
Abstract
Fibroblast growth factor (FGF) signaling is implicated in the control of pluripotency and lineage differentiation of both human and mouse embryonic stem cells (mESCs). FGF4 dependent stimulation of ERK1/2 signaling triggers transition of pluripotent ESCs from self-renewal and lineage commitment. In this study, Sprouty 1 (Spry1) expression was observed in undifferentiated mESCs, where it modulated ERK1/2 activity. Spry1 was confirmed as dispensable for the maintenance of self-renewal. However, suppression of Spry1 expression and subsequent activation of ERK1/2 signaling promoted neural differentiation and inhibited endothelial differentiation of mESCs. Moreover, evidence is presented which indicates that SHP2, a major determinant of balance between mESC self-renewal and differentiation, directly regulates Spry1 activity to modulate ERK1/2 signaling and lineage-specific differentiation in mESCs. Our results show that Spry1 has an essential role in the lineage specific differentiation of mESCs.
Introduction
T
Autocrine fibroblast growth factor 4 (FGF4)-induced ERK1/2 signaling also occurs in mESCs, instructing them to exit the ground state of self-renewal and initiate differentiation [4,5]. Notably, LIF-stimulated phospho-Stat3 activity is finely tuned with the phospho-ERK pathway in the ESC state via Zata-chain associated protein kinase-70, SHP1, and SHP2 to maintain the stemness of ESCs [6 –9]. Cell fate decision between self-renewal and differentiation is determined in stem cells by the balance between Jak/Stat3 and ERK1/2 signaling. When the ERK1/2 pathway is stimulated by extrinsic signals (eg, cytokines and growth factors) and intrinsic transcription factors, the self-renewal state of ESCs is disturbed, and ESCs initiate their differentiation by exiting from the self-renewal program [5,10]. Thus, the role of the ERK cascade may be to direct transition of mESCs into a state that is responsive to external differentiation stimuli.
These findings imply that the differentiation of ESCs is not a default pathway, but is actively instructed by the signals of the ERK1/2 pathway. Therefore, ESCs require an intricate regulatory network to maintain proper level of pluripotent differentiation capacity while they proliferate as self-renewing cells. FGF4 is produced by mESCs and can activate FGF/ERK signaling in an autocrine manner. FGF signaling activates the ERK1/2 pathway during mESC neuronal differentiation, and inhibition of either FGFR or ERK1/2 abolishes neuronal induction. The pro-differentiation autocrine FGF4/ERK signal is counterbalanced in the self-renewal state by the Jak/Stat3 pathway. However, the underlying mechanism regulating ERK1/2 activity in mESCs remains obscure.
The Sprouty (Spry) family of proteins is a modulator of receptor tyrosine kinase (RTK) signaling. Mammalian cells express at least 4 Sprouty isoforms (Spry1-4) whose activity is growth-factor and cell-context dependent [11,12]. Spry modulates RTK signaling mainly by repressing pathways that lead to ERK activation [12 –16]. The contribution of Spry proteins to the control of signaling pathways in a variety of cell types, including terminally differentiated cells, adult stem cells, and various cancer cells, is well established [17 –21]. Although ERK1/2 signaling is an essential regulator to maintain the pluripotency and initiate the differentiation of the cells, the expression and function of Spry proteins in ESCs has not been reported.
In this article, we report that Spry1 is constitutively expressed in mESCs and functions as a suppressor of the ERK1/2 signaling cascade until cells initiate differentiation. In addition, Spry1 activity in mESCs is regulated by SHP2 phosphatase, whose activity is known to balance the Jak1/Stat3 and ERK pathway in the self-renewal state of mESCs.
Materials and Methods
Reagents and cell culture
J1 mESCs (cat #SCRC-1010) were purchased from ATCC (
Genetic modification of mESCs
Spry1-overexpressing mES cell lines were generated by chromosomal integration of a Spry1 expression plasmid, which was constructed by cloning PCR-amplified Spry1 cDNA into pcDNA3.1 vectors (Invitrogen). shRNA plasmids targeting mouse Spry1 were purchased (RMM3981-97073145, RMM3981-98494969; Open Biosystems) to generate a stable Spry1-knocked-down cell line. Shp2 expression plasmid was constructed as previously described [8]. Nonspecific control siRNAs were purchased from Bioneer, and siRNAs targeting Spry1 were purchased from Dharmacom. mESCs were transfected with either siRNA or plasmid using lipofectamine 2000 (Invitrogen) following the manufacturer's instructions.
