaining.
Immunofluorescence
Fresh frozen liver tissues were subjected to 4–8 μm frozen sectioning. The sections were placed at RT for 30 min, fixed at 4°C in cold acetone for 10 min, and stored in liquid nitrogen. For IF staining, fresh frozen slides were thawed at room temperature for 5 min, washed 3-fold with PBS, and incubated for 30 min in 10% horse serum in PBS to suppress nonspecific binding of IgG. The sections were then incubated with mouse anti-OV6 mAb (R&D System Inc.) and with mouse anti-human-specific hepatocyte antigen, HepPar1 (Dako Italia S.p.A., Milan, Italy) and kept at 4°C overnight. After washing, a second blocking was performed, then the labeled secondary antibodies were added: FITC-conjugated or texas red-conjugated goat anti-mouse IgG (both from Santa Cruz Biotechnology Inc.) and incubated at room temperature in dark for 45 min. The sections were mounted with buffering glycerol 8:1 in PBS and observed under fluorescence microscopy. For negative control, the primary antibody was replaced by PBS.
RT-PCR analysis on liver tissues
To analyze the expression of hepatocyte functional markers in human-derived cells, RNA from liver of controls and transplanted animals was extracted and retro-transcribed according to standard protocols. Human-specific primers for ALB, αFP, and CK19 were used. Data were normalized to the expression of mouse 18S rRNA.
Results
Purified CD133+/CD34+/OV6low cells display characteristics of human hepatic progenitors and have high expansion potential
