
Abstract
Endothelial progenitor cells (EPC) represent a relatively rare cell population, and expansion of sufficient cell numbers remains a challenge. Nevertheless, human adipose-derived stem cells (hASC) can be easily isolated and possess the ability to differentiate into endothelial cells. Here, we propose the isolation and characterization of multipotent endothelial-like cells (ME-LC) with the capacity to maintain their vascular progenitor properties for long periods. hASC were isolated from lipoaspirates and cultured through distinct consecutive culture stages for 2 months to enrich ME-LC: first in Dulbecco's modified Eagle's medium–fetal bovine serum (stage I), followed by a stage of culture in absent of fetal bovine serum (stage II), a culture in SFO3 medium (stage III), and, finally, the culture of ME-LC into collagen IV-coated flasks in endothelial growth medium (EGM-2) (stage IV). ME-LC display increased expression levels of endothelial and hematopoietic lineage markers (CD45, KDR, and CXCR4) and EPC markers (CD34 and CD133), whereas the expression of CD31 was barely detectable. Reverse transcription (RT)-polymerase chain reaction assays showed expression of genes involved in early stages of EPC differentiation and decreased expression of genes associated to differentiated EPC (TIE-2, DLL4, and FLT-1). ME-LC formed capillary-like structures when grown on Matrigel, secreted increased levels of stromal cell-derived factor-1 (SDF-1), and showed the ability to migrate attracted by SDF-1, vascular endothelial growth factor, and hematopoietic growth factor cytokines. Importantly, ME-LC retained the capacity to differentiate into cardiomyocyte-like cells. We present a simplified and efficient method to generate large numbers of autologous ME-LC from lipoaspirates-derived hASC, opening up potential cell-based therapies for cardiovascular regenerative medicine.
Introduction
I
EPC are commonly characterized by the expression of the surface markers CD34 and CD133, lack of the hematopoietic marker CD45, and co-expression of CXCR4 and VEGFR2 (KDR/flk-1) [2,3]. EPC differentiation into mature endothelial cells (ECs) is accompanied by a loss of expression of CD133 and a concomitant increase in CD31 expression, CD144 (VE-cadherin), and other markers [4]. Furthermore, expression of certain genes is also used during the characterization of bona fide ECs. For instance, FLT-1, TIE-2, CCR7, and C-KIT are expressed on EPC, among others cell types, whereas CDK2, a cyclin-dependent kinase, is overexpressed on later stages of EPC differentiation and absent on early phases of EPC differentiation [5].
Blood-derived EPC or BM-derived stem cells have been used to improve myocardial perfusion and contractile function and enhance limb perfusion in patients [6,7]. However, there are still some drawbacks for their clinical utility such as the extremely low number of EPC in the bloodstream and the low availability and harvesting difficulties of BM-derived stem cells. This may be severely hampered in elderly patients or due to high morbidity associated with vascular disease [8].
In contrast, human adipose-derived stem cells (hASC) can be isolated in a greater number through a safe noninvasive routinely liposuction procedure. These hASC can also be expanded in culture and differentiate into different cell types, including ECs [9]. However, to use these progenitor cells clinically in regeneration of vascular and/or heart lesions, it is necessary to develop reliable and reproducible methods to isolate and expand these cells [10].
In the present study we propose a new approach of easy-to-derive large number of multipotent endothelial-like cells (ME-LC) from human adipose tissue with the capacity to display endothelial and cardiomyocytes-like properties in culture for long periods.
Methods
Isolation and culture of hASC from human adipose tissue
Subcutaneous adipose tissue was obtained from 15 different patients by a minimally invasive procedure after signed informed consent from all patients and approval from the Ethics Committee of the Clinic University Hospital of Málaga (Spain). This study conformed to the principles outlined in the Declaration of Helsinki. In each experiment we used at least 4 lipoaspirates. Isolation and culture of hASC was performed as described previously [11,12]. hASC were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma, St Louis, MO) containing 10% fetal bovine serum (FBS; Lonza, Basel, Switzerland) and 1% Penicillin–Streptomycin solution (Sigma) (DMEM-FBS).
Differentiation assays of hASC
hASC were plated at 2×103 cells/cm2 in DMEM-FBS and were allowed to adhere for 24 h. Culture medium was then replaced with specific differentiation inductive media. For adipogenic and osteogenic differentiation, cells were cultured for 2 weeks in Adipogenic Mesenchymal Stem Cells (MSC) Differentiation Bullet Kit and Osteogenic MSC Differentiation Bullet Kit (Lonza), respectively. For chondrogenic differentiation, cells growing in monolayer were cultured in NH ChondroDiff Medium (Miltenyi Biotec, Auburn, CA) for 3 weeks. Recent articles published by our group have demonstrated the capability of chondrogenic differentiation of hASC in monolayer cultures [12,13]. Differentiated cell cultures were stained with Oil Red O (Amresco, Solon, OH) for adipogenic differentiation, Alizarin Red (Lonza) for osteogenic differentiation, or Toluidine Blue (Sigma) for chondrogenic differentiation [14].
ME-LC isolation and expansion
hASC were split and seeded at 3×106 cells/T-75 tissue culture flask (BD Falcon, Franklin Lakes, NJ) in DMEM-FBS (stage I). After third or fourth cell-culture passage (2 weeks), the culture medium was replaced by serum-free medium (DMEM) to induce the development of multicellular aggregates that were termed “sphere cluster formations” (SCF) (stage II) (Fig. 1). SCF were scraped off after 3 weeks and seeded in a 6-well plate at a concentration of 10–15 SCF per well in DMEM-FBS for 48 h. Isolated cells were then subcultured in medium SFO3 [RPMI-1640: DMEM: F12, 0.1% bovine serum albumin, 50 μM 2-mercaptoethanol, and 1% Penicillin-Streptomycin (Sigma)] (stage III) [15]. Within the next 3 weeks, cells were scraped off and seeded into collagen IV-coated flasks and grown in EC medium, endothelial basal medium-2 (EBM-2; Lonza) containing 5% FBS, and human recombinant VEGF, hydrocortisone, human recombinant epidermal growth factor, human recombinant long R insulin-like growth factor-1 (R3-IGF-1), ascorbic acid, human recombinant basic fibroblast growth factor, and gentamicin sulfate-amphotericin-B (EGM-2; Lonza) (stage IV) (Fig. 1). Cells obtained from stages III and IV were considered as ME-LC. Human umbilical vein ECs (HUVEC) were also cultured in endothelial growth medium (EGM-2) as a control.