RNA extraction and real-time RT-PCR
Total RNA was extracted using TRIzol (Invitrogen), and 2–5 μg of total RNA was reverse-transcribed using the SuperScriptII™ First-Strand Synthesis System (Invitrogen) according to the manufacturer's instructions. Real-time RT-PCR was carried out using cDNAs with the Quantitect SYBR Green PCR kit (Qiagen). Reactions were carried out in triplicate using Exicycler™ 96 real-time quantitative thermal block (Bioneer). For quantification, target genes were normalized against the glyceraldehyde 3-phosphate dehydrogenase gene (Gapdh).
Immunoblotting
Immunoblotting was performed as previously described [23,24. Briefly, cells were washed twice with cold phosphate buffered saline, lysed with tissue lysis buffer (20 mM Tris-base, pH 7.4, 137 mM NaCl, 2 mM EDTA, 1% Triton X-100, 25 mM β-glycerophosphate, 2 mM sodium pyrophosphate, 10% glycerol, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, and 1 mM benzamidine), and centrifuged at 20,000×g to clarify the lysates. Whole-cell extracts were prepared, and 2,050 μg of protein was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and blotted using antibodies against Oct4 (sc-9081; Santa Cruz), Nanog (sc-30328; Santa Cruz), phospho-Stat3 (Tyr-705) (#9131; Cell Signaling Technology), nestin (sc-33677; Santa Cruz), VE-cadherin (sc-9989; Santa Cruz), Sox2 (sc-20088; Santa Cruz), Spry1 (sc-30048; Santa Cruz), phospho-ERK (M9692; Sigma), ERK2 (sc-154; Santa Cruz), and SHP2 (sc-280; Santa Cruz). Immunoreactivity was detected by enhanced chemiluminescence (Amersham).
Immunofluorescence staining
mESCs were immunostained as previously described [25]. Rabbit anti-mouse Oct4, nestin, or VE-cadherin antibody at 1:200 dilution and Alexa Fluor 488- (A21206; Molecular Probes) or Alexa Fluor 594-labeled anti-rabbit IgG secondary antibodies (A21207; Molecular Probes) at 1:300 dilution were used to detect Oct4, nestin, or VE-cadherin in the cells.
Teratoma formation
For teratoma formation assay, cells were trypsinized, and 5×105 cells were suspended in a DMEM/Matrigel solution (BD Biosciences Inc.) (1:1 ratio (v/v)). The cell/Matrigel suspension was then injected subcutaneously into NOD/SCID mice (Charles River Laboratories). Teratoma formation was examined for 6 weeks after the injection. The experiments were reviewed and approved by the Institutional Animal Care and Use Committee of CHA University. All procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85–23, revised 1996).
Results and Discussion
Spry1 regulates ERK1/2 phosphorylation in mESCs but does not affect self-renewal of mESCs
Counterbalancing the pro-differentiation autocrine FGF/ERK signal is crucial for mESCs to maintain their self-renewal and pluripotent nature. Therefore, FGF signaling should be tightly regulated at multiple levels in ESCs. Since the Spry family of proteins is a highly conserved group of negative feedback loop modulators of growth factor-mediated MAPK signals [24,26], we first examined the gene expression pattern of Spry1, 2, and 4, which are widespread in embryos and adults [11,27]. As shown in Fig. 1A, Spry1, 2, and 4 were expressed at different levels in the examined cells. Interestingly, the expression level of Spry1 was higher in differentiated ESCs (Fig. 1A). This result suggests the possibility that the role of Spry1 is related to the control of differentiation of mESC.
