
Abstract
Hair follicles form during embryonic development and, after birth, undergo recurrent cycling of growth, regression, and relative quiescence. As a functional mini-organ, the hair follicle develops in an environment with dynamic and alternating changes of diverse molecular signals. Over the past decades, genetically engineered mouse models have been used to study hair follicle morphogenesis and significant advances have been made toward the identification of key signaling pathways and the regulatory genes involved. In contrast, much less is understood in signals regulating hair follicle regeneration. Like hair follicle development, hair follicle regeneration probably relies on populations of stem cells that undergo a highly coordinated and stepwise program of differentiation to produce the completed structure. Here, we review recent advances in the understanding of the molecular signals underlying hair follicle morphogenesis and regeneration, with a focus on the initiation of the primary hair follicle structure placode. Knowledge about hair follicle morphogenesis may help develop novel therapeutic strategies to enhance cutaneous regeneration and improve wound healing.
Introduction
H
In postpartum humans and mammals, wound healing in response to skin injury does not provide the structure or function of the original tissue but instead results in the formation of scar tissue. This occurs despite the existence of cutaneous stem cells within the skin. Evidence suggests that this is largely due to the lack of inductive signals for cutaneous stem cells in the injured skin. Therefore, knowledge about follicle morphogenesis may help create a more appropriate microenvironment (niche) to initiate stem cell-based cutaneous regeneration.
Mesenchymal Induction of the Early Epidermis and the First Dermal Signal
At approximately mouse embryonic day (ED) 14.5, the placode for the primary hair is formed as the earliest hair follicle structure. Placodes, which are regularly spaced thickenings in the epidermis, histologically appear as small invaginations into the underlying dermis. Previous studies on epithelial–mesenchymal tissue recombination have shown that the initial signal for placode induction arises from the dermis [1]. Recognized as the “first dermal signal” it is believed to trigger the activation of promoters and repressors that compete with each other to establish the hair follicle initiation and regulate pattern formation. However, the composition of the “first dermal signal” has not been fully elucidated and the signal is likely temporospatially regulated. On the other hand, numerous activators and inhibitors for hair follicle induction have been detected [2], and some of the factors that exert significant effects on hair follicle morphogenesis, such as Wnt, BMP, Shh, Dkk-1/4, noggin, APCDD1, and Edar, have been studied extensively. Of the known signaling mechanisms involved in hair follicle development, the Wnt/β-catenin and Eda/Edar pathways are high in the hierarchy of placode activators and appear to play a critical role in initiating the structure [2,3]. Wnt activities are detected as early as ED 13.5, whereas Eda/Edar/NF-κB activities are not observed in the skin until ED 14.5 [3,4]. Additional data show that focal Wnt/β-catenin signaling occurs independent of Eda-A1/Edar/NF-κB signaling and that β-catenin is required for the activation of Eda-A1/Edar/NF-κB signaling in the developing skin epithelium [3]. Taken together, these studies indicate that Wnt signaling is essential for the induction of hair follicles [5 –7] (Fig. 1i).