Sorted cells were 98.6%±0.43% CD133+/CD34+ (n=7). A minor cell population (0.8%±0.05%) was also positive for the oval antigen marker OV6 (n=5) (Fig. 1A). Phenotypic characterization after in vitro expansion (Table 3) showed a surface-marker profile that was consistent with that reported for fetal liver cells [17,18]. After 2 weeks expansion, the presence of CD133+ cells still accounted for 10%–15% of the total, whereas CD34+ cells had declined much more markedly. A slight increase (from 0.8% to 1.3%) of OV6+ cells was observed. Cells to be differentiated were taken 8–10 days after initial seeding, when CD133+ still accounted for 22%–25% (Fig. 1B) and the proliferative potential was still high (doubling time <24h). CD133+ cells are a very small fraction of mononucleated cells present in human UCB (0.14%±0.4%). Cell growth and expansion in culture starts after 2–3 days from initial seeding and shows a maximum of proliferative potential between day 5 and 7. Taking the cells to be differentiated 8–10 days after initial seeding was a suitable compromise: it allowed for higher cell number and a good proliferative potential. The cells were taken 48 h from passage, when they were in exponential growth.
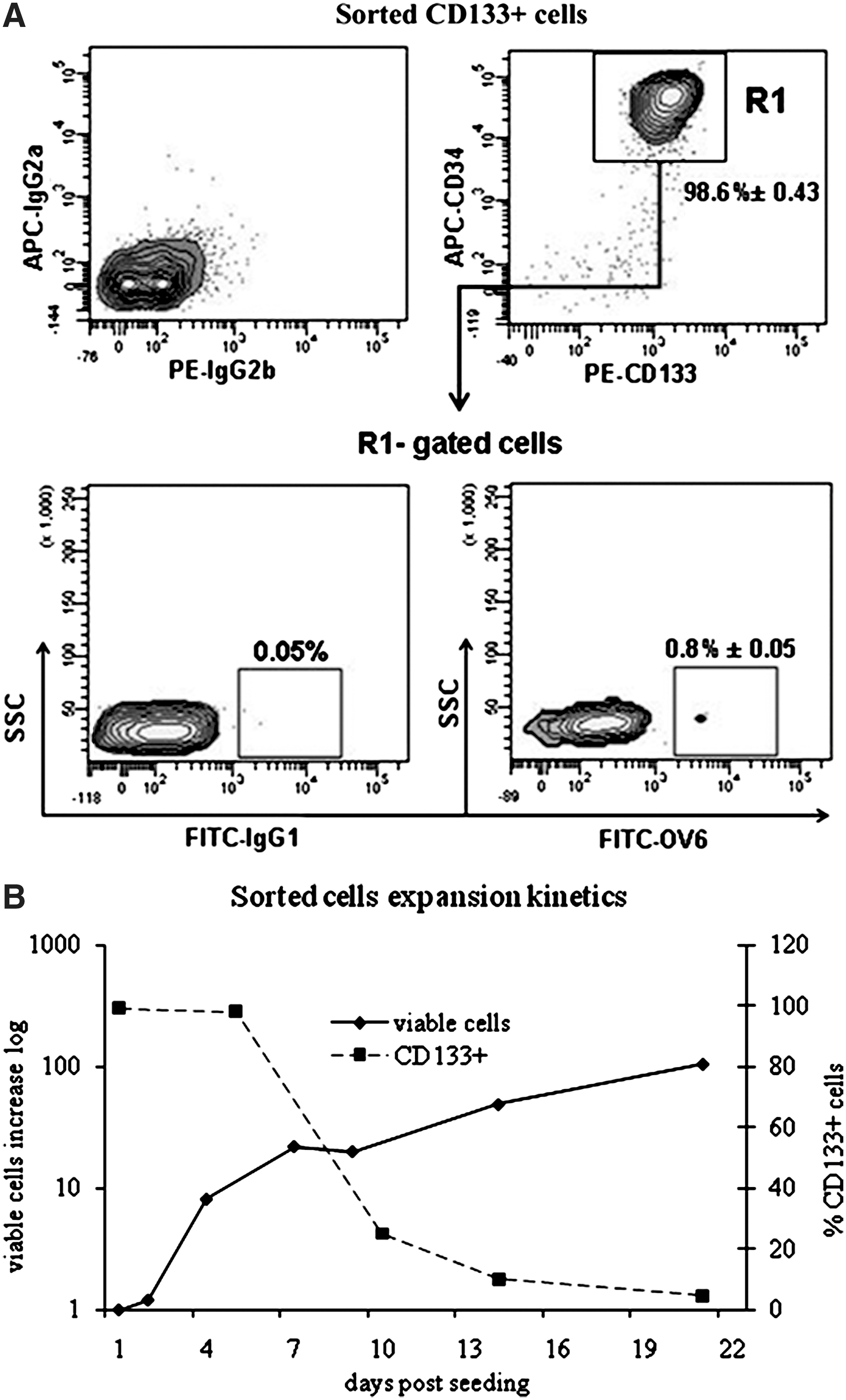
Morphological changes and hepatic lineage markers of differentiating cells
Differentiating cells undergo morphological changes
In the presence of hepatogenic medium, cells rapidly adhered to substrata and underwent changes in both morphology and size (Fig. 2A). After 1 week, a large proportion of the cells had acquired a small round birifrangent oval-shaped morphology or oval-flattened shape, which strictly resembled those described for hepatic progenitors in the human fetal liver [10]. These cells then evolved into an oval-like heterogeneous population. Morphological heterogeneity was maintained even after prolonged culture (2 months, data not shown), albeit a majority of cells displayed characteristics of hepatic progenitor cells.
