
Abstract
Mesenchymal stromal cells derived from the human amnion (hAMSC) currently play an important role in stem cell research, as they are multipotent cells that can be isolated using noninvasive methods and are immunologically tolerated in vivo. The objective of this study was to evaluate their endothelial differentiation potential with regard to a possible therapeutic use in vascular diseases. hAMSC were isolated from human term placentas and cultured in Dulbecco's modified Eagle's medium (DMEM) (non-induced hAMSC) or endothelial growth medium (EGM-2) (induced hAMSC). Induced hAMSC changed their fibroblast-like toward an endothelial-like morphology, and were able to take up acetylated low-density lipoprotein and form endothelial-like networks in the Matrigel assay. However, they did not express the mature endothelial cell markers von Willebrand factor and vascular endothelial-cadherin. Gene expression analysis revealed that induced hAMSC significantly downregulated pro-angiogenic genes such as tenascin C, Tie-2, vascular endothelial growth factor A (VEGF-A), CD146, and fibroblast growth factor 2 (FGF-2), whereas they significantly upregulated anti-angiogenic genes such as serpinF1, sprouty1, and angioarrestin. Analysis of protein expression confirmed the downregulation of FGF-2 and Tie-2 (27%±8% and 13%±1% of non-induced cells, respectively) and upregulation of the anti-angiogenic protein endostatin (226%±4%). Conditioned media collected from hAMSC enhanced viability of endothelial cells and had a stabilizing effect on endothelial network formation as shown by lactate dehydrogenase and Matrigel assay, respectively. In summary, endothelial induced hAMSC acquired some angiogenic properties but resisted undergoing a complete differentiation into mature endothelial cells by upregulation of anti-angiogenic factors. Nevertheless, they had a survival-enhancing effect on endothelial cells that might be useful in a variety of cell therapy or tissue-engineering approaches.
Introduction
M
The amnion is an especially promising source of cells for therapeutic use, as its feasibility in clinical applications has already been confirmed. Its first documented clinical use goes back to 1910, where it was applied as a surgical material in skin transplantation [12]. Since then, it has been used in a variety of clinical settings such as treatment of chemical burns, skin ulcers, and ophthalmology. Its beneficial effects are awarded to its anti-inflammatory, immunomodulatory, and scar formation-reducing properties, among others [13].
MSC isolated from the human amnionic membrane (hAMSC) have phenotypic and functional similarities to bone marrow-derived MSC. They have been successfully differentiated toward cells of the classical mesodermal (osteogenic, adipogenic, and chondrogenic), ectodermal (neurogenic), and endodermal (hepatogenic, pancreatic) lineages [10]. Similar to bone marrow derived MSC, they actively suppress T-lymphocyte proliferation [14 –16] and block differentiation and maturation of monocytes into dendritic cells in vitro [17]. After xenogeneic transplantation into neonatal swine and rats, hAMSC engraft without immunosuppression [18]. Very recently, it was shown that amnionic membrane application could reduce liver fibrosis in a bile duct ligation rat model [19] and improve cardiac function of ischemic rat hearts [20]. In addition, isolated allo- and xenogenic hAMSC could reduce bleomycin-induced lung fibrosis in a mouse model [21].
These studies suggest that hAMSC hold great promise for a potential use in cell therapy and tissue engineering. One of the major challenges in this field lies in the vascularization of engineered tissues and in finding a suitable cell population for the endothelialization of vascular grafts. MSC might be an interesting choice; however, their endothelial differentiation potential is still controversial. Although some groups have reported the differentiation of MSC originating from bone marrow or adipose tissue into endothelial-like cells [22,23], opposite results showed that MSC from bone marrow could not be differentiated toward the endothelial lineage [24,25]. Since the bone marrow and adipose tissue are highly vascularized, it is difficult to obtain cultures free of primary endothelial cells. It might be possible that the isolated MSC populations already contained some endothelial or endothelial progenitor cells before endothelial induction in vitro. Thus, instead of inducing endothelial differentiation of MSC, the chosen angiogenic culture conditions might have led to a selective proliferation of already existing endothelial cells. Here, the amnion shows an additional advantage: Being avascular, it allows an easy isolation of MSC cultures free of endothelial cells.
Based on the current controversial reports, in this study, we tested the endothelial differentiation potential of hAMSC after excluding the presence of endothelial cells. In addition to applying phenotypic characterization and functional studies, we evaluated the effect of angiogenic culture conditions on gene and protein expression of hAMSC using microarray analysis and angiogenic protein arrays. Further, we examined the paracrine effects of hAMSC on the viability and network formation of endothelial cells.
Materials and Methods
Isolation and culture of hAMSC
Human term placentas of normal pregnancies (range 38 to 42 weeks) were obtained after spontaneous delivery or cesarean section with informed consent. Approval of the Ethical Committee of the Medical University of Graz was granted (No. 21-079 ex 09/10).