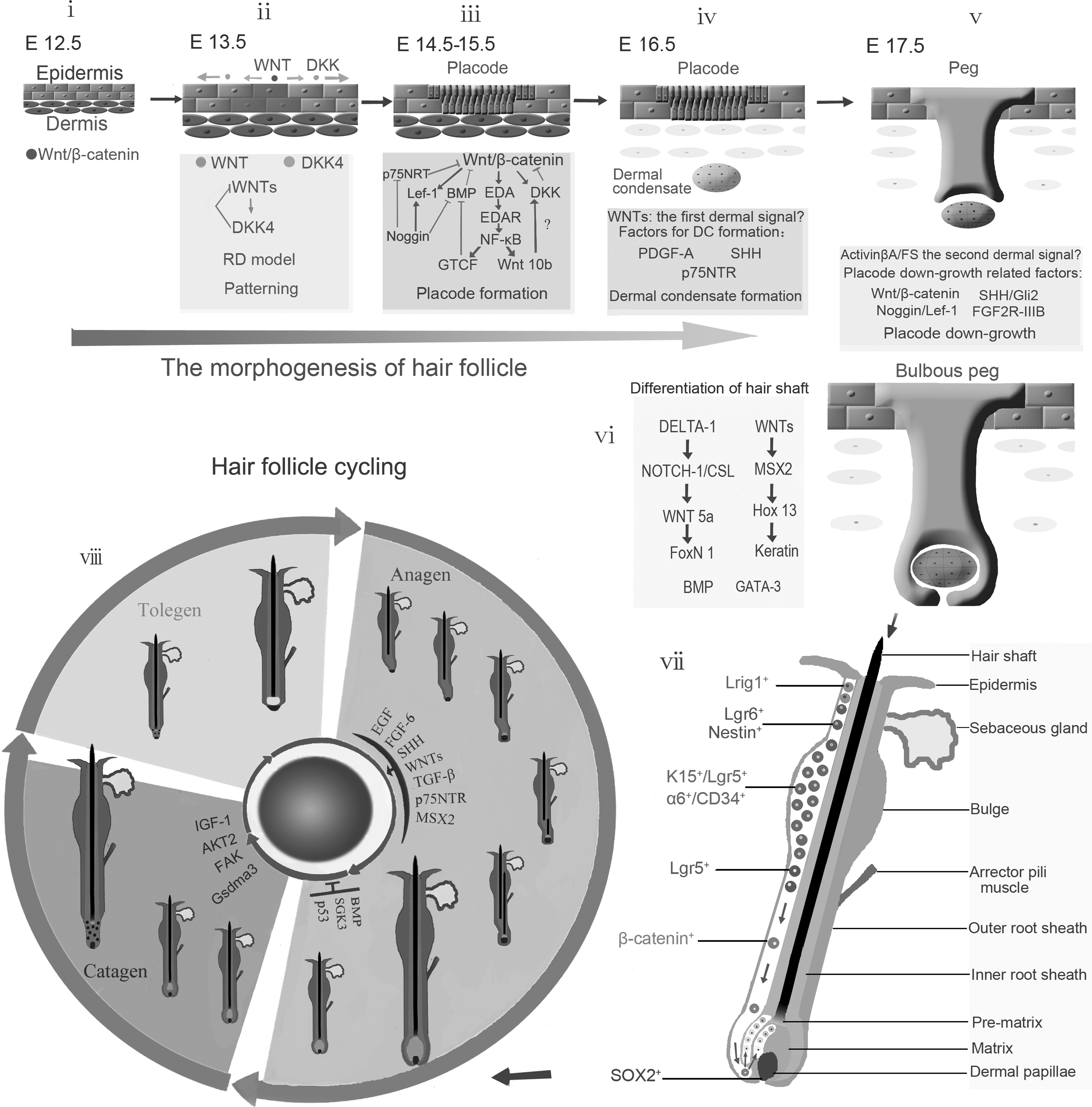
The morphogenesis and cycling of hair follicle.
Signal Pathways in Hair Follicle Initiation
Although Wnt signaling is important in the initiation of hair follicle development, it is modulated by other proteins. First, several proteins have been identified that interplay with components of the canonical Wnt/β-catenin signal pathway. β-Catenin/LEF1/TCF transcriptional complex, for example, activates numerous target genes [8]. Dkk-1/4, which acts as an antagonist for Wnt signaling, exerts its inhibitory function by binding directly to LRP5/6 coreceptors, resulting in their endocytosis and eventual degradation [9]. Interestingly, Dkk-4 is induced by canonical Wnt signaling. The Dkk-4 promoter interacts directly with the LEF-1/β-catenin complex, and therefore, it has been proposed that Dkk-1/4 act in a negative feedback loop to attenuate canonical Wnt signaling [10]. Unexpectedly, Dkk-4 is also identified as the direct target of Eda/Edar/NF-κB signaling, suggesting that Edar signaling is an indirect antagonist of canonical Wnt signaling [11]. ACPDD-1, which is a membrane-tethered glycoprotein and acts as a dimmer on the cell surface, is a novel Wnt inhibitor. APCDD-1 interacts with LRP5 and Wnt3A and prevents the formation of the Wnt receptor complex, thus interfering the biological effects of Wnt signaling. A mutation of Leu9Arg in the signal peptide of APCDD1 that perturbs its translation from the endoplasmic reticulum to the plasma membrane results in the deregulation of Wnt signaling and miniaturization of hair follicle, known as hereditary hypotrichosis simplex [12]. In addition to the Wnt signal, several other signals appear to be involved in hair follicle morphogenesis [2].