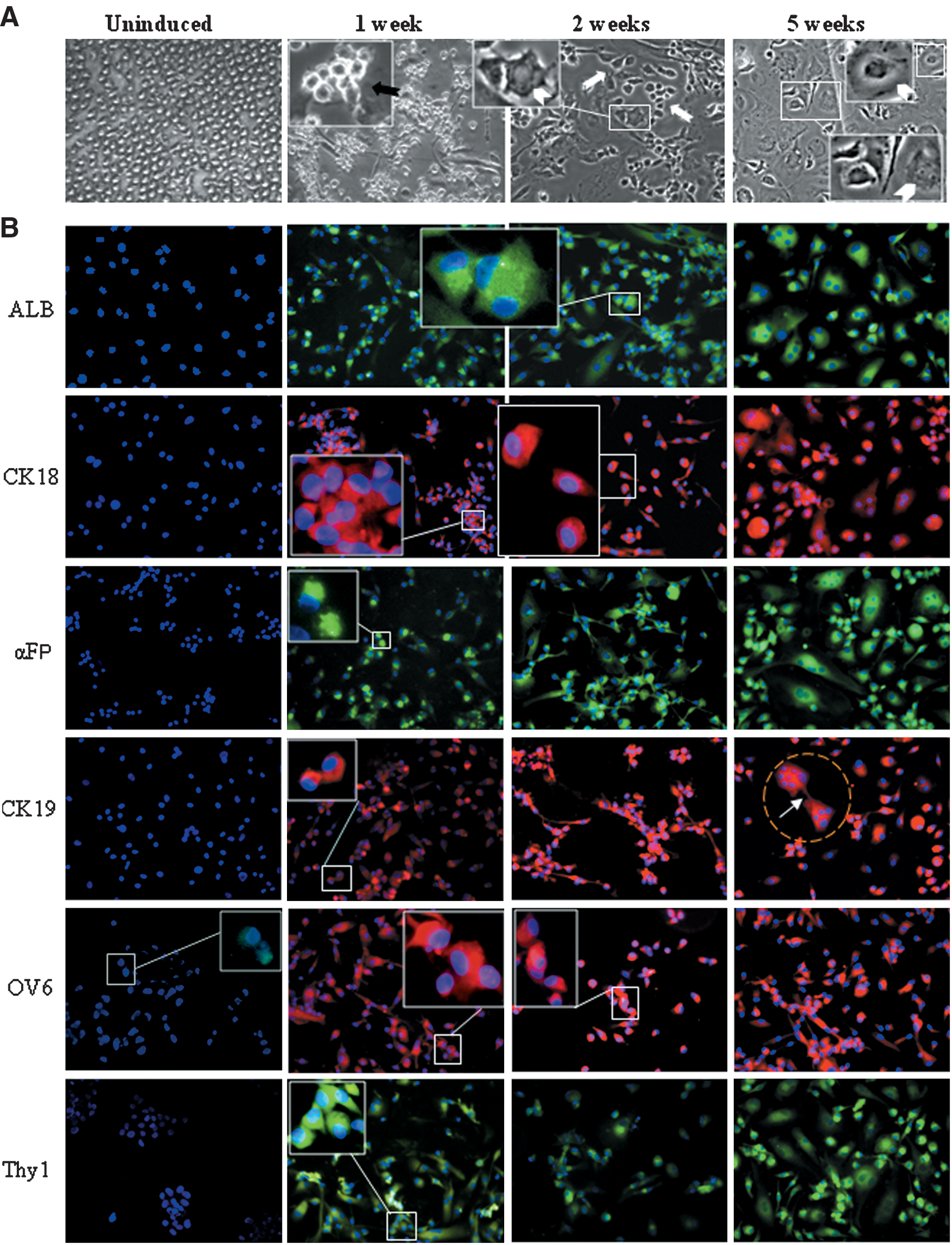
Differentiating cells undergo morphological changes and express hepatic markers.