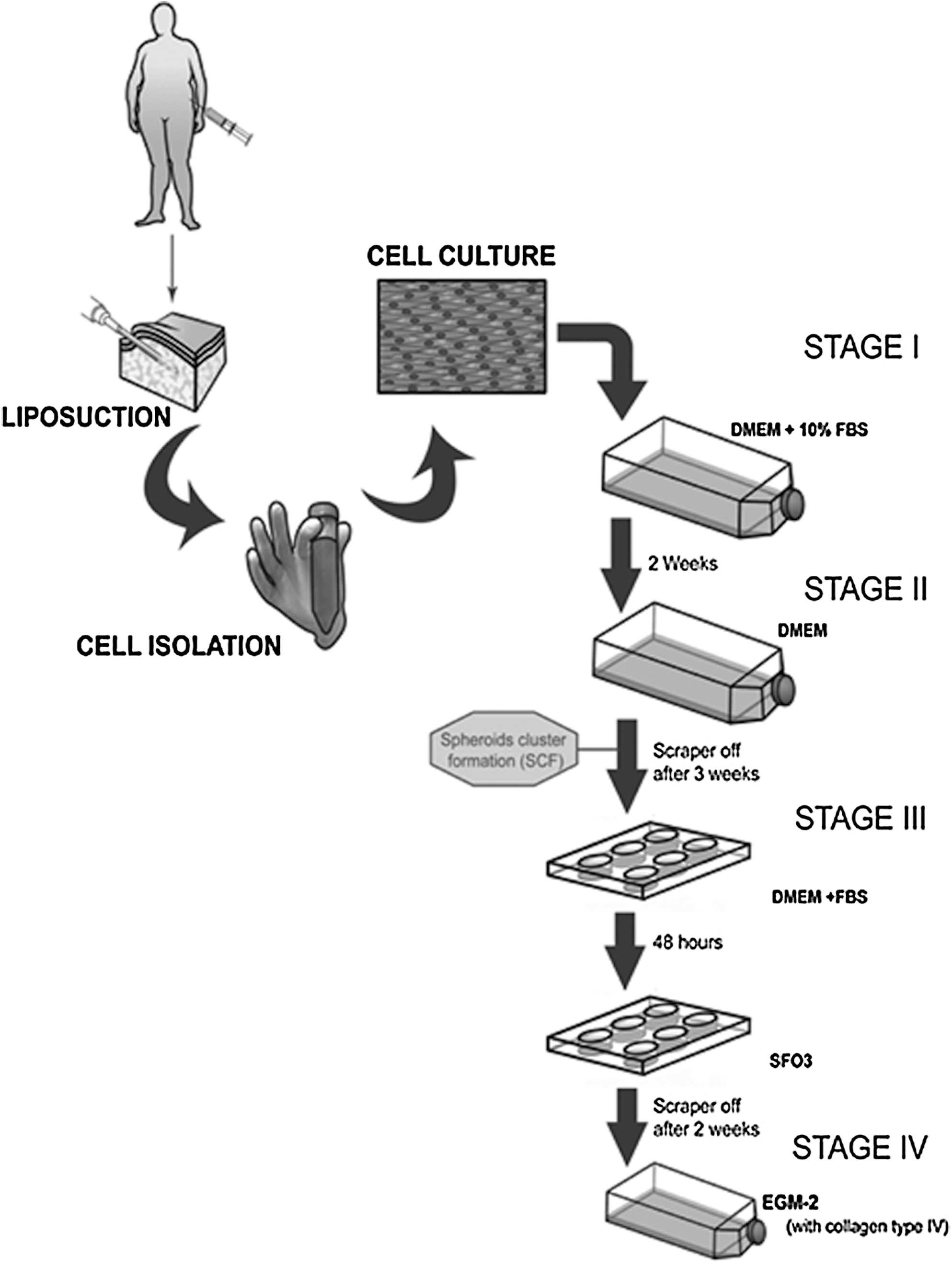
Cartoon representing the ME-LC isolation and purification procedure. hASC were isolated by enzymatic digestion from lipoaspirates obtained from patients by a minimally invasive surgery. Stage I: cells cultured in DMEM-FBS for 10–14 days; stage II: cells cultured in DMEM-FBS for 10–14 days and then 2–3 weeks in DMEM; stage III: cells scrapped off from the stage II and seeded in SFO3 for about 3 weeks; stage IV: cells scrapped off from the SFO3 culture medium and cultured into collagen IV-coated flasks in EGM-2 for at least 1 week. ME-LC, multipotent endothelial-like cell; hASC, hASC, human adipose-derived stem cell; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; SCF, sphere cluster formations.
Flow cytometry
Cells were trypsinized, washed, and resuspended in phosphate-buffered saline (PBS) with 2% bovine serum albumin (Sigma), and 2 mM ethylenediaminetetraacetic acid (EDTA, Sigma). Cells were incubated in the dark at 4°C for 45 min with the following fluorochrome-conjugated monoclonal antibodies: CD133-PE (Miltenyi), CD105-APC, CD90-FITC (eBioscience Inc., San Diego, CA), KDR-APC (R&D System, Minneapolis, MN), CD34-FITC, CD45-APC-Cy7, CD 73-PE, and CXCR4-PE (BD Biosciences, San Jose, CA). Cells were then washed in PBS and analyzed in a fluorescence-activated cell sorting (FACS) Canto II cytometer equipped with the FACS Diva analysis software (BD Biosciences) [16]. Data obtained were expressed as mean±standard error (SE) from 4 independent experiments performed in triplicate (P<0.05).
Gene expression profile
For reverse transcription-polymerase chain reaction (RT-PCR) analysis, total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. The cDNA reaction was performed using 0.5–2 μg of total RNA with primers from SuperScript II-kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Forward and reverse primer sequences and expected PCR product sizes for each specific gene are shown in Table 1. The PCR was performed with Reddy Mix PCR Master Mix (Thermo Fischer Scientific, Epsom, United Kingdom). After initial denaturation (2 min at 94°C) 35 cycles were performed (20 s 94°C, following by 1 min annealing and 51°C for FLT-1, TIE-2, DLL4, CDK2, and β-actin and 43°C for CXCR4, CD133, and Ccr-7 followed by 1 min extension at 72°C). The PCR products were run on 1% agarose gel and photographed under UV light [17,18].