Isolation of hAMSC was performed according to the protocol of Soncini et al. [26]. The amnion was manually separated from the chorion and washed with sterile 0.9% saline (Fresenius Kabi, Bad Homburg, Germany) supplemented with 150 IU/mL penicillin, 150 μg/mL streptomycin (both from PAA Laboratories, Pasching, Austria), and 0.4 μg/mL amphotericin B (Gibco, Invitrogen, Paisley, UK). The amnion was cut into small pieces and incubated in 2.5 U/mL dispase (BD Biosciences, Bedford, MA) at 37°C for 9 min. Subsequently, the amnion was transferred to Dulbecco's modified Eagle's medium (DMEM) low glucose (Gibco, Invitrogen) supplemented with 15% fetal bovine serum (FBS) Gold (Gibco, Invitrogen), 100 IU/mL penicillin, and 100 μg/mL streptomycin for 10 min. Next, the amnion was incubated with 1.0 mg/mL Collagenase A and 0.01 mg/mL DNase (both from Roche, Penzberg, Germany) for 2 h. After centrifugation for 3 min at 150 g, the supernatant was poured over a 100 μm cell strainer (BD Biosciences) and centrifuged for 10 min at 300 g. The cell pellet was washed in phosphate-buffered saline (PBS; Gibco, Invitrogen) and resuspended in either DMEM supplemented with 15% FBS, 100 IU/mL penicillin, and 100 μg/mL streptomycin (non-induced hAMSC) or endothelial growth medium-2 (EGM-2; Lonza, Walkersville, MD) for endothelial induction (induced hAMSC). EGM-2 contains 2% FBS, epidermal growth factor (EGF), hydrocortisone, vascular endothelial growth factor (VEGF), fibroblast-like growth factor-2 (FGF-2), insulin-like growth factor 1 (IGF-1), ascorbic acid, and heparin. Aliquots of the cell suspensions were spun down on slides immediately after isolation (cytospins). Remaining cells were grown on culture flasks coated with 1% gelatin (Sigma-Aldrich, St. Louis, MO) and harvested with accutase (PAA Laboratories). Medium was changed every 2–3 days.
Isolation and culture of placental endothelial cells
Endothelial cells from normal term human placentas were isolated as published earlier [27]. Briefly, after removal of the amnion, arterial chorionic blood vessels at the apical surface of the chorionic plate were resected. Vessels were washed with Hank's balanced salt solution (HBSS, Gibco) to remove residual blood. Endothelial cells were isolated by perfusion of vessels with HBSS containing 0.1 U/mL collagenase, 0.8 U/mL dispase (both from Roche), supplemented with 300 IU/mL penicillin, and 300 μg/mL streptomycin, prewarmed to 37°C. The perfusion time was limited to 7 min to avoid contamination with non-endothelial cells. The cell suspension was centrifuged (200 g for 5 min), the pellet was resuspended with EGM-MV medium (Lonza), and the cells were plated on culture plates pre-coated with 1% gelatin. The endothelial identity was confirmed by positive staining for the classical endothelial marker von Willebrand factor (vWF; immunoglobulin fraction, rabbit anti-human, 0.7 μg/mL; Dako, Glostrup, Denmark) and absence of markers against fibroblasts (CD90, clone ASO2, 0.1 μg/mL, mouse IgG1; Dianova, Hamburg, Germany), and smooth muscle cells (smooth muscle actin, clone 1A4, 0.2 μg/mL, mouse IgG2a and desmin, clone D33, 0.4 μg/mL, mouse IgG1, both from Dako).
Flow cytometry analysis
Flow cytometry analyses were carried out at the Flow Cytometry Core Facility at the Center for Medical Research (ZMF) and at the Institute of Cell Biology, Histology, and Embryology of the Medical University of Graz. The surface marker expression of hAMSC was analyzed with a FACSLSRII® instrument equipped with 355 and 405 nm UV lazers, a 488 nm argon ion lazer, and a 635 nm red diode lazer (Becton Dickinson, Franklin Lakes, NJ). hAMSC were washed and labeled for 30 min at 4°C at concentrations according to individual titration with monoclonal antibodies against CD105 (PE-labeled, clone 2H6F11, 1:33; Caltag Laboratories, Burlingame, CA), CD14 (FITC-labeled, clone MOP9, 1:100), CD34 (APC-labeled, clone 8G12, 1:100), CD44 (PE-labeled, clone 515, 1:33), CD45 (PE-Cy7-labeled, clone HI30, 1:100), CD73 (PE-labeled, clone AD2, 1:20), CD90 (APC-labeled, clone 5E10, 1:100), and HLA-DR (PerCP-labeled, clone 243 (G46-6), 1:100, all from BD Biosciences). Appropriate isotype-matched antibodies were used as negative controls (BD). Data from 10,000 viable cells were acquired. List mode files were analyzed with FCS Express Software (BD).
Immunohistochemistry/immunocytochemistry
Cryosections (5 μm) of human term placental samples were mounted on microslides (Assistent, Karl Hecht AG, Sondheim, Germany). hAMSC were grown on gelatin-coated glass chamber slides (Lab-Tek II, Nalgene Nunc International, Naperville, IL) and washed with PBS before being harvested. Cryosections and cells on cytospins and chamber slides were air-dried for at least 4 h and stored frozen. Before immunostaining, tissue and cells were fixed in acetone for 4 min. Slides were immunolabeled using the UltraVision LP Detection System (Thermo Scientific, Fremont, CA) according to the manufacturer's instructions. The following antibodies were diluted in antibody diluent (Dako) and applied for 30 min at room temperature: vWF (immunoglobulin fraction, rabbit anti-human, 0.7 μg/mL, Dako), vascular endothelial-cadherin (VE-cadherin; clone F-8, 0.33 μg/mL, mouse IgG1; Santa Cruz Biotechnology, Santa Cruz, CA), and VEGF receptor-2 (VEGFR-2; clone FLT-19, 10 μg/mL, mouse IgG1; Sigma-Aldrich). IgG controls and normal rabbit immunoglobulin fraction control (Dako) for vWF were used in the same concentrations as the respective antibodies. After 3 washing steps in PBS, slides were incubated with primary antibody enhancer for 10 min, followed by horseradish peroxidase-polymer for 15 min. The slides were washed again thrice in PBS, and immunolabeling was visualized by a 5 min exposure to 3-amino-9-ethylcarbacole (all from UltraVision kit, Thermo Scientific). The slides were counterstained with Mayer's hematoxylin (Merck, Darmstadt, Germany), washed in distilled water, and mounted with Kaiser's glycerol gelatin (Merck).