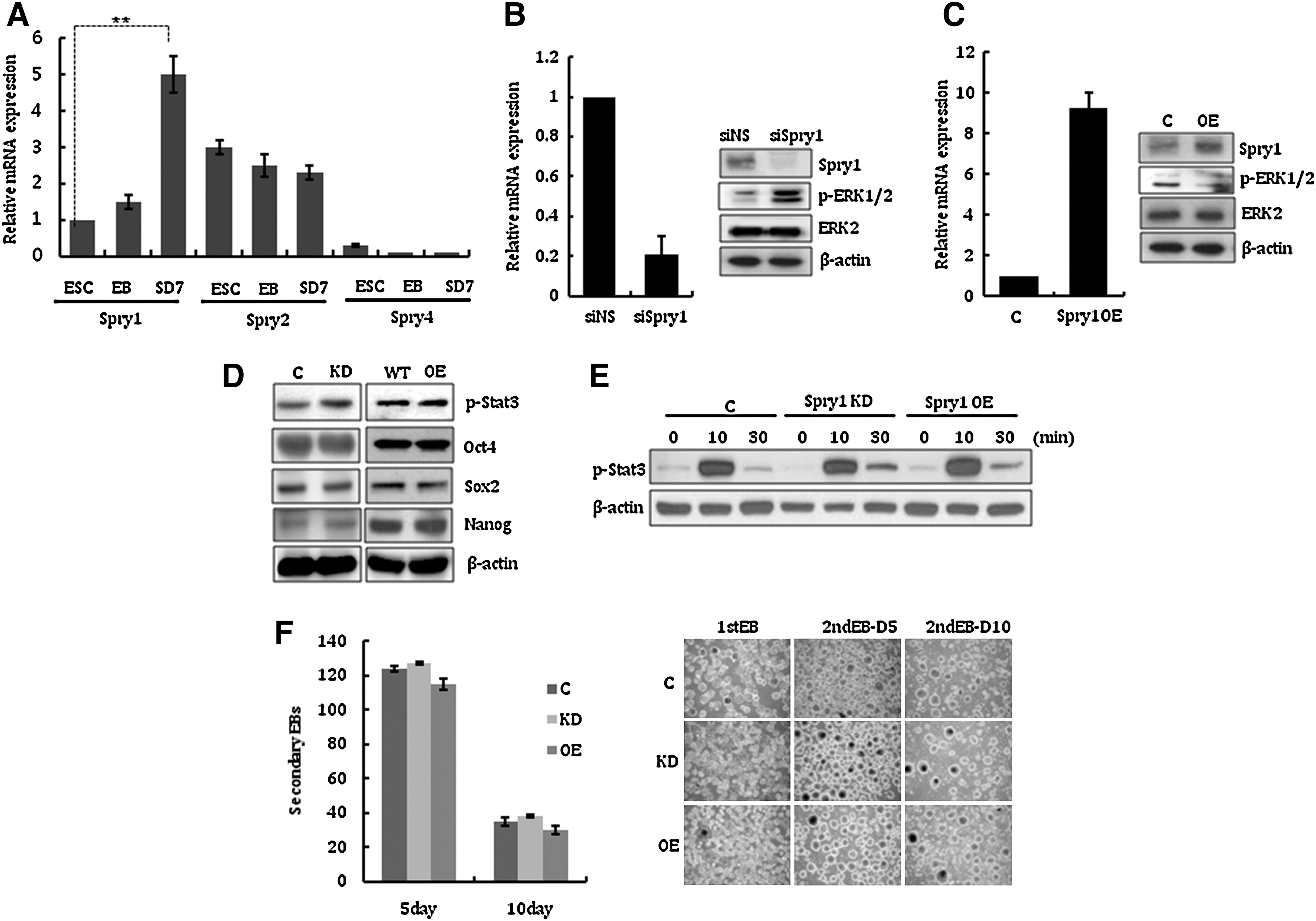
Spry1 expression in undifferentiated mESCs and its effect on their self-renewal.