Functional capillary formation assays
The ability to form capillaries in semisolid medium was tested by culturing trypsinized cells on Matrigel™-coated 96-well plates (BD Biosciences) in EGM-2 medium. Matrigel™ was thawed, used to cover the culture plastic (50 μL per well of a 96-well plate), and allowed to solidify for 1 h at 37°C. Cells from stage I to stage IV cultures were independently seeded. Outgrowths obtained from cultures at different stages of the endothelial isolation process were seeded on Matrigel™-containing plates at 5 to 20×103 cells per well and cultured in EGM-2 medium for 7 days. Four hours, 24 h and 7 days after the initial plating photographs were taken using a Leica DM 5500B (Leica, Solms, Germany) microscope equipped with the Meta Systems software [19]. Figures were processed with Adobe Photoshop 7.0. Cells were counted for the formation of capillary structures. The number of capillary-like structures was measured after 24 h and each cord portion between the ramifications was considered 1 capillary unit. Mean±SE values were obtained by evaluating the whole cultures of each well under the same conditions from 3 independent experiments performed in duplicate. A semiquantitative measurement of capillary formation on Matrigel™ was performed as described elsewhere (capillary formation index) [20,21], using HUVEC-like control.
Cytokine determination
The production of the chemokine SDF-1 in different cultures was assayed by enzyme-linked immunosorbent assay (ELISA). First, serum and other supplements were removed from the culture medium to avoid potential interference with the measurements. Cell culture supernatants were collected 24 h later and were used for the assay. ELISA was performed using the Human SDF-1 Kit (R&D System), according to the manufacturer's protocol, and the measurements of emitted signal at 450 nm were taken with the ELx800TM microplate absorbance reader (Bio-Tek Instruments GmbH, Bad Friedrichshall, Germany). All data about SDF-1 secretion were compared taking account the same number of cells in each culture stage (5×105 cells) and were obtained from 4 independent experiments performed in duplicate.
Migration assays
To determine cell migration, a modified Boyden chamber assay was performed using a 24-well microchemotaxis chamber (BD Biosciences). About 105 cells growing in collagen IV-coated flasks in EGM-2 medium (stage IV) were seeded onto the upper Boyden chamber in EBM-2 medium supplemented with 10% FBS. In the lower chamber, a culture medium containing 50 ng/mL of VEGF, 25 ng/mL of hematopoietic growth factor (HGF), and 100 ng/mL of SDF-1 was added. Cells were labelled with 5 μM of calcein AM (Invitrogen) and they were photographed using a confocal microscope (Leica DMI6000). The nonmigrating cells in the upper chamber were scraped off using blunt-ended forceps and swabs, and washed with PBS. Moreover, after 1–12 h incubation at 37°C in a 5% CO2 atmosphere, the cells were fixed in 2% paraformaldehyde for 5 min and washed in PBS. The fluorescence from the cells migrated to the lower chamber was measured using a fluorescence microplate reader (FLx800; Bio-Tek Instruments, Inc., Winooski, VT) from the bottom at 485/535 nm wavelength. The migrated cells were represented by the ratio of fluorescence as compared to the control.
Cardiac differentiation
ME-LC (stages III and VI) were seeded at 5–20×103 cells per well of a Matrigel™-coated 96-well plate in EGM-2 medium. Culture medium was replaced 2 weeks later by EBM-2 containing 5–10 or 15 μM of 5-azacytidine (5-aza) for 24–48 h. Culture medium was changed back to EGM-2 and cells were cultured for 3–4 weeks. Cells were then detached from Matrigel™ with dispase (BD Biosciences) and seeded in a 8-well chamber slide (Nunc, Rochester, NY) at 5–10×103 cells per well for 4 days in EGM-2 for immunofluorescence analysis.
Immunofluorescence
Cells were washed 3 times with PBS and fixed with 4% paraformaldehyde in PBS for 30 min at room temperature. Then, cells were permeabilized with 0.1% Triton X-100 for 15 min, washed 3 times with PBS, and blocked in 2% blocking buffer solution (Roche, Barcelona, Spain) for 1 h at room temperature. Cells were then incubated overnight with primary antibodies diluted 1:100 in blocking buffer solution at 4°C, washed 3 times in PBS, and then incubated for 2 h with secondary (FITC- or TRITC-conjugated) antibodies diluted 1:200 in blocking buffer solution. Afterward, they were washed 3 times in PBS and slides were mounted with 4′,6-diamidino-2-phenylindole-containing mounting solution (Ultra Cruz TM Mounting Medium; Santa Cruz Biotechnology, Santa Cruz, CA). Cells treated in the same way but incubated with isotype-matched control antibodies were used like negative control. Antibodies used were as follows: human desmin (Goat monoclonal; Sigma); human cardiac-specific troponin T (mouse monoclonal; Research Diagnostics, Flanders, NJ); and human sarcomeric α-actinin (mouse monoclonal; Sigma). Photographs were taken with a Leica DM 5500B fluorescent microscope equipped with Meta Systems software.
Statistical analysis
Statistical analysis was performed using the nonparametric Kruskal–Wallis H-test for independent experiments. Significant differences between groups were estimated using the Mann–Whitney U-test. For the statistical analysis SPSS 15.0 software program was used. Data are presented as mean±SE. P<0.05 was considered as statistically significant.
Results
hASC obtained from lipoaspirates possess MSC properties
hASC isolated from human lipoaspirates were maintained in culture for >10 weeks with no signs of senescence. FACS characterization showed that ex vivo cultured hASC expressed the surface markers CD105 (>99%), CD90 (>90%), and CD73 (>99%) but lacked expression for both hematopoietic and EC markers CD45, CD34, CD133, CXCR4, and KDR (Fig. 2A). Cells cultured in adipogenic medium acquired typical morphology of lipid-laden cells containing intracellular lipid-filled droplets, which stained positive for Oil Red O. Alizarin Red S staining demonstrated the presence of osteogenic differentiation, with the presence of mineralized nodules as shown in Fig. 2B. Chondrogenic differentiation was confirmed by toluidine blue staining showing accumulation of proteoglycans (Fig. 2B).

Phenotypic characterization and differentiation potential of hASC.
ME-LC isolation and expansion
ME-LC were isolated through a series of consecutive stages I to IV detailed in the methods section (see Fig. 1 for details). hASC obtained from lipoaspirates grew as a monolayer and displayed a fibroblast-like and spindle-shaped morphology when they were cultured in DMEM-FBS (stage I, Fig. 3A). When the culture medium was replaced by DMEM without serum, the cells began to form 3-dimensional cellular aggregates (termed SCF), which increased in size over time (stage II; Fig. 3B). We obtained between 50 and 70 SCF in each T-75 tissue culture flask. When these SCF were scraped-off and seeded in SFO3 medium, SCF began to connect each other and showed a variable morphology (stage III; Fig. 3C, D). At the end of stage III were obtained 3.9±2.1×105 cells in each well-plate from 10–15 SCF. For the stage IV, cells scraped-off from 2 wells were seeded in a T-75 tissue culture flask. Cells displayed an elongated morphology and were arranged in parallel or grew attached surrounding the SCF (stage IV; Fig. 3E, F). At this stage the doubling time of ME-LC was shorter than hASC, 2.7±0.1 days and 3.8±0.3 days, respectively.