Proliferation of hAMSC
hAMSC were either cultured under standard conditions in DMEM supplemented with 15% FBS on gelatin coated plates or under different endothelial induction conditions, consisting of culture in EGM-2 in the absence or presence of 50 ng/mL VEGF (VEGF165; ReliaTech, Wolfenbuettel, Germany), on gelatin, or fibronectin (1 μg/cm2; R&D Systems, Minneapolis, MN) coated plates. Cells were seeded in triplicate with a density of 104 cells/well in 6-well plates and harvested after 5 days. Cells were counted by automatic cell counting (Casy® Model TT; Schärfe Systems, Reutlingen, Germany), reseeded in a density of 104 cells/well, and again harvested after 4 d. Cumulative population doublings were calculated according to the formula (lnN−lnN 0)/ln2, with N=amount of harvested cells and N 0=amount of seeded cells. For the determination of the cumulative population doublings over several passages, cells were harvested at 90% confluence.
DiI-Ac-LDL-uptake assay
hAMSC were seeded on gelatin-coated glass chamber slides and cultured in DMEM with 15% FBS (non-induced hAMSC) or in EGM-2 (induced hAMSC) for at least 7 days. Subsequently, cells were incubated with 10 μg/mL acetylated low-density lipoprotein labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindo-carbocyanine perchlorate (DiI-Ac-LDL; BTI Biomedical Technologies, Stoughton, MA) for 4 h according to the manufacturer's instructions. Cells were fixed with 4% paraformaldehyde, stained with DAPI (1:2000; Invitrogen, Eugene, OR), and observed with a Leica DM600B fluorescent microscope (Leica, Wetzlar, Germany) connected to an Olympus DP72 digital camera (Olympus, Tokyo, Japan). Placental endothelial cells (PlEC) served as positive controls.
Matrigel assay
hAMSC were cultured under standard (non-induced) or angiogenic (induced) conditions for at least 10 days. Then cells were seeded in a 96-well plate precoated with 40 μL Matrigel (BD Biosciences) according to the manufacturer's instructions at a density of 104 cells/well in 100 μL EGM-2. Cells were observed using a Cell-IQ Analyzer 2004-01 (Chip-Man Technologies, Tampere, Finland). PlEC served as positive controls. Videos were prepared with Cell-IQ Analyzer Pro-Write v.AN 2.0.1 (Chip-Man Technologies).
Design of microarray experiments and RNA isolation
hAMSC were isolated from 3 different placentas, and aliquots were cultured either in DMEM+15% FCS (non-induced hAMSC) or EGM-2 (induced hAMSC) on gelatin-coated culture flasks until cells reached 90% confluence (about 5–8 days). Then, the cells were reseeded and cultured in the respective media for 14 days before RNA isolation. Total RNA was isolated using RNeasy Mini Kit (Qiagen, Hilden, Germany). The integrity of each RNA sample was determined using an Agilent 2100 Bioanalyzer (Agilent, Foster City, CA), and only RNA samples with integrity values of 9.5–10 were used for hybridization.
Hybridization and data analysis of microarrays
Total RNA was labeled using the Affymetrix GeneChip® Whole Transcript Sense Target Labeling Assay and hybridized to GeneChip Human 1.0 ST arrays as described by the manufacturer (Affymetrix, Santa Clara, CA). Hybridizations were carried out at the Molecular Biology Core Facility at the Center of Medical Research at the Medical University of Graz. Briefly, 100 ng of total RNA were reverse transcribed to cDNA using random hexamers tagged with a T7 promoter sequence. Double-stranded cDNA was subsequently used as a template in an in vitro transcription reaction followed by cDNA synthesis, fragmentation, and labeling through a terminal deoxynucleotidyl transferase. The hybridization cocktail was incubated overnight at 45°C while rotating in a hybridization oven. After 16 h of hybridization, arrays were washed and stained in an Affymetrix GeneChip fluidics station 450, according to the Affymetrix-recommended protocol. Arrays were scanned on an Affymetrix GeneChip scanner. CEL files were imported into Partek Genomic Suite v6.4 software (Partek, Inc., St Louis, MO) and robust multi-chip average normalized (including background correction, quantile normalization across all arrays, median polished summarization based on log transformed expression values). For statistical analysis, a paired-sample t-test was performed between the treatment groups. Differentially expressed genes were selected by P<0.005 (heat map, Tables 1 and 2) or P<0.05 (Venn diagram) and a fold change (FC) ≥2. Expression of genes above the background was determined by a signal intensity level >5 after background correction.
FC, fold change.
FC, fold change.
The according data has been deposited in NCBI's Gene Expression Omnibus (GEO) [28] and is accessible through GEO Series accession number GSE28385 (
Protein isolation and angiogenic protein array analysis
hAMSC were isolated from 5 different placentas and either cultured in DMEM with 15% FBS (non-induced hAMSC) or in EGM-2 (induced hAMSC) on gelatin-coated culture flasks until cells reached 90% confluence (about 5–8 days). Then, the cells were reseeded and cultured in the respective media for 14 days. Before protein isolation, cells were washed twice in PBS and lysed with RIPA-buffer (Sigma-Aldrich) containing 4% complete protease inhibitor cocktail (Roche, Mannheim, Germany) for 5 min. Lysates were clarified by centrifugation at 300 g for 10 min at 4°C. Supernatant was used immediately or stored at −80°C for further analysis. Total protein concentration was determined by Lowry protein assay. Protein of 5 hAMSC isolations was pooled, and a total of 250 μg was applied to the Human Angiogenesis Antibody Array C1000 (RayBiotech, Norcross, GA). This array contains 43 different angiogenic proteins spotted in duplicates onto 2 membranes. Membranes were processed according to the manufacturer's instructions. Chemiluminescent imaging was performed using the FluorChemQ system, signal densities were analyzed with AlphaView software version 2.0.1.1 (both from AlphaInnotech, Cell Biosciences, Santa Clara, CA), and ratios of the respective protein and internal standard densities were determined. Protein expressions by induced hAMSC are presented as percentages of the expression of non-induced cells (set to 100%).