Differentiation toward hepatic lineage
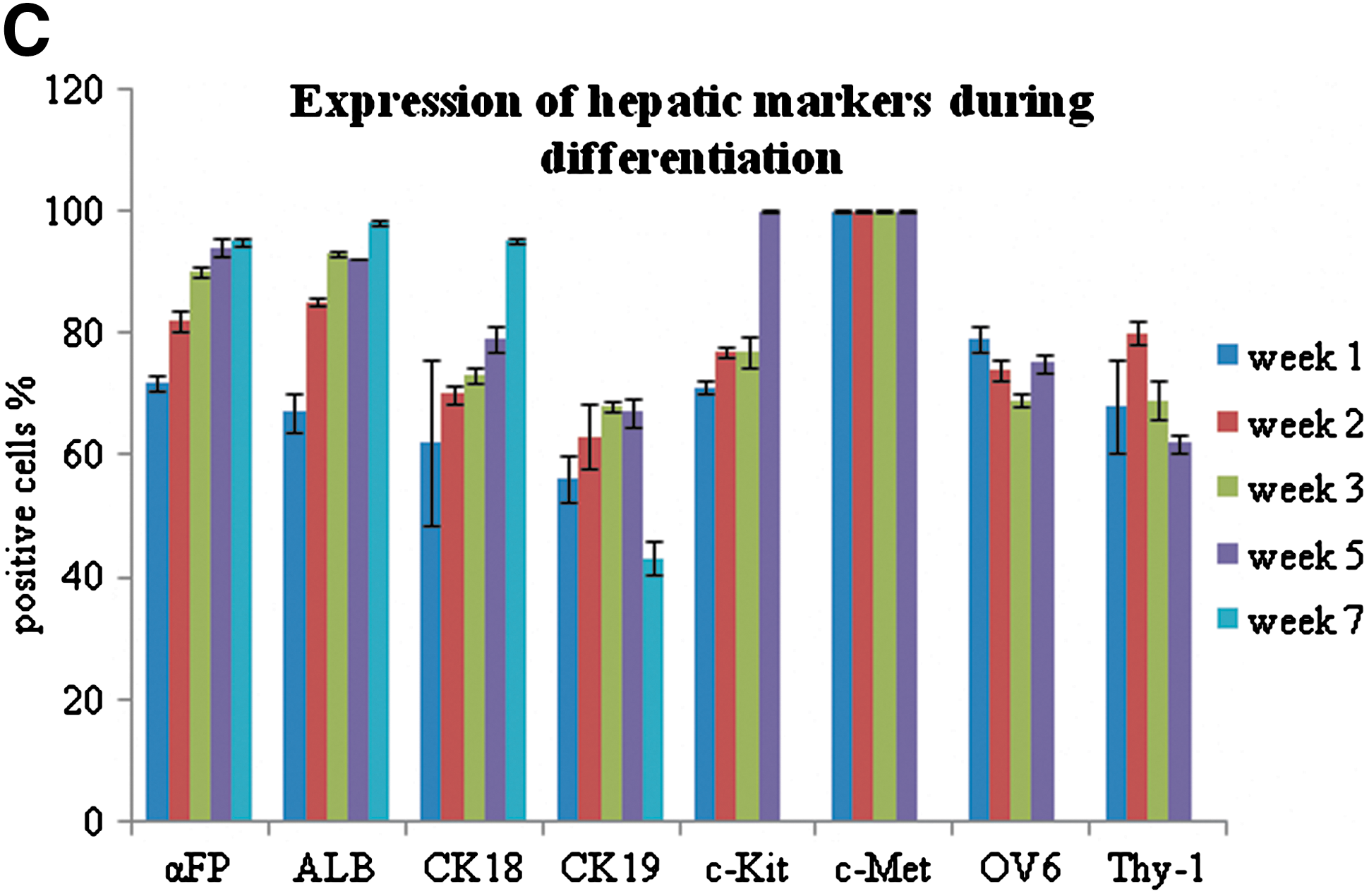
Early (αFP, CK19, OV6) and late (ALB, CK18) markers of the hepatic lineage were analyzed (Fig. 2B, C). After induction, a rapid onset of cells expressing hepatic markers was observed. At week 1, >50% of cells had stained positive for hepatic markers. By week 2, >80% of cells were ALB and αFP positive, and roughly 70% stained positive for CK18 and 60% for CK19, with a slight excess of the former. The difference in percentage of cells expressing these 2 CKs increased in later times and was particularly emphasized at week 7, when CK19+ cells had decreased to 43%, whereas CK18+ had reached to 95%.
Of note, after only 1 week of induction, roughly 80% of cells stained positive for OV6, and this range of positivity was maintained throughout the induction treatment. As just stated, at the beginning of induction, the OV6+ cells accounted for about 1% of the total. After 48 h of induction, about between 8% and 10% of the cells expressed OV6 (data not shown) and at 1 week the vast majority was OV6+. Similarly, Thy-1, which was only expressed in 0.1% of cells before induction, reached 70% positivity after 1 week and co-expressed with OV6 (data not shown). About 70%–80% of cells stained for c-Kit between 1 and 3 weeks and 100% as c-Met at 5 weeks.