Changes in cellular morphology and distribution in different culture stages.
ME-LC isolated after several culture stages express markers associated to vascular progenitors coupled to a decreased expression of the mesenchymal marker CD90
Flow cytometry was carried out in each of these culture stages to assess the presence or absence of hematopoietic, endothelial, and mesenchymal markers. The expression of the hematopoietic markers CD133, CD34, CD45, KDR, and CXCR4 significantly varied throughout the isolation process (P<0.05, Mann–Whitney U-test) (Fig. 4A). Initially, the cultures were negative for all markers during stages I and II. When the cells were cultured in SFO3 medium (stage III), they slightly upregulated these markers to a some extend: 14.1%±1.7% for CD133, 18.3%±4.9% for CD34, 21%±3.7% for CD45, 27.3%±5.5% for KDR, and 13%±1.65% for CXCR4. In stage IV, when the cells had been cultured in EGM-2 medium, the expression of these markers dropped (Fig. 4A).

Fluorescence-activated cell sorting and RT-PCR analysis of endothelial, hematopoietic, and mesenchymal markers throughout ME-LC isolation stages.
High levels of expression of the MSC markers CD105, CD73, and CD90 were observed throughout the different stages of the ME-LC isolation procedure (Fig. 4B). A Mann–Whitney U-test indicates significant (P<0.05) differences among the expression of the 3 markers (Fig. 4B). Interestingly, CD90 expression decreased significantly in cells cultured in SFO3 medium (stage III: 41%±15.8%) in comparison with cells in the stage I (94.1%±2.2%), stage II (80±8.6), and stage IV (87.5%±2.7%).
Figure 4C shows a comparison between marker expression of cells at stages I, III, and IV versus HUVEC, which were used as control of mature ECs. HUVEC and ME-LC showed expression of the progenitor markers (CXCR4 and KDR). CD133 and CD34 were found expressed in ME-LC but not in HUVEC. Expression of the MSC marker CD90 decreased in both ME-LC cultured in SFO3 and HUVEC as compared with cells at stage I. Finally, the endothelial marker CD31 was highly expressed in mature HUVEC (63%±11.4%) but it was barely expressed in stages I, III, and IV (Fig. 4C). All together, these data suggest that ME-LC isolated from hASC cultures express markers resembling a vascular progenitor phenotype.
Expression levels of genes related with EPC were assessed by RT-PCR. Cells cultured from stage I to IV maintained the expression of genes such as CD133, CXCR4, CDK2, and FLT1. A weak expression of CCR7 and CDK2 genes was detected in cells at stage III (Fig. 4D). Expression of TIE2 was only detected at stage I, but not in subsequent stages. Finally, expression of DLL4 (a Notch ligand) was found highly expressed at stage I. Its expression, however, decreased at from stage II onward and was completely lost at stages III and IV (Fig. 4D). These gene expression data suggest the endothelial progenitor phenotype of ME-LC.
ME-LC enhance functional capillary-like structures formation in a Matrigel assay
As shown in Fig. 5A, cells from cultures at stage I and II were not able to form any capillaries over 7 days. On the other hand, cells previously grown in SFO3 or EGM-2 (stages III and IV, respectively) displayed a large number of capillary-like structures as early as 4 h after being seeded on Matrigel™ and the appearance of capillary-like structures increased overtime. After 7 days in culture, a well-established cellular network was present in all the cultures (Fig. 5A). As a positive control, the capability of HUVEC to form capillary-like structures in Matrigel™ was also assessed. The results showed the appearance of these capillary-like structures after 4 and 24 h in Matrigel™. However, these structures disappeared when HUVEC were cultured for 7 days likely due to their very mature nature (Fig. 5A).
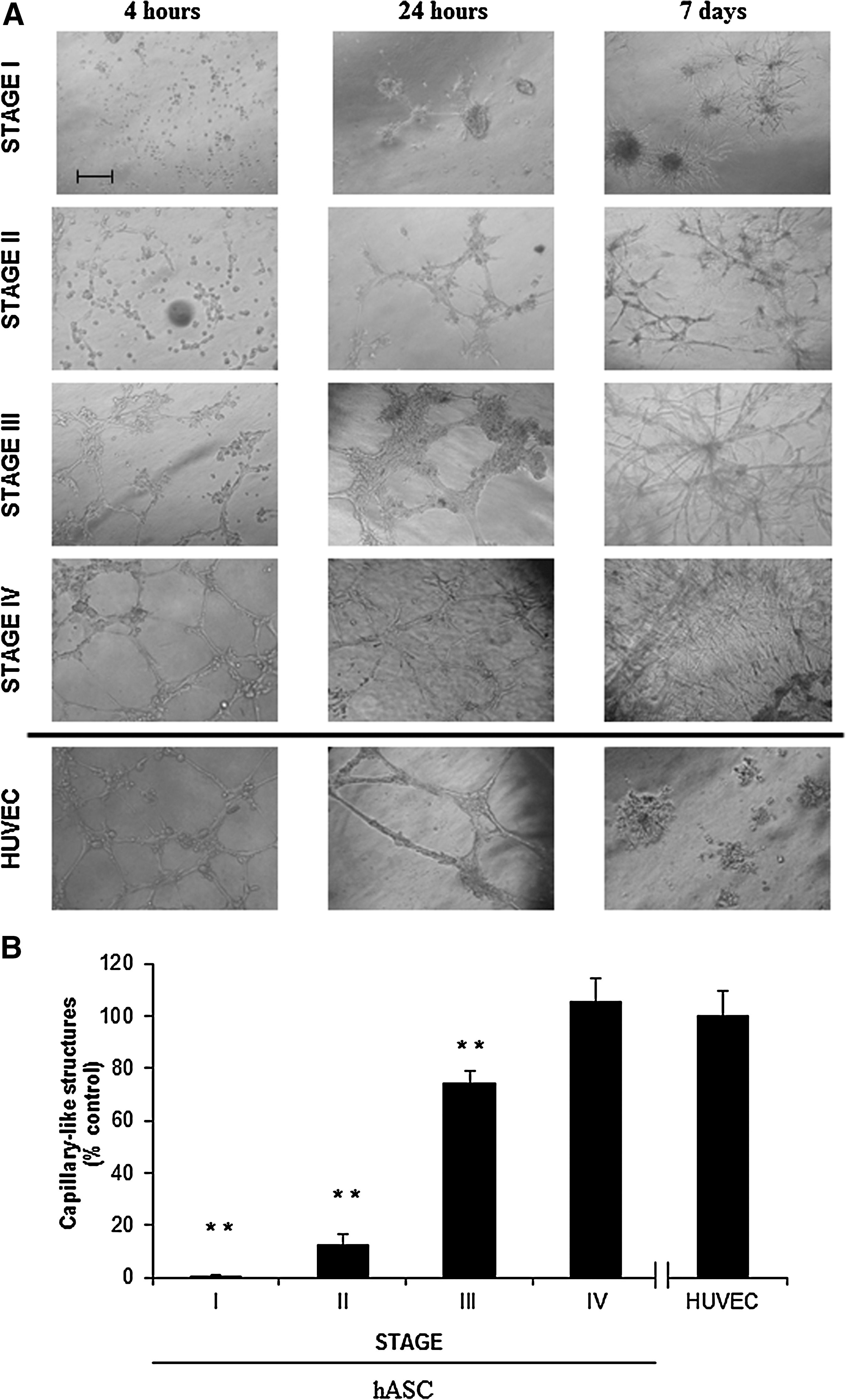
Capillary network formation assay.