To investigate the potential function of Spry1 in mESCs, we first examined whether Spry1 modulates the phosphorylation of ERK1/2 in mESCs. As expected, transfection of mESCs with siRNA targeting Spry1 induced phosphorylation of ERK1/2, which suggests that Spry1 functions to suppress the activity of ERK1/2 signals in mESCs (Fig. 1B). Consistently, phospho-ERK1/2 levels in mESCs were suppressed when Spry1 was overexpressed by the transfection of a Spry1 expression plasmid (Fig. 1C). We next generated stable mESC lines in which Spry1 expression was knocked down (Spry1 KD) or overexpressed (Spry1 OE). Using a set of Spry1 shRNA plasmids, 2 mESC clones (KD4 and KD5) were successfully established. In these clones, Spry1 expression was suppressed by ∼80% compared with control mESCs, and phospho-ERK1/2 level was subsequently increased (Supplementary Fig. S1; Supplementary Data are available online at
To examine whether altered Spry1 expression affects mESC self-renewal, we examined the expression of undifferentiated ESC markers p-Stat3, Oct4, Sox2, and Nanog. Figure 1D shows that suppression or overexpression of Spry1 does not disturb the expression of self-renewal markers. Consistent with this result, the phosphorylation status of Stat3 in LIF-stimulated mESCs, which had been starved of LIF and FBS serum for 24 h, was similar between Spry1-modulated mESCs and control mESCs (Fig. 1E). The efficiency of secondary EB formation, which reflects the ability of mESCs to maintain an undifferentiated state [28], was comparable, regardless of Spry1 expression level (Fig. 1F). However, when EBs were spontaneously differentiated by RA, Spry1 KD EBs differentiated rapidly compared with control, whereas Spry1 OE EBs could not differentiate properly (Supplementary Fig. S2). These results suggest that Spry1 functions as a counterbalance to suppress pro-differentiating signals in mESCs under self-renewal culture conditions, although it is dispensable to maintain self-renewal of undifferentiated mESCs.
Spry1 modulates neural and endothelial differentiation of mESCs
When the ERK1/2 signaling pathway is stimulated by FGF4, mESCs exit from the self-renewal state and initiate lineage commitment [5]. Since ERK signals should be activated to commit neural induction of mESCs [5], we reasoned that Spry1 expression level would change on mESC neural differentiation. The expression of Spry1 was first examined in mESCs undergoing neural differentiation. As expected, Spry1 expression decreased when mESCs were cultured in neural induction media (Fig. 2A). Compared with control cells, Spry1 KD mESCs produced a large number of nestin-positive and Oct4-negative neural precursors by day 5 (Fig. 2B). A selective inhibitor of ERK1/2, U0126, blocked the enhanced neural differentiation capacity of Spry1 KD