At week 1, the cells expressing OV6, Thy-1, and the other markers involved in ductular reaction show a higher increase with regard to the cells that express more specific hepatocytic and ductal linage markers, ALB, CK18, and CK19. This suggests that the activation of hepatic progenitors is the first response to the hepatogenic stimulus and is rapidly achieved after induction, whereas the definitive commitment toward the mature hepatic cells, hepatocytes, or cholangicytes, needs longer time (Fig. 2C).
Hepatic proteins and liver genes
Hepatic proteins expression
(Fig. 3A). At early times, intracellular αFP and CK19 reacted faintly, reflecting the scant production of these 2 proteins at the initial differentiation stage [13,19]. Surprisingly, OV6 protein, in spite of the high cell positivity showed with IF, was not detectable at week 1. We see only 2 possible explanations for such a discrepancy: it could be due either to reduced sensitivity of the antibody in WB or to maturation process and/or conformational changes of the protein during differentiation, thus affecting its exposure and detection. Further, OV6 antigen has been demonstrated to share common epitopes with CK14 and CK19 [20]. Thus, the observed variability of OV6 band pattern could result from the detection of both CK14 (56 kDa) and CK19 (44 kDa) by the antibodies directed against OC antigen 6. At 2 weeks, it seems that CK14 is predominant, whereas at 5 weeks CK19 is predominant, in agreement with what was found by Haruna and co-workers for hepatic progenitor cells in developing human liver [8].

Differentiation commitment.
RT-PCR and quantitative PCR reveal increasing expression of liver genes during induction
ALB, αFP, and CK18 mRNAs were detectable from week 1 onward, whereas hepatocyte nuclear factor 4, a marker of mature hepatocytes, was only detected later (Fig. 3B). The quantitative analyses showed a 3.62- and a 4.4-log increase in ALB and CK18, respectively (hepatocytic markers), and >2.5-log increase in CK19 and αFP (biliary markers), after 5 weeks of hepatogenic culture (Fig. 3C). This demonstrates the persistent expression of markers in both compartments during induction and differentiation. All markers showed parallel slopes.
Oval markers and functional activity of induced cells
Differentiating cells are bipotential and express oval markers
To ascertain whether hepatocytic and cholangiocytic markers were co-expressed in the same cell, double-staining IF was performed. After 3 weeks, the majority of cells maintained their bipotential and co-expressed markers for hepatocytic and biliary lineages (Fig. 4A), whereas the presence of cells ALB+/CK19- and CK18+/ αFP suggested a minor subset definitely committed toward hepatocytic lineage. The oval markers, OV6, αFP, CK19, and the hematopoietic markers involved in mobilization and oval-cell activation in rodents, c-Kit, c-Met, and Thy-1, were always co-expressed (Fig. 4B). Taken together, these results strongly support the hypothesis of a population of oval-like iBHPs.

Co-expression of hepatic lineage and ductular reaction markers.
Induced cells display functional activity
Differentiation efficiency was also evaluated at the functional level. After 2 weeks of hepatogenic induction, differentiating cells were able to incorporate acetylated low density lipoprotein (Fig. 5A), and cytochrome P450 activity was detectable, with a slight increase in presence of PB (Fig. 5B). Maximum cytochrome P450 activity was observed at week 3. Glycogen production and storage, as determined by periodic acid-Schiff staining, was evident in induced cells from week 2 (Fig. 5C).