The number of capillary-like structures was counted in every Matrigel™-coated well after 24 h of culture such as is described in the Methods section. As the hASC were cultured throughout the distinct stages they gradually gained ability to form capillary-like structures (P<0.05). By stage IV, the number of capillary-like structures was similar in comparison with the data rendered by HUVEC, used as control (Fig. 5B).
Increased release of the angiogenic cytokine SDF-1 by ME-LC
The presence of the SDF-1 cytokine in the medium was analyzed by ELISA at different time points. There was a significantly increased of SDF-1 (P<0.05) in supernatants harvested at stage III (583±67 pg/mL) and IV (1420±225 pg/mL) in comparison with the SDF-1 levels at stages I and II (148±20 pg/mL and 148±37 pg/mL, respectively). Moreover, when cells at stage IV were maintained for further 2 weeks in EGM-2 medium, the concentration of SDF-1 increased drastically (4113±170 pg/mL) (Fig. 6A).
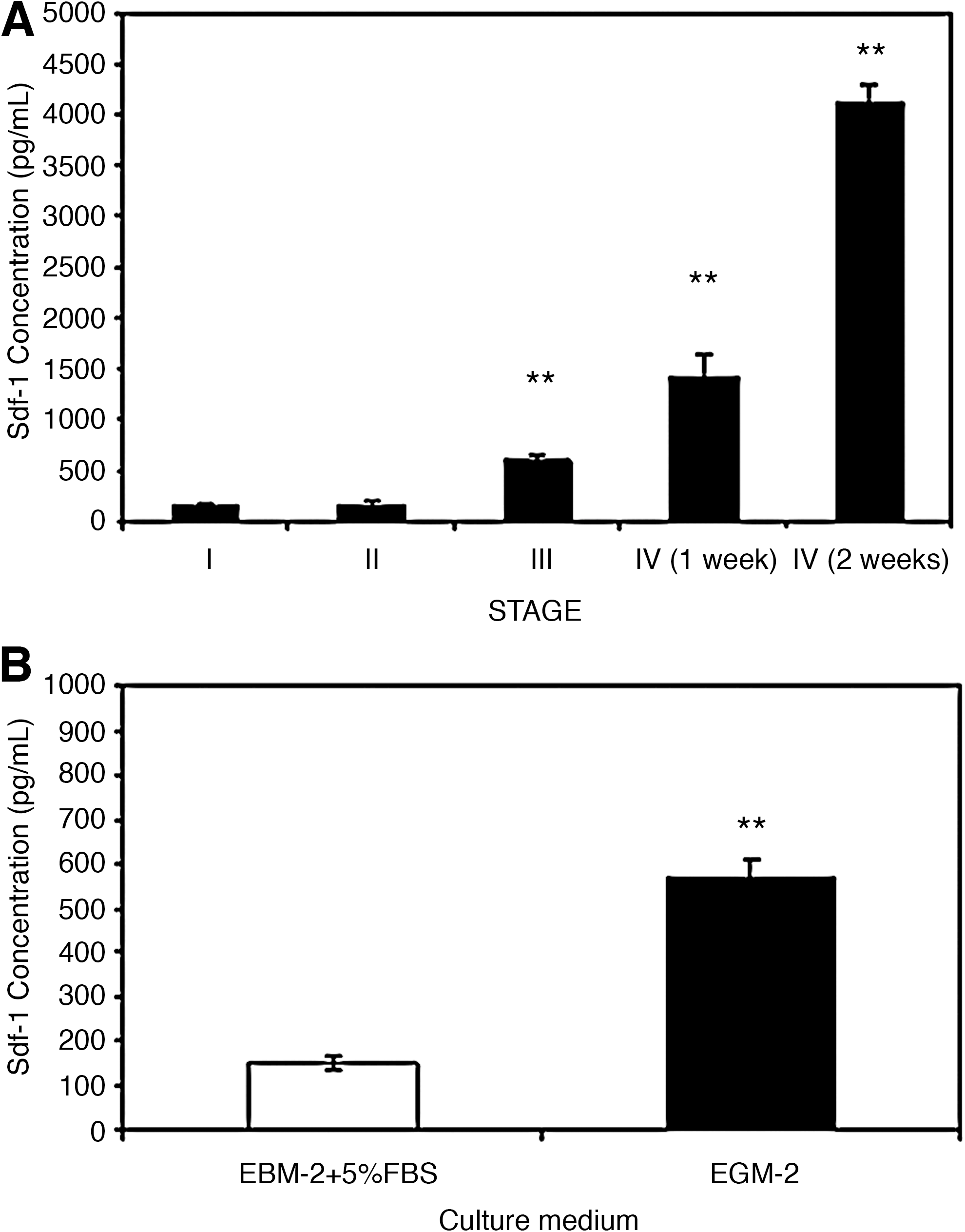
SDF-1 detection in culture supernatants.
Finally, to determine the influence of endothelial growth factors on ME-LC included in a model of extracellular matrix, we cultured ME-LC for 2 weeks on Matrigel™-coated plates on EGM-2 medium versus EBM-2 medium supplemented with 5% FBS. The SDF-1 levels in the supernatants were 148±15 pg/mL in cells cultured in EBM-2 supplemented with 5% FBS and 568±40 pg/mL for cells cultured in EGM-2 (Fig. 6B).