cells, which suggested that Spry1 regulated neural differentiation of mESCs mainly via the ERK1/2 signaling pathway. Quantitative analysis of nestin mRNA and immunoblot analysis of nestin and Oct4 protein at day 5 after neural induction confirmed the immunocytochemical staining results (Fig. 2C). In contrast to Spry1 KD, the majority of Spry1 OE cells did not express nestin but retained expression of the pluripotent marker Oct4 under neural differentiation conditions (Fig. 2D). These Oct4-positive cells remained viable and proliferative, similar to those under mESC culture conditions, for at least 7 days (data not shown), indicating that Spry1 overexpression blocks mESCs from entering neural lineage commitment. Nestin mRNA and protein expression patterns at day 5 after neural induction were consistent with immunocytochemical staining results (Fig. 2E).
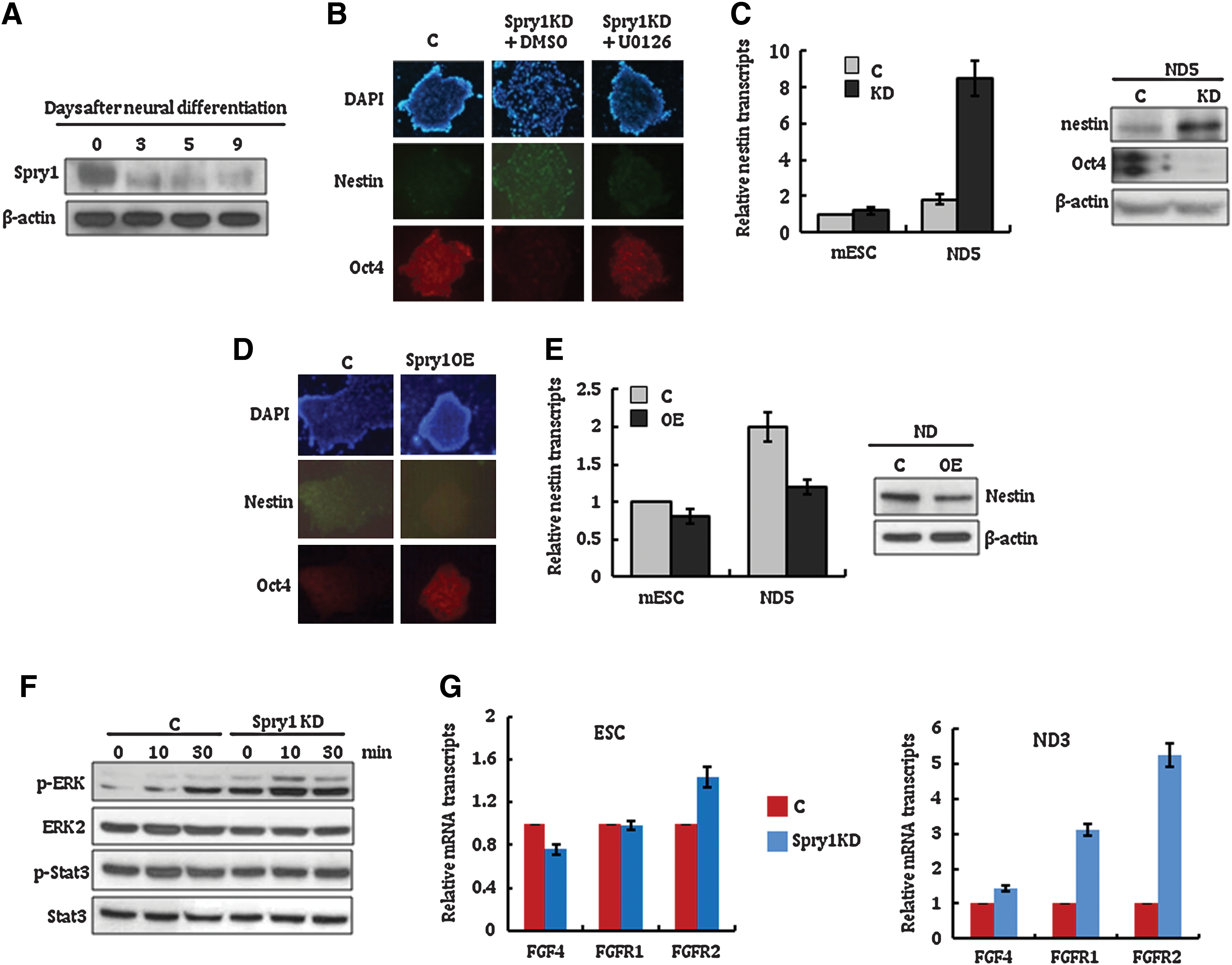
Suppression of Spry1 stimulates neural differentiation of mESCs.
To understand the underlying mechanism of Spry1 activity during the neural differentiation of mESCs, the FGF4 responsiveness of Spry1 KD mESCs was then examined. As shown in Fig. 2F, LIF-starved Spry1 KD cells rapidly phosphorylated ERK1/2 on basic FGF stimulation compared with control mESCs, whereas phosphorylation of Stat3 was comparable between the cells. This result suggests that suppression of Spry1 induces an acute mESC response to the basic FGF stimulus, which leads mESCs to rapidly transition from self-renewal to neural commitment. Interestingly, FGF receptor-1 and receptor-2 expression levels increased significantly in Spry1 KD mESCs at 3 days after neural differentiation conditions (Fig. 2G). The increased expression of FGF receptor-1 and receptor-2 in Spry1 KD was sustained for 7 days after neural differentiation of mESCs (Supplementary Fig. S3). This finding implies that transcription regulation of FGF receptors is one of the mechanisms by which Spry1 modulates the ERK1/2 pathway in mESCs. Understanding the underlying mechanism used by Spry1 to modulate FGF receptors will greatly expand our knowledge of the regulatory cascade of the ERK1/2 signaling pathway in mESCs.
To investigate the effect of Spry1 on other lineage differentiation, the expression of Spry1 was analyzed during endothelial cell differentiation. Contrary to neural differentiation, Spry1 expression was noticeably increased 3 days after endothelial cell differentiation (Fig. 3A). Consistently, phosphorylation of ERK1/2, which is inhibited by the Spry1, was significantly decreased (Fig. 3A). Self-renewal factors such as Sox2, phospho-Stat3, and Stat3 level was decreased as endothelial differentiation of mESCs proceeded, which suggests that the expression of self-renewal factors and Spry1 is independently regulated (Supplementary Fig. S4).