Preparation and analysis of hAMSC-conditioned media
To prepare hAMSC-conditioned medium (CdM), confluent non-induced and induced hAMSC were washed with PBS and then incubated with EGM-2 for 48 h. Control medium (EGM-2) was prepared in parallel in culture flasks without cells. On harvest, hAMSC-CdM and control medium were centrifuged at 300 g for 10 min and then stored at −80°C.
For determining the effect of hAMSC-CdM on endothelial cell viability, lactate dehydrogenase (LDH) activity was analyzed in supernatants of PlEC that were cultured in 96-well plates for 96 h either in CdM or in the respective control medium. LDH activity was measured using an LDH Cytotoxicity Detection Kit (Takara Bio, Inc., Shiga, Japan) according to the manufacturer's instructions. Experiments were performed with non-induced and induced hAMSC from 2 and 4 different isolations, respectively, using triplicates per experiment. Statistical analysis was performed using paired Student's t-test. Data were expressed as mean±standard deviation. P values of <0.001 were considered statistically significant.
To test the effect of hAMSC-CdM on network formation of endothelial cells, PlEC were resuspended in hAMSC-CdM or in control medium EGM-2 and cultured on Growth Factor Reduced Matrigel (BD Biosciences) according to the manufacturer's instructions. Cells were observed using a Cell-IQ Analyzer 2004-01, and videos were prepared with Cell-IQ Analyzer Pro-Write v.AN 2.0.1 (both from Chip-Man Technologies).
Results
Immunophenotypic characterization of hAMSC
Flow cytometry revealed the expression of common MSC surface markers. hAMSC were positive for CD90, CD73, CD105, and CD44 while being negative for CD14, CD45, HLA-DR, and CD34 (Supplementary Fig. S1; Supplementary Data are available online at
Expression of endothelial markers by hAMSC in situ and in vitro
hAMSC did not express the mature endothelial cell markers vWF and VE-cadherin in situ (tissue sections), directly after isolation (cytospins), or in vitro (cells in culture), which confirms the amnion to be an avascular tissue. However, a subpopulation of hAMSC expressed the endothelial precursor cell marker VEGFR-2. PlEC served as positive controls (Fig. 1).