ME-LC are able to migrate in response to angiogenic cytokine stimuli
To investigate the cell migratory response of ME-LC toward different angiogenic cytokines such as SDF-1, VEGF, and HGF, we used a modified Boyden chamber with cells cultured in EBM-2 medium supplemented with 10% FBS for 12 h (Fig. 7A). SDF-1, VEGF, or HGF cytokines induced cell migration of ME-LC from the upper compartment through the pores of the membrane into the lower compartment (Fig. 7B, C). Negative controls were performed adding cytokine-free medium into the lower compartment.
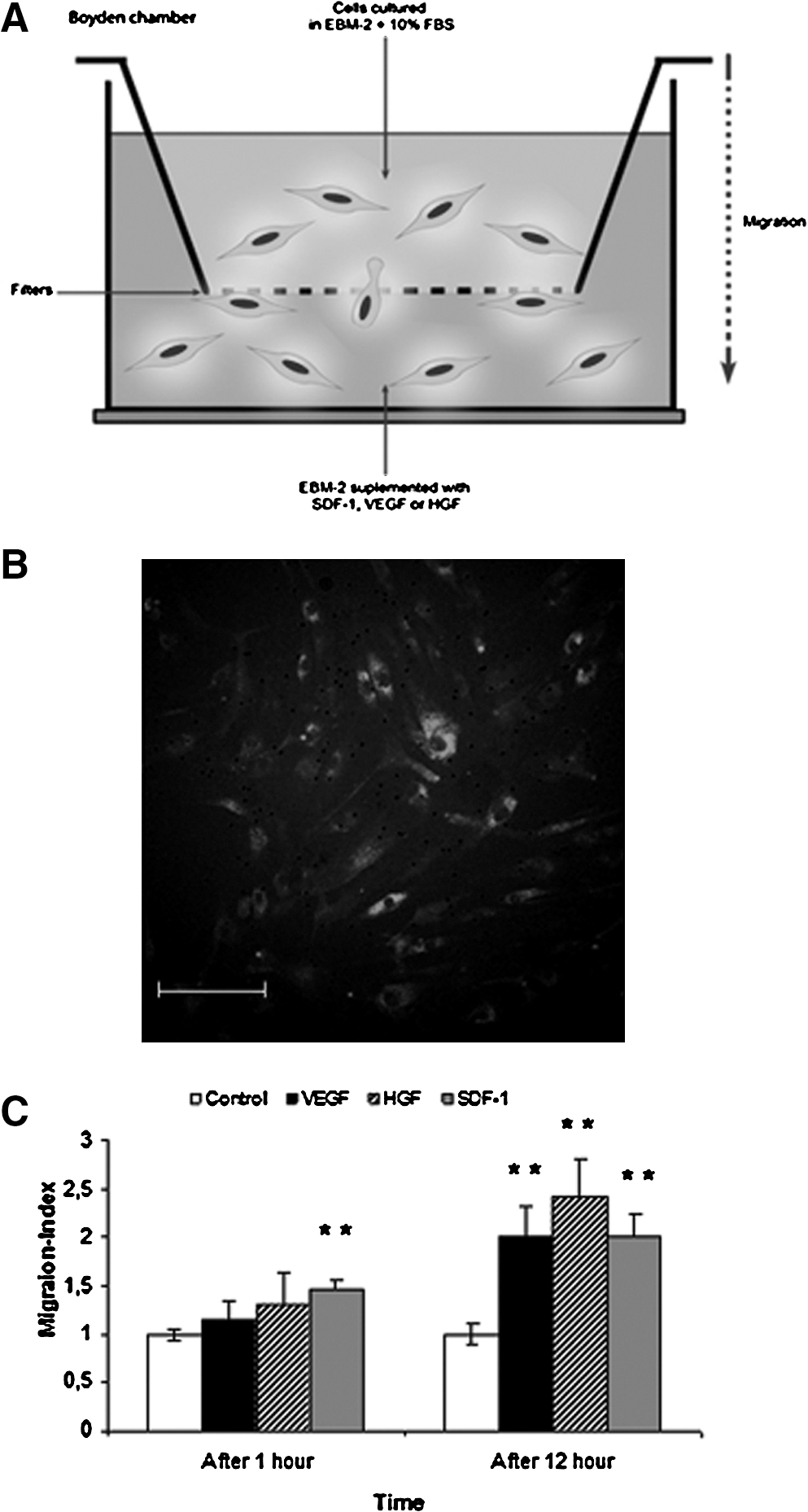
Migration capacity of ME-LC.
ME-LC have the ability to differentiate into a cardiac phenotype
Finally, we tested the potential of the ME-LC to differentiated toward a cardiomyocyte phenotype after exposure to 5-aza [12]. Morphological changes and expression of cardiac-specific markers were determined after 3 weeks of culture. Upon 5-aza treatment, ME-LC changed their morphology. Treated ME-LC were wider and displayed branching fibers easily observed by phase-contrast microscopy (Fig. 8A). Immunocytochemistry analysis revealed the expression of typical cardiomyocyte markers such as Troponin-T, Desmin, and α-Actinin in the cytoplasm of the 5-aza-treated cells. Interestingly, these markers colocalized (Fig. 8B). Moreover, cells that stained positive for these cardiac markers showed a parallel and interconnected distribution with the presence of bi- and multinucleated cells. Control ME-LC nontreated with 5-aza were negative for all the cardiomyocyte markers examined (Fig. 8C).