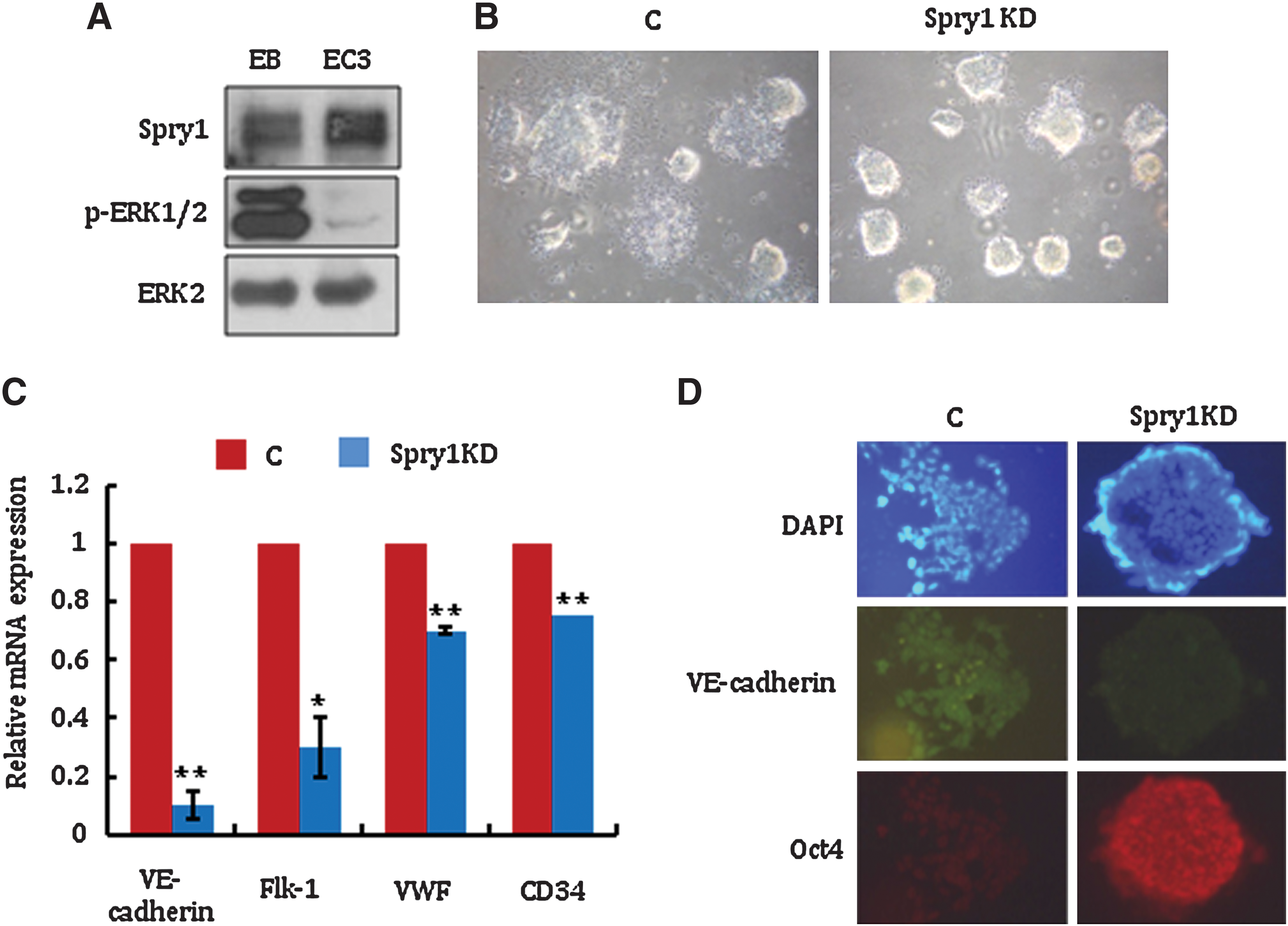
Suppression of Spry1 inhibits endothelial differentiation of mESCs.
Interestingly, Spry1 KD mESCs sustained EB morphology in endothelial cell differentiation culture media (Fig. 3B). The expression level of endothelial markers such as VE-cadherin, Flk-1, von Willebrand factor, and CD34 was significantly lower in Spry1 KD compared with control cells, which supported the defective endothelial differentiation of Spry1 KD cells (Fig. 3C).