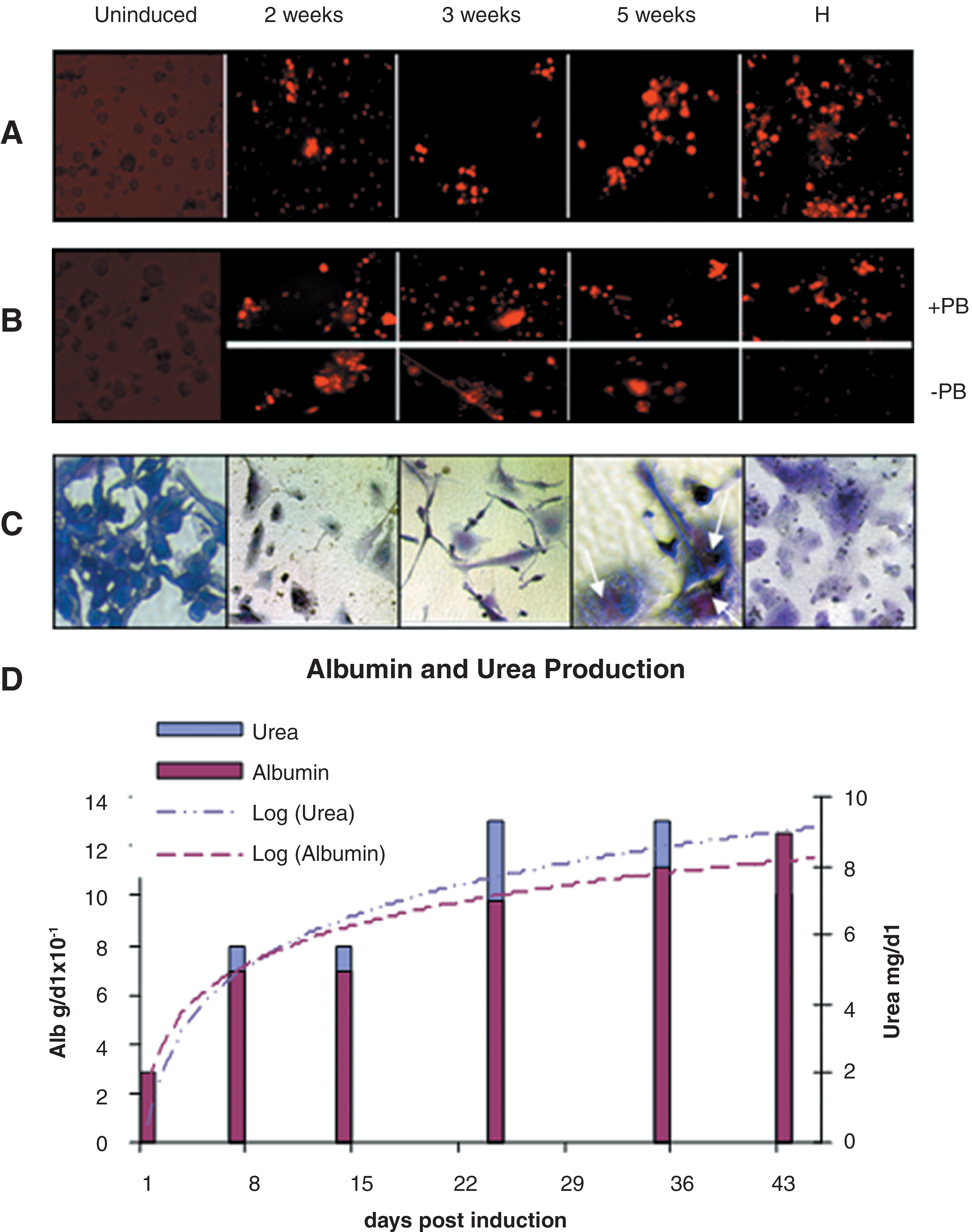
Functional characterization of differentiating cells.
Colorimetric assays for ALB and urea confirmed their production, release, and accumulation in the medium in a time-dependent manner after induction (Fig. 5D).
NOTE: identical results on cell induction, differentiation and hepatic functionality were obtained using cryopreserved cells (data not shown).
Liver engraftment of iBHPs in SCID mice
iBHPs are able to engraft injured liver in SCID mice
To investigate the liver engraftment capacity of iBHPs, 1 week-induced early activated cells were injected into SCID mice (n=4) that had N-acetyl-paraminophen-induced acute liver injury [21,22]. One animal died 10 days after transplantation. After 21 days, the 3 remaining mice were sacrificed. Examination of human-derived cells within recipients' livers by immunohistochemistry (IHC) showed several clusters of human HLA-class I antigen-positive cells localized subendothelially in the periportal areas. No human cells were observed within livers of the control group (Fig. 6A–D). IF on fresh-frozen tissue sections confirmed the IHC data and showed several clusters of cells that were highly positive for the HepPar1 hepatocyte antigen and for oval marker OV6 around the bile ducts and along the bile canaliculi in transplanted animals (Fig. 6E–O), whereas no positivity was detectable in controls (Fig. 6P, Q).