Expression of endothelial markers. hAMSC do not express vWF or VE-cadherin in situ (tissue sections) and in vitro (cells in culture). However, a subpopulation shows expression of VEGFR-2 in situ (see arrows) and in vitro (see inset for higher magnification). PlEC served as positive controls. Scale bar: 50 μm. hAMSC, human amnion-derived mesenchymal stromal cells; vWF, von Willebrand factor; VE-cadherin, vascular endothelial-cadherin; VEGFR-2, vascular endothelial growth factor receptor; PlEC, placental endothelial cells. Color images available online at
Proliferation of hAMSC upon endothelial induction
We tested the effect of different endothelial culture conditions on the proliferation of hAMSC to evaluate optimal culture conditions. Non-induced hAMSC (cultured in DMEM+15% FBS on gelatin) showed cumulative population doublings of 2.5±0.1 after 5 days and 2.9±0.8 after 9 days. Endothelial culture conditions highly increased the proliferation potential of hAMSC. The mean cumulative population doublings under endothelial conditions were 7.3±0.2 and 13.1±0.3 after 5 and 9 days, respectively. There were no distinct effects among the different endothelial conditions consisting of culture on fibronectin or gelatin and in the presence or absence of 50 ng/mL VEGF in EGM-2 (Fig. 2). For further studies on endothelial induction, the use of gelatin-coating and EGM-2 medium without additional VEGF was chosen. Supplementary Fig. S2 shows that under these conditions the cumulative population doublings of hAMSC were clearly higher over several passages compared with non-induced hAMSC.
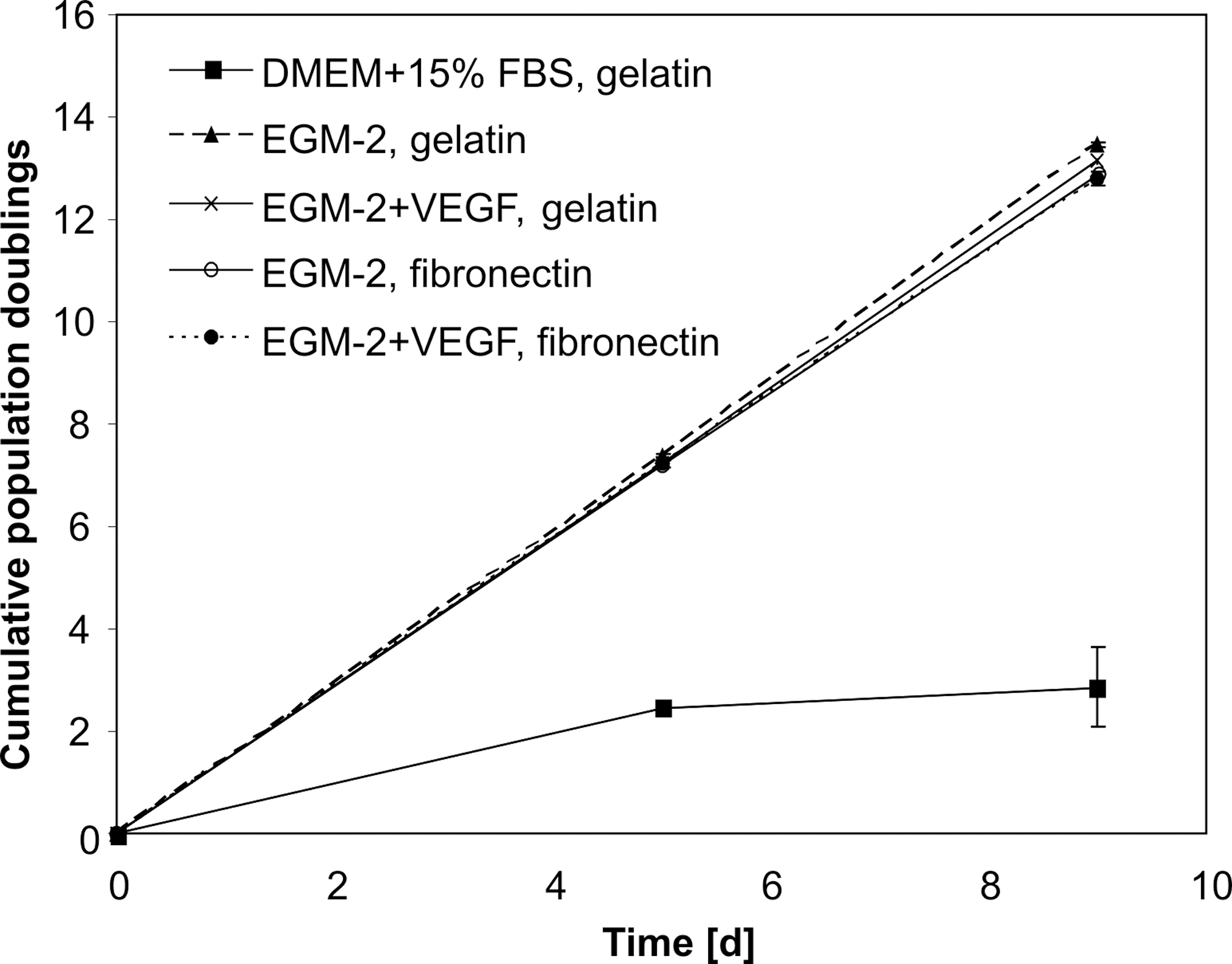
Cumulative population doublings of hAMSC. Cells were either cultured in DMEM+15% FBS on gelatin or under different endothelial conditions (EGM-2±50 ng/mL VEGF on gelatin or fibronectin coating). EGM-2, endothelial growth medium-2; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum.
Change of morphology and uptake of DiI-Ac-LDL upon endothelial induction
Non-induced hAMSC showed a fibroblast-like morphology (Fig. 3A). After culturing the cells in EGM-2 for a minimum of 5 days, they changed their morphology to a cobblestone-like phenotype (Fig. 3B), similar to PlEC (Fig. 3C).
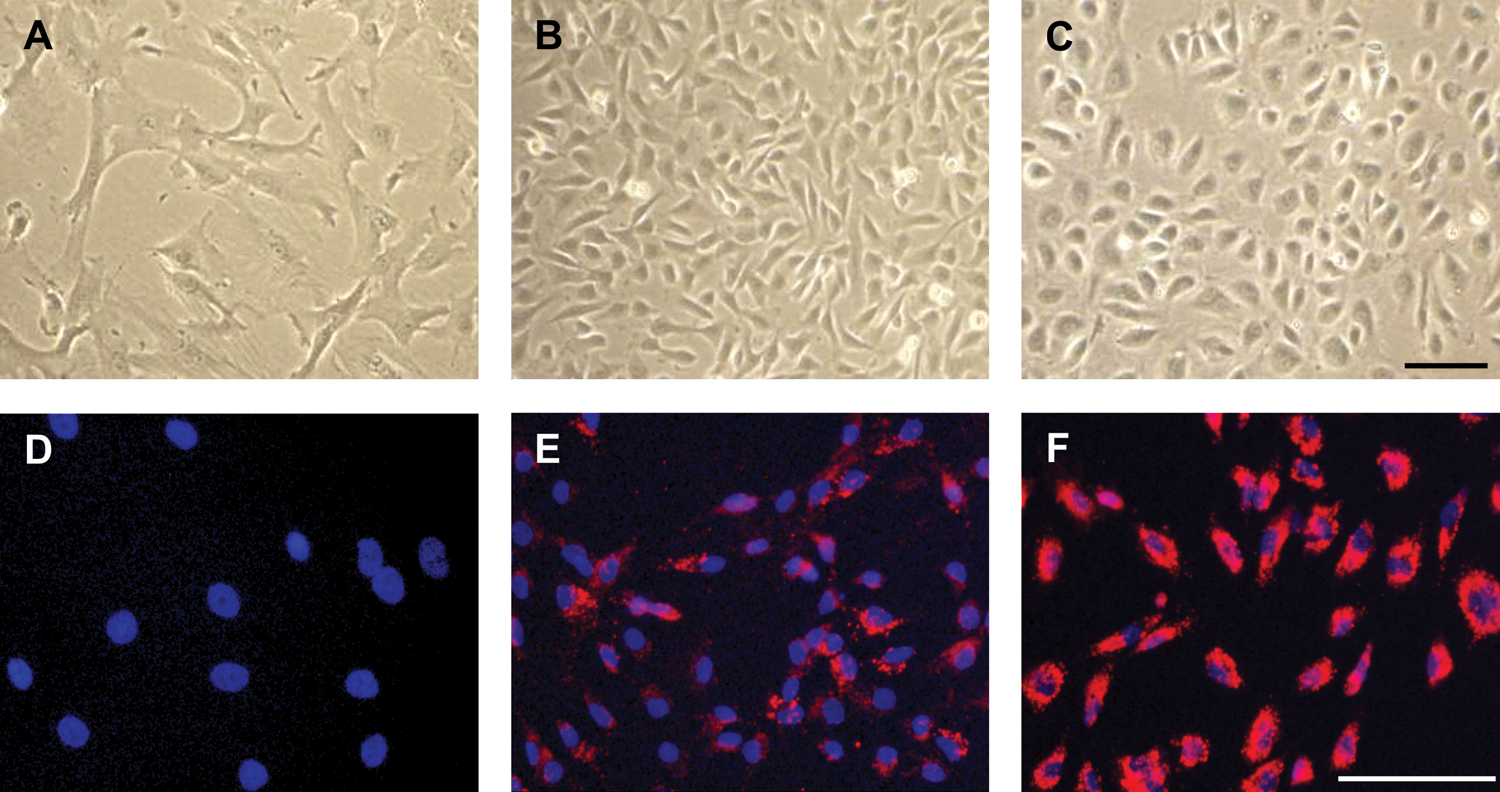
Morphology and DiI-Ac-LDL uptake by non-induced and induced hAMSC compared with PlEC. Noninduced hAMSC show a fibroblast-like morphology
The uptake of DiI-Ac-LDL is specific for endothelial cells and macrophages and occurs via a scavenger receptor. Non-induced hAMSC did not take up DiI-Ac-LDL (Fig. 3D). After induction with EGM-2, hAMSC internalized DiI-Ac-LDL with varying intensity (Fig. 3E). PlEC served as positive controls (Fig. 3F).