To further confirm the effect of Spry1 on mESC endothelial cell differentiation, VE-cadherin expression was compared in control and Spry1 KD cells by immunocytochemical staining. As shown in Fig. 3D, most Spry1 KD cells were Oct4 positive and VE-cadherin negative, whereas control cells were VE-cadherin positive and Oct4 negative in endothelial differentiation media. Interestingly, addition of U0126 to the endothelial differentiation media did not restore the differentiation capacity of Spry1 KD cells (data not shown). Unlike neural differentiation, endothelial differentiation regulated by Spry1 appears to be independent of ERK1/2 signaling. Either FGF receptor-1 or receptor-2 expression levels were not changed significantly in Spry1 KD mESCs under endothelial cell differentiation conditions (Data not shown). This result further supports that FGF-ERK1/2 pathway does not play a major role in the Spry1-mediated endothelial differentiation of mESCs. The molecular mechanism by which Spry1 regulates mESC endothelial differentiation remains unknown. This altered differentiation capacity of Spry1 KD or OE cells was further analyzed by comparing teratoma formation. Consistent with in vitro assay, normal teratoma were well developed from control mESCs in all 6 SCID mice, whereas small-sized teratoma was formed in only one out of 6 SCID mice after 6 weeks of injection of Spry1 KD or OE mESCs (data not shown). Taken together, these observations suggest that Spry1 regulates neural and endothelial lineage commitment during mESC differentiation.
Spry1-mediated modulation of ERK1/2 activity is regulated by SHP2 in mESCs
We next investigated how Spry1 activity is regulated in mESCs. In cultured Drosophila cells and the developing eye, SHP2, a cytoplasmic tyrosine phosphatase, associates in a complex with Spry1, and purified SHP2 protein dephosphorylates the tyrosine residue critical for Spry1 activity [29]. Further, SHP2 functions as a molecular switch to determine mESC fate between self-renewal and differentiation, mainly through bidirectional modulation of ERK and Stat3 signals [9,28,30]. Therefore, we hypothesized that ERK signal is regulated in mESCs by a SHP2-Spry1 double-negative feedback circuit. To determine the regulatory relationship between SHP2 and Spry1 during ERK signaling, the effect of SHP2 on the suppressive activity of Spry1 in ERK1/2 phosphorylation was analyzed in mESCs. If Spry1 is a substrate of SHP2, then overexpression of Shp2 should rescue the suppression of ERK1/2 phosphorylation caused by Spry1. As expected, phosphorylation of ERK1/2, which was decreased