Edar signaling appears also to be essential for hair follicle formation. Edar signaling is mediated by extracellular ligand Eda and its receptor Edar [13], which is a member of the growing TNF superfamily [14,15]. Mutations of 1 of the 2 genes cause identical ectodermal dysplasia phenotypes in mice and humans [16]. In humans, the mutation characterized with a pathophysiological phenotype includes sparse head hair, abnormal teeth, and defects of sweat glands, termed HED. The Eda family of ligands includes 2 closely related isoforms and trimeric type II membrane proteins [17]. Eda-A1 and Eda-A2 have been shown to bind to and activate 2 different receptors, Edar and X-linked Eda-A2 receptor (Xedar), respectively [18]. The activated Edar or Xedar then leads to activation of NF-κB transcriptional factors, which are transported to the nucleus and manipulate the activity of corresponding target genes [19]. The Edar intracellular region contains a death domain. Once the Edar is activated, a death domain adapter protein named Edar-associated death domain (Edaradd) is recruited to the death domain of Edar and links the receptor to downstream signaling pathways. Edaradd is encoded by the mouse crinkled locus [20]. Loss or gain of Edar signaling affects the initiation of several hair follicle types [17]. The importance of Edar signaling in hair follicle formation was systemically studied with the mutant EdaA1 (tabby), EdaR (downless), EDARADD (crinkled), and IκBαDN knock-in mice (suppressed NF-κB activity), which all induced identical ectodermal dysplasia phenotype analogous to human HED, including no placode formation for primary guard hairs and lack of hairs behind the ears and on the tail [2]. Additionally, morphological analyses showed that in mice with defective EdaR and suppressed NF-κB activity, placode development was initiated, but arrested at the preplacode stage 0/1. Further studies suggested that NF-κB was essential and sufficient to induce Shh expression, which activates Gli2 in the skin and mediates mitogenic signals of Shh by transcriptional activation of cyclin D1 and cyclin D2, resulting in the proliferation of target cells [21]. Shh is not required for initiating hair follicle development, but is essential for controlling placode ingrowth and morphogenesis of the hair follicle [4,22]. These findings support the speculation that Shh mediates the NF-κB-transmitted Eda A1/EdaR signaling to control postinitiation hair placode ingrowth. In contrast, XEDAR-deficient and EDA-A2 transgenic mice exhibit normal hair follicles and other appendages, indicating XEDAR signals are dispensable for the development of ectoderm-derived appendages [23]. Interestingly, a recent study suggests that EDA2R is involved with androgenetic alopecia (AGA), but further studies are needed to clarify the role of these 2 genes and their potential interactions in the development of AGA [24,25].
Troy is a member of the TNF receptor superfamily and is homologous with Edar in its ligand-binding domain [26]. Lymphotoxin-alpha has been identified as a functional ligand of Troy [27]. Further analyses reveal that Troy signaling acts redundantly with Edar signaling in regulating the initiation of hair follicle development [28]. Intriguingly, Lgr4 [leucine-rich repeat-containing G-protein-coupled receptor 4 (GPCP4)] has been postulated as a novel gene that regulates the development of hair follicles [29]. Lgr4K5 knockout (KO) mice showed impaired hair placode formation with sparse head hair and focal alopecia behind their ears. This phenotype was similar to those observed in tabby (Eda) and downless (Edar) mice. Further analyses of mice lacking Lgr4 showed reduced expression of hair follicle-related genes including Lef-1, Edar, and Shh. In addition, cells lacking Lgr4 showed higher phosphorylation levels of smad1/5/8, suggesting potential interactions between the BMP signal and the Lgr4 signal during placode formation [30].