Expression of mature endothelial markers upon endothelial induction
Endothelial culture conditions did not result in the appearance of vWF or VE-cadherin positive cells, even when EGM-2 was supplemented with concentrations of VEGF of up to 100 ng/mL and cells were grown on fibronectin. Further, neither extension of the induction period to 3 weeks nor culturing the cells under low oxygen concentrations (2%) could induce expression of vWF or VE-cadherin genes or proteins (data not shown).
Gene expression changes upon endothelial induction
To identify the effect of endothelial culture on gene expression changes of hAMSC, a microarray was performed. Of the 28,870 transcripts analyzed, 16,761 genes were expressed (signal level >5 after background correction). After filtering with P<0.005, 200 genes were found to be differentially regulated with a FC >2, of which 92 were upregulated and 108 downregulated on endothelial induction (Fig. 4A). Genes were ranked by FC and screened for angiogenic functions. The first 25 genes that were up- and downregulated under angiogenic conditions are shown in Tables 1 and 2. Selected genes of interest are displayed in a Venn diagram (Fig. 4B). The according data has been deposited in NCBI's GEO and is accessible through GEO Series accession number GSE28385 (

Gene expression changes on endothelial induction.
Interestingly, induced hAMSC significantly downregulated typical pro-angiogenic genes such as tenascin C (FC−27.9), Tie-2 (−16.8), VEGF-A (−5.7), CD146 (−3.8), and FGF-2 (−2.2, P<0.05), whereas they upregulated genes with anti-angiogenic functions such as serpin peptidase inhibitor F1 (serpin F1, FC 31.4, P<0.01), the FGF-2 signaling antagonist sprouty1 (FC 10.6), and angioarrestin [angiopoietin (ANGPT)-like 1, 8.2]. The only factor with a possible pro-angiogenic function that we found to be upregulated was platelet-derived growth factor-D (PDGF-D, 26.6).
The genes of the MSC markers CD90, CD73, and CD105 were expressed equally by non-induced and induced hAMSC (FC >-2 and <2, signal level >5). In addition, both expressed VEGF-B and -C, placental growth factor (PIGF), VEGFR-1 and 2, angiopoietin-1 (see Fig. 4B), neuropilin 1 and 2, tissue inhibitors of metalloproteinases 1 and 2 (TIMP1 & 2), and the pericyte markers NG2 and PDGF receptor-β (PDGFR-β).
Expression of angiogenic proteins by hAMSC upon endothelial induction
hAMSC were analyzed for the presence of angiogenic proteins using the Human Angiogenesis Antibody Array C1000 (Fig. 5). Non-induced cells expressed high levels of the pro-angiogenic factor FGF-2. After endothelial induction, its expression decreased to 27%±8% compared with non-induced cells. Additional angiogenic factors that were downregulated include interleukin-8 (IL-8) (21%±5%), matrix metalloprotease-1 (MMP-1) (31%±1%), Tie-2 (13%±1%), and urokinase-type plasminogen activator receptor (UPAR) (23%±1%). On the contrary, the expression of the anti-angiogenic protein endostatin increased to 226%±4% as a result of angiogenic induction. In addition, epidermal growth factor (EGF) was upregulated to 236%±99%.