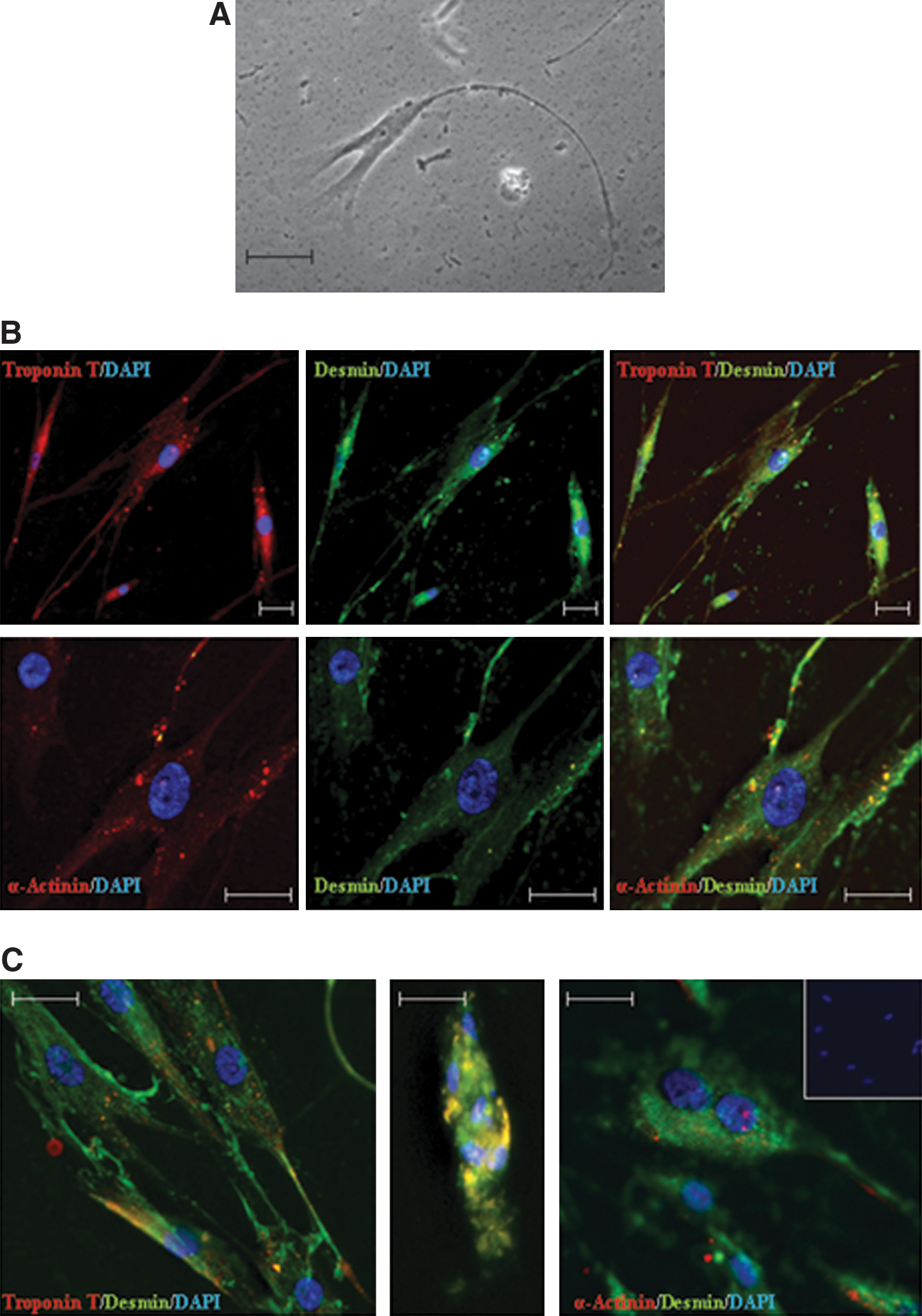
Cardiomyocyte differentiation potential of ME-LC.
Discussion
Cardiovascular diseases, such as the ischemic heart disease and peripheral arterial occlusive disease, cause an elevated morbidity and mortality in developed countries. Currently, several clinical trials use different strategies for cell delivery and a diverse cell sources for transplantation [6 –8]. In this respect, regenerative therapy to treat endothelial tissue damage has focused on the use of autologous stem cells, mainly EPC harvested from blood, BM, or umbilical cord blood [22]. Nevertheless, the scarcity of EPC in adult tissues makes its therapeutic use a challenge. As an alternative, the endothelial differentiation potential of MSCs has been explored by stimulating MSCs with angiogenic growth factors and it was proved that the differentiated MSCs were able to integrate into new blood vessels in vivo [23].
Recently, it has been shown that adipose tissue contains a population of adult multipotent cells with extensive proliferative capacity in vitro which are able to differentiate into several lineages, including ECs, smooth muscle cells, and cardiomyocytes [13,24,25]. In fact, there are preclinical studies supporting the capability of MSCs obtained from BM, umbilical cord blood, or adipose tissue to differentiate into endothelial mature cells [26,27]. However, it was reported that mature ECs can proliferate in vitro although they gradually loss their proliferative potential hampering their clinical application [28]. From a therapeutic standpoint, it becomes necessary the isolation of sufficient numbers of progenitor cells capable of maintaining their angiogenic potential in vitro for long periods. Here, we present a simple and reproducible approach to maintain ME-LC isolated from subcutaneous adipose tissue. Moreover, we tried to induce cardiomyocytic differentiation to demonstrate the capacity of ME-LC to differentiate into both endothelial and cardiomyocyte-like cells, which could have advantages in the stem cell-based cardiovascular therapy.
Phenotypic characterization of hASC isolated from lipoaspirates showed a high expression of mesenchymal-specific surface markers such as CD105, CD73, and CD90, and barely expressed hematopoietic stem cells (HSC) or EPC markers (CD45, CD34, CD133, CXCR4, or KDR). Moreover, hASC possessed the ability to differentiate into various lineages as previously shown [29]. hASC were cultured along several stages (stage I to IV, Fig. 1). Culture in serum-free media for 3 weeks resulted in the appearance of SCF that increased in number and size throughout the subsequent culture stages. Previously, Hirashima et al. [15] demonstrated that a chemically defined serum-free culture system, including 2-mecarptoethanol, had the ability to support the proliferation of ECs and their progenitors from mesoderm cells. When cells were cultured in SFO3 medium (stage III) and in EC medium (EGM-2; stage IV), both termed ME-LC, increased their expression of EPC and hematopoietic markers (CD34, CD133, KDR, CXCR4, and CD45) [30]. The coexistence of hematopoietic and endothelial markers in the ME-LC is indicative of a phenotype resembling early vascular progenitors since it has been reported the existence of a bipotent precursor cell, termed the hemangioblast, capable of giving rise to both hematopoiesis and vascular endothelium [31]. In contrast, mature ECs HUVEC were negative for CD34 and CD133 progenitor markers and strongly positive for CD31, a mature endothelial marker. These antigens (CD34 and CD133) are lost upon differentiation of endothelial progenitors to endothelium [30]. CD34 expression in hASC is correlated with replicative capacity, differentiation potentials, expression profiles of angiogenesis-related genes, and immaturity or stemness of these cells [32]. Similar results were obtained by Howson et al. [33] using postnatal aorta to develop culture conditions for the isolation of nonendothelial mesenchymal cells with long-term maintenance in an undifferentiated state. Under serum-free conditions vascular progenitor cells obtained were CD34+/CD31−, grew forming spheroids, and were identified as pericyte progenitor cells [33]. However, another study showed that using the same serum-free media, cells obtained from lipoaspirates had increased proportion of Flk-1+ (KDR) marker and were negative for CD34, CD45, and CD133. When these cells were seeded in EC differentiation medium for 3 days, the expression of Flk-1+ decreased over time, whereas the expression of mature EC markers increased [27]. In contrast to our study, they cultured the cells during shorter period in serum-free media and in EC medium, likely accelerating endothelial maturation due to serum components, which have been proved to induce cell maturation [34].