BMPs play pivotal roles in hair follicle development. BMPs compose a large subgroup within the transforming growth factor-β (TGF-β) gene superfamily [31,32]. TGF-β receptors consist of 2 subfamilies, type I and type II receptors. There are 3 type I receptors (BMP type I A receptor, BMP type I B receptor, and ACVRI) and 3 type II receptors (BMPRII, ActRIIA, and ActRIIB) [32]. Upon ligand binding, receptor-regulated Smads form heterodimers with Smad4 and then translocate into the nucleus to regulate the expression of TGF-β target genes [33]. In mice, inhibition of BMP signaling by overexpressing the BMP antagonist noggin results in a marked increase in size of the anagen hair follicle and the replacement of zig-zag hairs by awl-like hairs [34]. By contrast, constitutive deletion of noggin is characterized by the lack of the induction of secondary hair follicles [35,36]. In chicks, bead-mediated delivery of BMPs inhibits local feather formation [37]. In addition, several inhibitors of BMPs, expressed in developing skin, including Noggin, Smad7, and Sostdc1/Ectodin/WISE, are themselves transcriptional targets of BMP, likely acting as feedback inhibitors of the signaling pathway [38]. So it is evident that BMPs play a crucial role in hair follicle morphogenesis and tend to be dominant negative regulators for hair follicle formation [2].
Coordinated Signals in Hair Follicle Patterning
In mice, hair follicles are formed in response to a sequence of inductive waves and are spaced in regular patterns. Numerous studies have been carried out to understand the mechanisms underlying hair follicle patterning with a focus on the RD model [39,40]. The RD model is based on the positive and negative feedback regulation of an activator/inhibitor pair, together with a faster diffusion and decay of the inhibitor compared with the activator [41]. As canonical Wnt signaling is essential and sufficient for the induction of hair and feather follicles and appears to be the most upstream regulator of hair follicle morphogenesis [6,42], Wnt signaling represents an appealing candidate for the primary signal that manipulates follicle distribution. Dkk-1/4 exert their inhibitory property of Wnt signaling by binding to the LRP5/LRP6 component of the Wnt receptor complex [9]. Expression of the Dkk-1 is directly controlled by secreted Wnts [43]. Dkk-4 is also regulated by canonical Wnt signaling, as 5 LEF/TCF consensus binding motifs are found within 700 base pairs upstream of the transcriptional start site of DKK-4. In addition, Dkks should defuse faster than Wnts, as Wnt proteins are about 20% to 60% larger than Dkks. Hence, Wnts and their inhibitors Dkk-1/4 satisfy the criteria of the RD model. Further analyses based on a combined experimental and computational modeling approach validate that Wnt and its inhibitor Dkk interplay in an RD manner as primary determinants of murine hair follicle spacing [44] (Fig. 1ii).
Hair follicle patterning is accomplished with additional and more complicated molecular interactions besides the Wnt-Dkk RD model. The interplay between Edar and BMPs is also responsible for the distribution of hair follicles. Edar signaling is essential for hair follicle placode formation and rapid positive-feedback Edar signaling is coupled with the induction of BMPs, which in turn inhibits Edar signaling. Connective tissue growth factor (CTGF), with upregulated expression in follicles, is another target of Edar signaling. CTGF binds to and inhibits BMPs in a manner analogous to that of Noggin [45]. Thus, inhibitory activities of BMPs for Edar signaling are compromised in the follicle as BMPs are inhibited by CTGF. As a result, BMPs can only exert their inhibitory activity at a distance from follicles. Consequently, the hair follicle placodes is formed with a certain interval between each other and organized in a relatively regular pattern. In conclusion, Edar–BMPs interactions (activation–inhibition) direct hair follicle patterning by modulating signal receptivity, rather than restricting the localization of an inducing ligand [38].