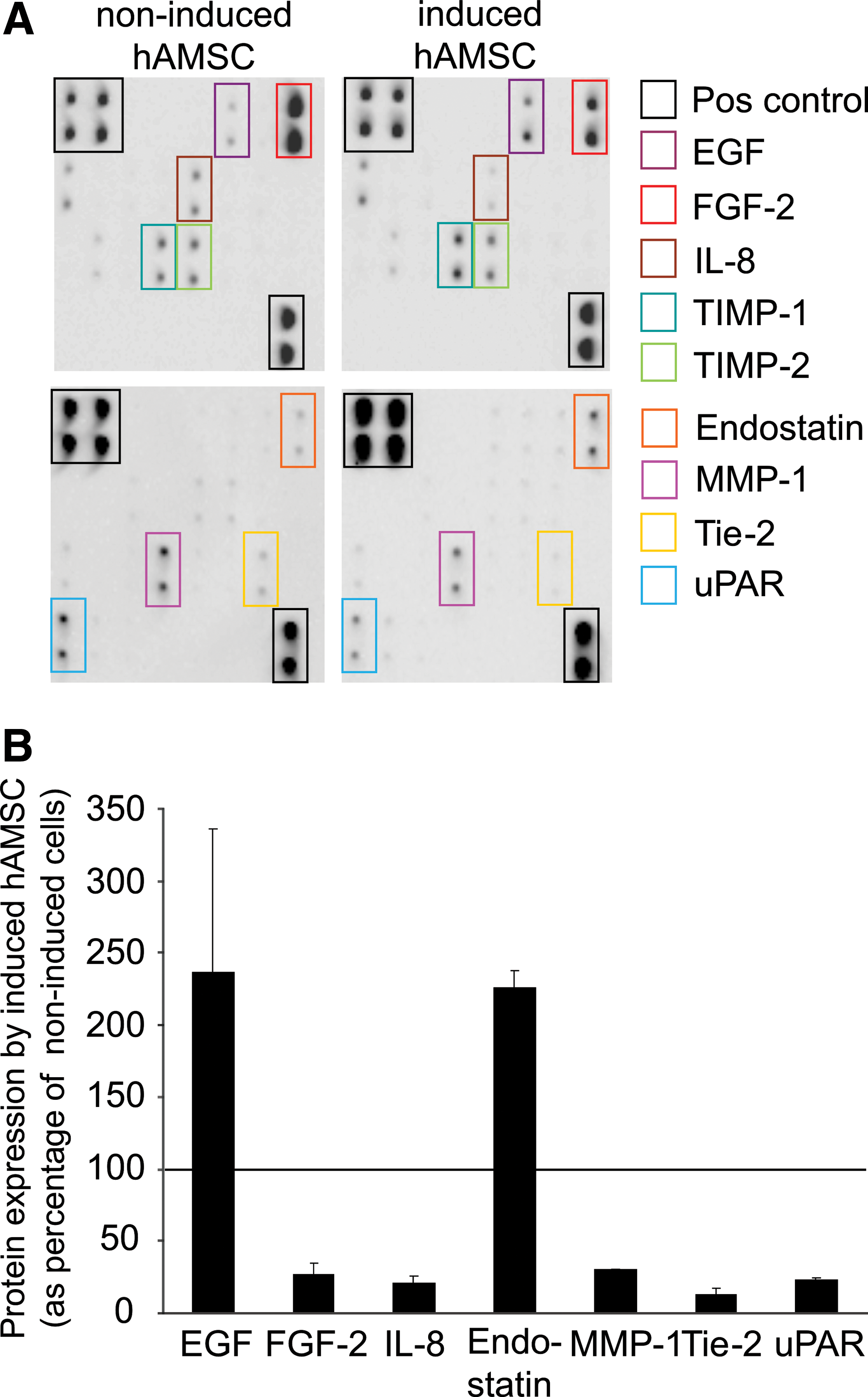
Expression of angiogenic proteins.
Angiogenesis assay–network formation
The Matrigel assay is a commonly used method to evaluate network formation by endothelial cells and was applied to investigate whether induced hAMSC were also able to form networks.
After culture under standard (non-induced) or endothelial (induced) conditions for at least 10 days, hAMSC were seeded on Matrigel in EGM-2. Non-induced hAMSC formed network-like structures within 6 h; however, these networks disintegrated fast (Fig. 6A-D, see also Supplementary Video S1). Induced hAMSC (Fig. 6E–H, Supplementary Video S2) formed networks similar to PlEC (Fig. 6I–L, Supplementary Video S3). While the endothelial networks remained static, networks of induced hAMSC became more wide-spread over time. In addition, branches formed by PlEC showed signs of degeneration (dark cell clusters in Fig. 6K, L) already after 24 h, whereas networks formed by induced hAMSC remained viable for about 48 h. However, cells within these networks remained negative for vWF (Fig. 6M, N). PlEC served as positive control (Fig. 6O, P).

Network formation on Matrigel. hAMSC were cultured under standard
Effect of hAMSC-CdM on endothelial cells
LDH is an enzyme that is released into the culture medium by damaged cells [29]. When PlEC were cultured in the presence of CdM collected from induced hAMSC, the activity of LDH in the culture supernatants was significantly reduced to 48.7%±14.9% of control, thus, endothelial cell viability was clearly enhanced (Fig. 7A). The addition of induced hAMSC-CdM to PlEC supported the formation of network-like structures in the Matrigel assay. While networks formed by endothelial cells in control medium had disintegrated after 72 h (Fig. 7B, see also Supplementary Video S4), networks formed by cells cultured with induced hAMSC-CdM were still stable (Fig. 7C, see also Supplementary Video S5). Under the same conditions, CdM collected from non-induced hAMSC neither enhanced endothelial cell viability (Fig. 7D) nor supported endothelial network formation (Fig. 7E, F).

Effect of hAMSC-CdM on EC. Induced hAMSC-CdM significantly reduces LDH activity in PlEC supernatants, shown as percentage of control after 96 h of culture
Discussion
In this study, we investigated the endothelial differentiation potential of MSC isolated from the amnionic membrane of human term placentas. We could show that the amnionic mesenchyme is free of mature endothelial cells as documented by the absent staining with the classical endothelial markers vWF and VE-cadherin. Interestingly, a subpopulation of hAMSC is positive for VEGFR-2. Together with its ligand VEGF, VEGFR-2 plays an important role during early placental and embryonic vascular development. Its expression has been documented in vasculogenic and angiogenic precursor cells found in placental villi [30]. Mice deficient in VEGFR-2 (VEGFR-2-/-) died in utero as a result of an early defect in the development of hematopoietic and endothelial cells [31]. Therefore, the presence of VEFGR-2 in the amnionic mesenchyme suggests an endothelial progenitor potential of hAMSC.
Careful immunocytochemical analysis for the expression of vWF and VE-cadherin on freshly isolated cells was performed to exclude a contamination with endothelial cells before endothelial induction. All isolations in this study were free of endothelial cells.
On endothelial induction with EGM-2 containing 2% FCS, EGF, hydrocortisone, VEGF, FGF-2, and IGF, hAMSC showed some endothelial-like characteristics: They changed their fibroblast-like to a more endothelial cell-like morphology and acquired the ability to take up Ac-LDL. In addition, only induced hAMSC were able to form long-lasting networks similar to endothelial cells in the Matrigel assay. Non-induced hAMSC cultured in DMEM with 15% FBS for at least ten days initially also formed network-like structures in Matrigel. This could be explained by the fact that before the application in the Matrigel assay, these cells were resuspended in EGM-2. Therefore, the network formation may be due to a short-term stimulatory effect of abundant growth factors present in both the Matrigel and in EGM-2. However, these networks were very unstable and disintegrated within 6 h.