by Spry1 transfection, was increased by the cotransfection of Spry1 and Shp2 (Fig. 4A). When control and Spry1-overexpressing mESCs were transfected with Shp2 expression plasmid and then starved of LIF and FBS, FBS stimulation induced a rapid increase of ERK1 phosphorylation in both control and Spry1 OE cells (Fig. 4B). We next tested whether Spry1 interacts with SHP2 in mESCs using coimmunoprecipitation analysis, and confirmed that Spry1 was associated with SHP2 in mESCs (Fig. 4C).
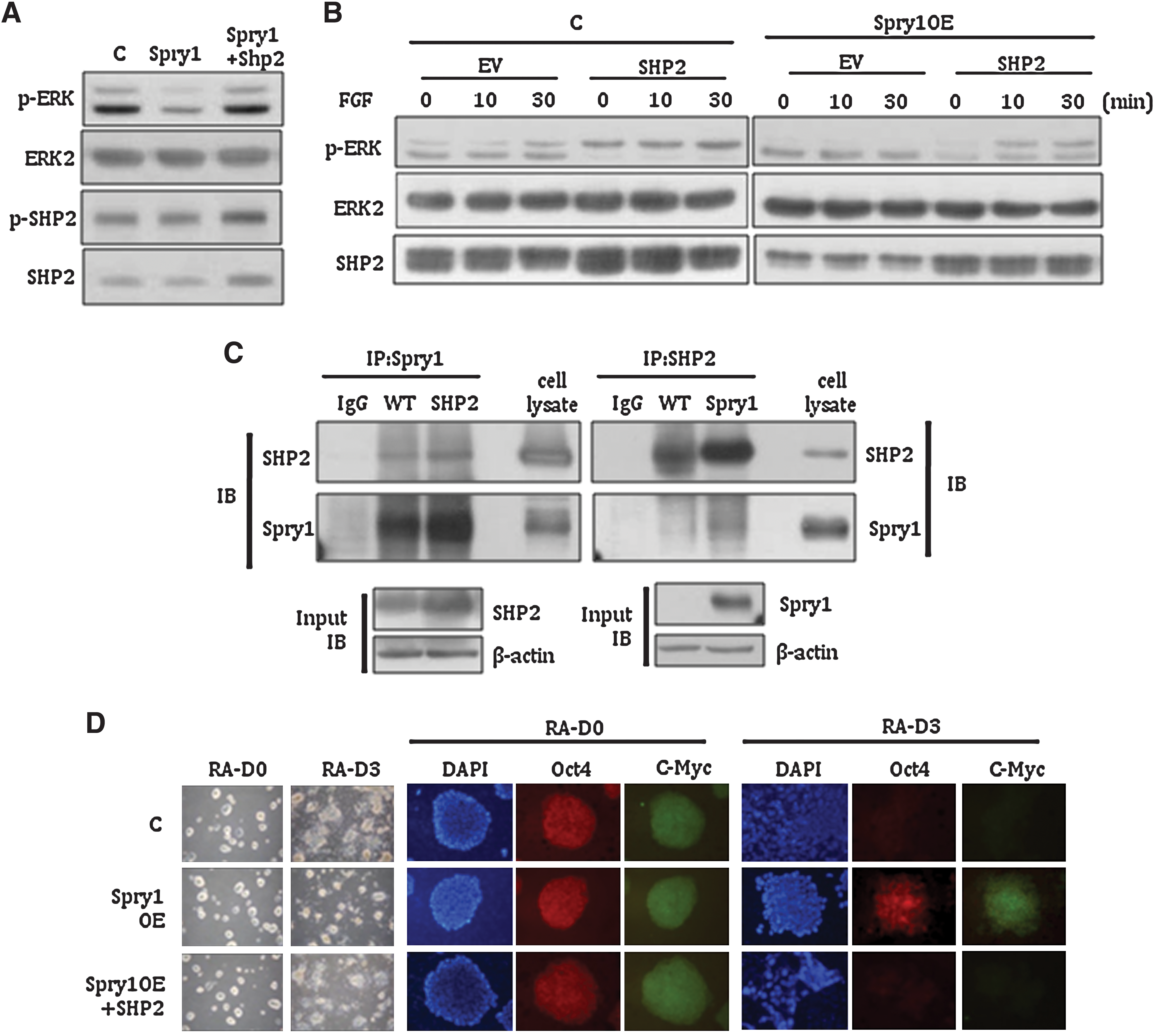
Spry1-mediated modulation of ERK1/2 activity is regulated by SHP2 in mESCs.
Finally, we examined whether the defective differentiation capacity of Spry1-overexpressing mESCs is rescued by Shp2 overexpression. Spry1 OE mESCs transfected with Shp2 expression plasmid differentiated comparably to control cells under spontaneous differentiation conditions (Fig. 4D). Consistently, immunocytochemical analysis of Oct4 expression suggested that the sustained self-renewal capacity of Spry1 OE mESCs under differentiation culture conditions was switched to a pro-differentiation program by the overexpression of Shp2 (Fig. 4D, right panel).
SHP2 is known to promote ESC differentiation mainly through bi-directional modulation of ERK and Stat3 pathways, which suggests that mESCs maintain pluripotency through a finely tuned balance of positive and negative effectors. Our study suggests that Spry1 activity is regulated by SHP2, possibly through direct interactions between the 2 proteins. Unlike SHP2, however, the modulation of Spry1 expression does not affect the self-renewal capacity of mESCs. This finding implies that SHP2 regulates ERK1/2 signaling through multiple pathways that may or may not be interconnected with self-renewal. Our results suggest that Spry1-mediated transfer of SHP2 signal to the ERK1/2 pathway and self-renewal mechanism are operated independently.
In summary, this study reveals a role for a known negative regulator of ERK1/2 in mESC lineage differentiation. The identification of Spry1 as a mediator of SHP2 and ERK1/2 signals provides definite insights into how the differentiation of ESCs is orchestrated. Improved understanding of the role played by Spry1 in lineage-specific differentiation also opens up additional avenues for understanding the pluripotent nature of ESCs.
Footnotes
Acknowledgments
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korean government (MOST) (2010-0020566, 2010-0003254 and 2010-0016803).
Author Disclosure Statement
No competing financial interest exists.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.