The determination of interfollicular fate is equally crucial for the patterning of epidermis. BMP signaling exhibits inhibition for follicle or bud formation in the adjacent interfollicular or interbud tissue, but whether it promotes interfollicular or interbud fate and patterning is unclear [46]. Keratinocyte growth factor (KGF) and epidermal growth factor (EGF) signals have been identified as candidates for blocking hair follicle induction and promoting interfollicular epidermal fate in the developing mouse skin [47], which are speculated by promoting cell growth and proliferation in the interfollicular epidermis [48,49]. EGF signaling is mediated by EGF receptor (EGFR) and its associated ligands including TGF-α, heparin-binding EGF-like growth factor, amphiregulin, betacellulin, epiregulin, and epigen [50]. KGF signaling is transmitted via KGF and its receptor FGFR2(IIIb) [51]. Immunofluorescence analysis shows that the expression levels of EGFR and FGFR2(IIIb) are low in hair follicle placodes. Constitutive EGF and KGF signaling can inhibit hair follicle morphogenesis. Similarly, EGF- and KGF-treated embryonic skin lacks placodes and dermal condensates, coupled with enhanced interfollicular epidermal phenotype. Thus, KGF and EGF signals promote epidermal fate at the expense of hair follicle development [47]. Therefore, balanced signals for follicular fate and interfollicular fate may be crucial for the development of hair follicles in normal density.
The Morphogenesis of Hair Follicles
The placode is the earliest morphological structure to form during hair follicle development. It is formed as the consequence of focal epithelial proliferation induced by signals arising from the underlying mesenchyme, which is called the “first dermal signal” [1]. Wnt/β-catenin signaling is essential for the initiation of the placode, as conditional ablation of β-catenin in mouse skin epithelium prevents hair placode formation in the embryo [52], and ectopic Dkk1 expression results in “nothing happens without Wnt” in terms of epithelial appendage formation—no hair follicles, no teeth, no sebaceous glands [2,6]. EdaA1/EdaR/NF-κB signaling is also needed for placode formation of guard and zig-zag hairs. Its role in the placode induction is probably through the effect of NF-κB in cell proliferation and survival. Interestingly, a recent study showed that sustained epithelial β-catenin activity resulted in excessive induction of hair follicles even in the absence of Eda/Edar/NF-κB signaling, thus suggesting that Wnt/β-catenin is upstream of Eda and plays a predominant role in hair follicle induction [53]. Once the placode is induced, various factors are expressed in it, including fibroblast growth factors (FGFs), TGF-β2, Delta1, Noggin, Follistatin (FS), Gremlin, MSX1, and MSX2 and all these factors promote hair follicle development. Conversely, the BMPs including BMP2, BMP4, and BMP7, which are expressed in the mesenchyme or placode in response to Edar and Wnt/β-cantenin, act as inhibitors for hair follicle formation. Thus, hair follicle morphogenesis involves a complex interplay between the Wnt and BMP signaling pathways.