Human-derived cells in the liver parenchyma of liver-injured severe-combined immunodeficient mice injected with iBHPs.
Engrafted cells express human hepatic genes
RNA was extracted from livers of recipient and control group and assayed for RT-PCR. Transcripts for human ALB, αFP, and CK19 were present in all SCID-injured mice transplanted with iBHPs. No transcript for human proteins was present in the control group (Fig. 7).

Expression of human hepatic proteins in iBHPs engrafted livers. RNA was extracted from livers of injured animals, and the expression of human-specific ALB, αFP, and CK19 was analyzed. Amplification products for ALB and αFP were present in all transplanted mice (#7, #8, #9), CK19 transcripts were present in mice #7 and #8; only in traces in mouse #9. No transcripts were present in control mice (Ctrl).
Mice serum hepatic enzymes were also analyzed. A better recovery of hepatic functionality was observed in iBHPs injected animals, in particular, AST serum levels were significantly decreased compared with controls (p<0.05) (Table 4).
P<0.005.
iBHP, induced bipotent hepatic progenitors.
Discussion
Successful isolation and culture of hepatic progenitor cells opens up new perspectives for cell-based therapies and other biotechnological applications [23]. In our study, we demonstrate that UCB-derived CD133+/CD34+/OV6low cells are inducible in vitro to differentiate into iBHPs that express genes and proteins of hepatic and biliary markers, ALB, CK18, αFP, CK19, and the oval markers OV6, c-Met c-Kit, and Thy-1. In humans, hepatic progenitors similar to rodent OCs are postulated to represent the adult liver stem compartment, but a well-defined phenotype still remains elusive. The search for isolation and characterization of human liver progenitor cells (LPCs) is still the subject of intensive investigations [12,24,25]. The marker currently regarded as the best, OV6, seems to recognize epitopes on CKs and cannot be used for immunoisolation [26]. Despite extensive studies, the origin of LPCs remains controversial; both hematopoietic and hepatic sources have been hypothesized. Corcelle et al. [27] identified 2 distinct cell populations: one of hematopoietic origin that expresses OV6/CK19/Rab3b and is activated in the periportal region; the other, which resides in the liver, expresses the hematopoietic stem cell (HSC) markers c-Kit, but not Thy-1. Only this latter population was able to proliferate up to 7 weeks and to form clusters that expressed c-Kit, Thy-1, and ALB. We purified from cord blood a stem-cell subset that contained a small fraction of OV6+ cells. This finding raises the question of whether CD133+ cells “include” an LPC reservoir and may represent an expandable source of hepatic progenitors. This hypothesis is supported by the presence of several oval-shaped cells, by the rapid increase of OV6-positive cells early after hepatic induction (about 100-fold in 1 week) (Fig. 2; Table 3), and by their persistence throughout the differentiation treatment. In the human liver, the OV6 antibody recognizes cells with a progenitor-like phenotype with the capacity to differentiate into OV6-positive ductal cells or lobular hepatocytes [28]. iBHPs constantly express high levels of c-Kit and c-Met that are well established and generally acknowledged markers involved in OC proliferation [29]. The cell population characteristics were similar to those observed by Corcelle et al. for liver-residing OC [27]. Thy-1, whose positivity supports a relationship with OC activation [11], very scantily expressed in mononuclear cells and in sorted cells (0.1%), rapidly increased on induction and co-expressed with OV6. Within OV6+/c-Kit+/c-Met+ cell population, the presence of mature hepatic differentiation markers at later times, that is, ALB, hepatocyte nuclear factor 4, glucose-6-phosphatase, and of the ductal progenitor markers CK19 and αFP, characterizes the maintenance of a “bipolar” nature within these cells.