Interestingly, both ME-LC and HUVEC expressed high levels of CXCR4 and KDR. Phenotypically, MSCs are identified by the absence of CD45, CD34, and other hematopoietic-associated markers [3]. In addition, the MSC marker CD90, whose expression was practically absent in HUVEC, was the marker whose expression most significantly decreased when ME-LC were cultured in SOF3 medium (stage III), suggesting endothelial differentiation. In agreement, it has been shown an annihilation of CD90 antigen expression on mesenchymal stromal cells by angiogenic stimulation in vitro [35].
In contrast, CD90-positive cells recovered at stage IV in which cells reached a high rate of proliferation. It has been shown that the expression of CD90 on EPC and pericytes may be indicative of their angiogenic potential and capacity for proliferation [36].
Gene expression profile confirmed the endothelial progenitor phenotype with the expression and maintenance of specific genes involved in self-renewal, cell cycle promotion, and antiapoptotic such as has been recently showed in cord blood-derived EPC [5]. Both CXCR4 and CD133 vascular genes were expressed at similar levels throughout the distinct culture stages. Nevertheless, genes expressed in differentiated EPC such as Cdk2 and Flt-1[5] showed a weak expression level in ME-LC cultured in SOF3 medium. Another interesting result was the disappearance of Tie-2 gene expression, which has been previously reported as angiogenic factor clearly induced in the differentiated ECs [37]. Constitutive Ang1–Tie2 signaling is thought to maintain the quiescent endothelial phenotype in vivo [38]. In addition to ECs, Tie2 is expressed in a subpopulation of HSC being, in part, responsible of maintaining a quiescent state in the BM niche [39]. Also, our results showed that the expression of the Notch ligand, delta-like 4 (DLLl4), was downregulated in ME-LC, which correlated with the early formation of large number of capillary-like structures and the late formation of a vascular network. Recent studies have demonstrated that DLLl4 limits the angiogenic potential in developing blood vessels and the loss of DLL4 results in an arterial hyperbranching phenotype [40]. Interestingly, in contrast to HUVEC, only ME-LC cultured in SFO3 (stage III) and EGM-2 (stage IV) media were able to strongly enhance capillary tubes formation after 7 days. It has been shown that VEGF stimulation of HUVEC induces Dll4 expression, which reduced vessel sprout length in a 3D tubulogenesis assay, confirming that DLL4 signaling inhibits angiogenesis. DLL4 expression seems to acts as a switch blocking EC proliferation and allowing induction of a more mature differentiated phenotype [41].
Several studies have identified several molecules such as VEGF and SDF-1 as key regulators of the proliferation, chemotaxis toward ischemic tissues, and differentiation of EPC [42]. hASC secrete multiple potentially synergistic proangiogenic growth factors, including VEGF, HGF, and chemokine SDF-1, which are likely to play a pivotal role for the hASC-mediated angiogenesis [43]. In fact, SDF-1 has been shown to enhance neovascularization by accelerating EPC recruitment into ischemic foci [42]. In our study, ME-LC showed an increased secretion of SDF-1 in comparison with hASC, even when they were seeded into Matrigel™. In addition, the role of SDF-1 to induce migration of CD133+/CD34+/KDR+ cells has been shown [30]. In the present study, we demonstrate how the ME-LC, which express the SDF-1 receptor, CXCR4, were able to migrate toward an SDF-1 gradient. These data suggest the potential homing of ME-LC to sites of vascular injury for tissue repair.
Most importantly, we show that after exposure to 5-azacytidine ME-LC retained the capacity to differentiate into cardiomyocyte-like cells with the acquisition of a cardiogenic phenotype and the expression of cardiomyocyte-specific markers. Morphological changes consisted of the appearance of wide, branching, and multinucleated cells resembling cardiac muscle cells [44]. The expression of cardiomyocyte markers such as troponin-T and α-sarcomericactinin [44] and the presence of a desmin filaments network, which take part in regulating the mesodermal specification into cardiomyocytes [45], support the cardiomyocyte differentiation from ME-LC.
In summary, our studies indicate that subcutaneous adipose tissue may be a useful source of autologous ME-LC with the capacity to maintain their vascular progenitor properties. The culture method described in the present study may be used to isolate, maintain, and propagate these cells with an increased expression of specific endothelial progenitor markers. This methodology could be applied to MSCs from other origins such as BM-derived stromal/stem cells. However, the interest of our study was the use of hASC isolated from liposuction, which is a less invasive method than BM aspiration and allows the collection of a high rate of progenitor cells. This can overcome the limited proliferation potential of mature ECs and EPC, which hampers their clinical use. ME-LC increased the secretion of SDF-1, formed vascular-like structures, and displayed the ability to migrate toward a cytokine gradient. Moreover, ME-LC retained the capacity to differentiate into cardiomyocyte-like cells, showing expression of typical cardiomyocyte markers. This property suggests the potential of ME-LC to recellularize damaged tissue or strengthen the post-infarct scar as well as inducing neovascularization of the affected area, which could have advantages in the stem cell-based cardiovascular therapy. It has been demonstrated the ability of cardiac stem cells to differentiate into the ECs, contributing to neovascularization in the process of tissue remodeling and/or regeneration [46]. Furthermore, recent studies showed interest in the cardiac and endothelial capacity of stem cell, providing important tools for the study of differentiation in vitro and future stem cell therapy for ischemic cardiomyopathy [47,48]. Future studies should explore further the functional in vitro and in vivo mechanisms of ME-LC in cardiovascular diseases along with their potential therapeutic impact, and our experimental data suggest that these cells may prove to be a valuable tool for vascular and cardiac repair.
Funding
This work was supported in part by grants from the Instituto de Salud Carlos III (Fondo de Investigación Sanitaria grant number PI10/02295), the Consejería de Salud (Junta de Andalucía grant number PI-0384/2008), the Consejeria de Economía, Innovación y Ciencia (Junta de Andalucía excellence project number CTS-6568) and the University of Granada (GREIB translational project number GREIB.PT_2010_09). Research in P.M.'s Lab was supported by grants from the Instituto de Salud Carlos III (grant number PI070026), Ministry of Science and Innovation (MICINN; grant number PLE-2009-0111), and by the Junta de Andalucía (grant number CICE-P08-CTS-3678). C.B. is supported by the ISCIII-FIS (grant number CP07/00059).
Footnotes
Acknowledgments
We gratefully acknowledge Manuela Expósito from FIBAO for excellent technical assistance with statistical studies. We also thank Emma Gutierrez González for helping in the artwork.
Author Disclosure Statement
No competing financial interests exist.