Placode is formed as an epidermal response to the first dermal signal. In return, placode emits epidermal signals to the mesenchyme and stimulate the later to condensate. In addition, the second dermal signals from the condensate instruct the placodal epithelial cells to proliferate and invaginate into the dermal condensate [1]. The epithelial–mesenchymal signaling interaction is crucial for hair follicle formation. Wnt signaling likely acts as the epidermal signal to induce the dermal condensate [54]. The dermal condensate fails to develop in the absence of epithelial β-catenin [52]. Platelet-derived growth factor-A (PDGF-A) is also required for the development of hair follicles, and mice lacking PDGF-A are characterized with small dermal papillae, dermal sheath (DS) abnormalities, and thin hair. Additionally, PDGF-A is expressed in the placode, whereas its receptor is expressed in the dermal condensate [55]. Thus, Wnt and PDGF-A molecules are strong candidates of the first epithelial signal. Immunological studies or in situ hybridization show that fibronectin, β1 integrin, and Notch-1 are expressed in chick skin DCs [56]. Previous reports have implicated important roles for integrins in dermal condensate formation. It is hypothesized that integrins exert effects in the migration of dermal fibroblasts to the appendage via engagement on fibronectin. Notch signaling has also been suggested to play a role in bud and hair follicle formation, and ligand/Notch interactions induce the Notch intracellular domain (NICD) to be released and translocates into the nucleus where NICD forms a transcriptional complex with nuclear factors. It is speculated that the interaction between NICD and integrins switch integrins to a high-affinity state and hence stabilize the DC [56]. p75 neurotrophin receptor (p75NTR) is expressed in dermal condensate in developing hair follicles, and loss of function mutations in p75NTR gene affects the rate of hair follicle morphogenesis, suggesting that neurotrophins may be involved in the early stages of follicle formation [57]. Syndecan-1 is another factor that is specifically expressed in dermal condensate during murine hair follicle morphogenesis, but further analysis shows that syndecan-1 is not required for follicle initiation and development [58] (Fig. 1iii).
Once the condensate is formed it in turn sends signals to nascent follicle keratinocytes to stimulate their proliferation and down-growth into the underlying dermis and to form more mature follicles. The signal arising from dermal condensates is termed as second dermal signal.
The components of the second dermal signal have not been fully elucidated. Hair germs comprising epidermal placodes and associated dermal condensates were detected in both wild-type and Shh−/− embryos, but progression through subsequent stages of follicle development was blocked in mutant mice. Thus, the “second dermal signal” is likely to be activated by Shh [59]. Further studies suggest that Shh is regulated by Wnt/β-catenin signaling [60]. The downstream proteins that respond to Shh appear to be Gli1 and Pct1, as the levels of Gli1 mRNA were markedly reduced in the epithelial and mesenchymal components of Shh−/− primary hair germs and at the same time the expression of Ptc1 decreased in Shh mutant hair germs [61]. In addition, hepatocyte growth factor/scatter factor might also play a role in the “second dermal signal” between the follicular mesenchyme and epithelium [62] (Fig. 1iv, v). Once the primary hair follicle structure is induced, it subsequently differentiated into outer-root sheath (ORS), inner root sheath and hair shaft. Hair follicle differentiation is regulated by multiple signaling pathways. Among these signals, Homeobox C13 (Hoxc13), regulate the activity of Foxn 1, has been shown to be essential for proper hair shaft differentiation [63]. Heparanase 1, whose expression was primarily located to the inner root sheath of the anagen phase human hair follicle, has been suggested to be involved in inner root sheath differentiation. Further, inhibition of heparanase in cultured hair follicles induced a catagen-like process [64]. Dlx3 is another crucial transcriptional regulator in hair follicle formation and regeneration, and the selective ablation of Dlx3 in the epidermis result in complete alopecia due to a lack of hair shaft and inner root sheath formation. Importantly, Dlx3 has been identified in the downstream of Wnt signaling pathway but as an upstream regulator of several other transcription factors in hair differentiation such as Hoxc13 and Gata3 [65,66] (Fig. 1vi).