Progenitor cells isolated from human livers and from canals of Hering are c-Kit positive, suggesting that the c-Kit/SCF system play a role in mobilization and activation of OCs [30]. The c-Met/HGF pathway is important for DNA synthesis after liver injury [31] and in promoting hepatic differentiation [32]. In this context, our model, persistently co-expressing OV6, c-Kit and c-Met, with high levels of αFP and CK19, markers of ductal OCs [33 –35], accounts for a system that is able to achieve in vitro bipotent progenitors. Co-expression of hepatocytic (ALB, CK18) and biliary (αFP, CK19) lineage markers, with markers from HSCs (c-Kit, Thy-1), is typical and strongly matches the hypothesis of a progenitor population similar to rodent OCs [36]. At late times, in fully committed cells, marker expression becomes hepatocytic-specific (ALB+/CK19-; CK18+/αFP-). Our observations are consistent with a heterogeneous population of iBHP at various stages of differentiation, in which the maturation process is linked to loss of expression of progenitor markers (Fig. 4). Differentiation is normally linked to loss of cell proliferation; thus, the persistence of dividing cells within differentiating iBHPs population suggests the presence of a small subset retaining proliferative potential. Notwithstanding, iBHPs also display the metabolic activity of fully differentiated hepatocytes: production and secretion of ALB, αFP, and urea; synthesis and storage of glycogen; uptake of low density lipoprotein; and the specific response of CYP2B6 to PB induction.
The most important property establishing a liver stem cell model for hepatic regeneration is to select cells capable of efficiently reconstituting the damaged liver. Progenitor-dependant liver regeneration is a multistep process in which the interplay of cell types and factors involved play a pivotal role [7]. In our model, we found that the activation of hepatic progenitors is the first response to the hepatogenic stimulus and is rapidly achieved after induction. By injecting early activated bipotent progenitors, we aimed at supplying cells actively expressing factors of the initial step/s of activation/differentiation, able to stimulate in recipient animals the inner molecular signaling of regenerative process, to promote the correct engraftment and functional integration of transplanted cells, and to permit the onset of the liver regeneration. When transplanted into a mouse model of acute liver injury, iBHPs were able to escape the systemic blood and to colonize the damaged liver, where they concentrated in the bile ducts and canaliculi of the periportal areas. Cells engrafted with the appropriate integration into the host liver suggest a regenerative potential [22]. The expression of αFP and CK19, and the presence of OV6-positive cells in livers of recipient animals, may indicate bile duct development in addition to ALB secreting hepatocyte-like cells [37], which confirms the oval-like bipolar nature of iBHPs cells. In addition, the significative reduction of serum transaminases in the transplanted group with regard to control also suggests a functional benefit of iBHPs. The in vitro response of cord blood CD133+/CD34+/OV6low cells to hepatogenic microenvironment modulations mimics hepatic progenitor activation in vivo and triggers the hypothesis that this cell population truly encompasses immature precursors which can be activated to proliferate and differentiate into functional bipotent progenitors capable of properly engrafting damaged livers. These cells allow for a human model for in vitro progenitor-cell activation and may represent a powerful tool for the comprehension of the molecular mechanisms that lead to ductular reactions in human. Moreover, the high reproducibility and the possibility to achieve iBHPs also from cryopreserved cells make this model a potential candidate for future use in cell-based liver-directed therapies.
Footnotes
Acknowledgments
We are grateful to Dr. Elena Alfani for her assistance in quantitative PCR analysis and to Dr. Paola Lanza for IHC technical assistance. The authors thank Dr. Griselda Santacroce and Dr. Elisabetta Tarquini for their friendly cooperation and Professor Bruno Azzarone for his helpful critical comments of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
Financial Support
This research was supported by: Istituto Superiore di Sanità and Health Ministry Grant for the National Program on Stem Cell: ISS-CS 68; CNR-Ricerca Spontanea a Tema Libero (RSTL) N. 526 and N. 790 to Dr. A. Lisi; CNR Short Term Mobility Fellowship 2009- Prot. N. 001594 to Dr. A. Crema and CNR Short Term Mobility Fellowship 2009- Prot. N. 0030416 to Dr. D. Fioretti.