Even though hAMSC were responsive to the angiogenic factors present in EGM-2, they did not differentiate into mature endothelial cells. Induced hAMSC still expressed the common MSC markers CD90, CD73, and CD105. None of the angiogenic conditions used by previous studies [22,23] or addition of high concentrations of VEGF (100 ng/mL) and the use of fibronectin, an extracellular matrix protein known to promote endothelial differentiation [32], led to an expression of vWF or VE-cadherin (genes or proteins). Even cells forming the network-like structures in the Matrigel assay remained negative for vWF. Also, endothelial induction under 2% oxygen did not promote endothelial differentiation, although low oxygen conditions are known to induce angiogenesis [33] and have a pro-angiogenic effect on MSC [34].
So far, only one other study has investigated the endothelial differentiation potential of hAMSC [35]. This study reported that culture in DMEM supplemented with 2% FBS and 50 ng/mL VEGF led to a slightly increased expression of VEGFR-2 as well as appearance of vWF-positive cells. The discrepancies between the results of this previous study and our data might be due to differences in cell isolation protocols. In contrast to our findings, in this former study, non-induced hAMSC also formed stable networks in Matrigel. Thus, maybe a subset of endothelial-like cells was present within the primary isolated cell population, possibly caused by an incomplete separation of the amnion from the underlying vascularized chorion. This hypothesis is supported by our Matrigel data, which clearly showed that non-induced hAMSC cultures devoid of endothelial cells did not generate stable networks.
We assume that hAMSC resist a differentiation into mature endothelial cells which would be in accordance with the fact that amnionic membrane is one of the few avascular tissues that maintains its avascularity even though it is next to a highly vascularized tissue (the chorion) [36].
Our microarray analysis did not reveal upregulation of endothelial-specific genes in angiogenic-induced hAMSC. Under standard conditions, hAMSC express a variety of pro-angiogenic genes. Tenascin C, a large extracellular glycoprotein, has been shown to promote endothelial cell elongation and sprouting in bovine aortic endothelial cells and human umbilical vein endothelial cells [37,38]. Tie-2 (or TEK) is a tyrosine-kinase transmembrane receptor that is predominately expressed by endothelial cells. Its ligands are the ANGPT family members ANGPT 1, 2, and 4, which are secreted proteins with different functions in angiogenesis [39]. Both VEGF-A and FGF-2 are very potent inducers of angiogenesis [40,41]. However, these pro-angiogenic genes were downregulated in endothelial culture conditions, that is, in the presence of abundant angiogenic growth factors. Instead, genes with anti-angiogenic functions were upregulated: Serpin F1, angioarrestin, and sprouty1. Serpin F1, or pigment epithelium-derived factor, is one of the most effective natural angiogenesis inhibitors [42]. Angioarrestin, also known as ANGPT-like 1, and Sprouty1 have been shown to inhibit different angiogenic processes [43 –45].
The gene expression results were confirmed on the protein level using an angiogenic protein antibody array. Again, FGF-2 and Tie-2 were downregulated on endothelial induction, whereas the anti-angiogenic protein endostatin was clearly upregulated compared with non-induced controls. The expressions of VEGF-A and angioarrestin in this array were too low compared with the internal positive control to allow reliable quantification.
It seems as if hAMSC use this upregulation of anti-angiogenic and concomitant downregulation of pro-angiogenic genes and proteins as an autoregulatory mechanism to protect themselves against a differentiation into mature endothelial cells. However, they do not adopt anti-angiogenic properties toward endothelial cells. On the contrary, CdM collected from induced hAMSC even had a positive effect on endothelial cells as shown by enhanced viability and stabilized network formation.
These results are consistent with studies showing that MSC from bone marrow promote angiogenesis and support blood vessel formation [46 –51]. Au et al. could demonstrate that MSC stabilized engineered blood vessels and kept them functional for 130 days in vivo. The authors showed that MSC did not differentiate into endothelial cells but instead acted as pericyte-like cells [52].
Therefore, we assume that even though hAMSC are not ideal as a substitute for endothelial cells, they might be valuable in a variety of cell-therapeutic or tissue-engineering approaches where they can promote the survival of endothelial cells, the stabilization of pre-existing vessels, and the revascularization of ischemic tissues. A concrete example would be the endothelialization of vascular grafts. Here, PlEC could be used for the seeding of the luminal surface. A co-application of hAMSC as supporting stromal or mural cells might enhance the viability of the endothelial cells and, therefore, the patency of the vascular graft.
From our results, we conclude that an endothelial induction of hAMSC is advantageous for an application in therapeutic treatments, as it promotes a survival-enhancing effect on endothelial cells. Further, careful analyses of primary cell isolations are mandatory to exclude misleading results due to a contamination with endothelial cells.
In future studies, it needs to be determined how a co-application of PlEC and hAMSC could be used in vascular therapies. Banking of both cell types may provide a convenient source for autologous therapy and for matching recipients with histocompatible donors.
Footnotes
Acknowledgments
The authors thank the research nurses Bettina Amtmann, Sandra Eppich, and Petra Wagner of the Clinic of Obstetrics and Gynecology for placenta collection, and Kerstin Hingerl, Rudolf Schmied, and Monika Siwetz from the Institute of Cell Biology, Histology, and Embryology, Medical University of Graz, Austria, for their valuable technical assistance and expertise.
This work was supported by the Franz-Lanyar-Foundation (Projects # 339 and # 349) to I.L. J.D.F. and J.K. were funded by the Medical University of Graz within the Ph.D. program Molecular Medicine.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.