Hair Follicle Regeneration During Cycling
The hair follicle undergoes cyclical regeneration, which involves at least 10 epithelial and mesenchymal cell lineages, and persistently goes through 3 phases: anagen, catagen, and telogen [67]. Hair shaft is formed by rapidly proliferating matrix keratinocytes in the bulb located at the base of the growing follicle. The duration of the anagen phase greatly affects the length of the hair. Matrix cells eventually stop proliferating, and hair growth ceases in the catagen phase, when the lower follicle regresses. After the telogen phase, the lower hair-producing portion of the follicle regenerates, starting the new anagen phase [68]. Previous studies have identified many molecules that regulate the cycling process. Wnt [69,70], Shh [22,70], and KGF (also known as FGF7) [71,72] have been found to induce the anagen phase, whereas serine/threonine-protein kinase (SGK)3 and Msx2 are known to maintain the anagen phase [73 –75]. EGF and FGF [76 –78], TGF-β-family pathway members such as TGF-β1 and the BMP type IA receptor (BMPRIA) [79,80], brain-derived neurotrophic factor (BDNF), and possibly p75NTR [81,82] are involved in the transition of the anagen to catagen phase. Moreover, many other studies suggest that more factors might be functionally involved in the hair follicle cycling process, including Akt2 [74], vitamin D receptor (VDR) [83], the hairless protein (Hr) [84], circadian clock gene products [85,86], focal adhesion kinase (FAK) [87], vanilloid receptor-1 (TRPV1) and TRPV3 [88], insulin-like growth factor-I (IGF-1) [89], Gsdma3 [90], cyclooxygenase-2 (COX-2) [91], heat shock protein (HSP), p53 [92], and alkaline phosphatase (ALP) [93] (Fig. 1viii).
It has been proposed that ligands and antagonists derived from hair follicles and skin macroenvironment combine to produce unique signaling profiles that regulate hair follicle progression through cycling stages. Similar to that in embryonic hair development, Wnt (activator)/BMP (inhibitor) pair, among numerous activating and inhibitory signaling pathways for hair follicle regeneration, has been suggested to play a predominant role [94,95]. The relative activator/inhibitor strengths are likely to control the initiation and progression of hair follicle cycling.
Stem Cells for Hair Follicle Regeneration
The regeneration of organs is a common and widespread adaptive capability among metazoan creatures. Many larval and adult animals are able to regenerate large sections of their body plan after amputation, and this usually restores the structures that were removed by the operation. One well-known research model is that adult urodeles can regenerate their limbs by local formation of a mesenchymal growth zone or blastema [96]. Unfortunately, the regeneration potential tends to discount in great extent for mammalians and humans. Adult skin consists of a keratinized stratified epidermis and an underlying thick layer of collagen-rich dermal connective tissue providing support and nourishment [97]. Hairs and glands, appendages of skin, are derived from the epidermis but project deep into the dermal layer. When an adult human skin tissue is wounded deep into the level of hair bulbs in the dermis that causes a complete loss of hair follicles, the repairing epithelium does not regenerate hair follicles and other epidermal appendage structures [98].
Several studies have indicated that cutaneous stem cells mainly reside in the bulge region of the hair follicle. Lineage analysis has shown that all epithelial cells within the adult follicle originate from bulge cells [68]. The neural stem cell marker, nestin, has been considered as a marker for hair follicle pluripotent stem cells and their immediate, differentiated progeny [99,100]. The precise locations of the nestin-expressing cells in the hair follicle vary with the hair cycle. In mid- and late-anagen, nestin-expressing cells are located in the upper ORS as well as in the bulge area and colocalized with keratin 15 (Krt-15, also known as K15) [101]. The results suggest that nestin-expressing cells in the hair follicle bulge probably contribute to progenitors in the follicle ORS. K15 is highly expressed in the bulge, but lower levels of expression can be detected in the basal layers of the lower follicle ORS and the epidermis. When K15-positive budge cells were subcutaneously engrafted with neonatal murine dermal cells, they reconstituted all components of the cutaneous epithelium [102], suggesting that K15-postive budge stem cells have the full potential to form epithelial appendages. However, a subsequent study showed that stem cells from the upper portion of the follicle, but not stem cells from the budge, migrated into the skin wound and formed new hair follicles; moreover, the Wnt signal was identified to be critical for the follicle neogenesis, and neutralization of Wnt7 abolished the regeneration [7]. In consistence with this observation, Lgr6+ cells, which resided in a previously uncharacterized region directly above the follicle bulge, was suggested to permanently contribute to cutaneous regeneration, including hair neogenesis [103] (Fig. 1vii). These results suggest that a stem cell population capable of de novo hair follicle regeneration may exist in the hair follicle above the level of bulge [104] (Fig. 1vii).
CD34 and α6-integrin have long been considered as markers for murine hair follicle stem cells. The markers recognized 2 cell populations in the hair follicle budge, α6lowCD34high and α6highCD34high cells, and both cell populations displayed features of stem cells when they were cultured outside of their native niche and exposed to proliferation-inducing conditions. Either cell population exhibited the ability to form new follicles when the cells were engrafted with newborn dermal cells into nude mice [105], suggesting that integrin α6+/CD34high may represent stem cells with hair follicle regeneration potential. Leucine-rich repeats and immunoglobulin-like domains protein (Lrig)1 is a marker of human interfollicular epidermal stem cells and is involved in maintaining stem cell quiescence. Lrig1-expressing cells can give rise to all of the adult epidermal lineages in skin reconstitution assays [106]. Lgr5, a marker of intestinal stem cells, is expressed in actively cycling cells in the lower ORS of anagen hair follicles. Lgr5high cells isolated from telogen mouse skin formed large colonies with high efficiency. In skin reconstitution assay, Lgr5high keratinocytes constituted the most potent population in regenerating hair follicles, which exhibited 90% superior growth efficiency than follicles initiated by CD34+ cells. Further study suggests that Lgr5+ cells provide a constant flux of stem cells downward toward the dermal papilla during the growth phase of the hair follicle, and these cells seem to exert a elevated β-catenin expression [107] (Fig. 1vii).
Taker together, previous studies have identified several stem cell populations in the hair follicle, which express distinct surface markers and are located in different areas of the hair follicle. It is unclear whether they represent different types of stem cells in the hair follicle or stem cells in different differentiation stages. Future skin reconstitution assays with defined cell types will help identify essential stem cells for hair follicle neogenesis, and thus hair follicle regeneration will become a more controllable procedure [108].
Dermal Papilla Cells Are Essential for Hair Follicle Regeneration
Previous studies have indicated that the absence of hair follicle regeneration in the healing wound is due to a lack of proper inductive signals from the underlying wound dermis [109 –111]. Dermal papilla cells (DPCs), which are specialized cells derived from the mesenchyme, have been known to provide unique and critical signals for hair follicle development [109,112,113]. When engrafted with embryonic mouse epidermis into the skin, adult DPCs are capable of inducing hair follicle formation [114,115]. Further studies suggest that skin-derived precursors from follicle DPCs and lower DS cells are capable of and responsible for the differentiation into dermal cell types within the intact skin and the regulation of hair follicle regeneration [116]. Thus, follicle bulge cells and DPCs serve as epidermal and dermal precursors and are responsible for maintaining, repairing, and regenerating epidermis and dermis, respectively. However, it is unclear whether other cell types derived from the dermal mesenchyme are necessary for perfect cutaneous regeneration. In addition, the molecular mechanisms underlying the regulatory activities of DPCs have not been fully understood. A previous study showed that DPCs selectively expressed Wnt-5a, but not Shh [117], suggesting a role of Wnt signal in DPC-mediated follicle neogenesis. Indeed, forced expression of Wnt-5a in the deeper wound induced the formation of epidermal glandular structures, which projected deep into the newly formed dermis, and some of them resembled hair follicles in morphology [118]. Consistent with that found in embryonic hair follicle development, Wnt signals appear to be central in hair follicle neogenesis. However, Wnt signals alone seem not to be sufficient for perfect regeneration of hair follicles and other epidermal appendages structures, and signals derived from other cells in the mesenchyme are likely to be necessary.
The inductive property of DPCs appears to rely on BMP signals. Loss of BMPR1A in DPCs resulted in the loss of signature characteristics of DPCs in vitro and hair follicle inductive property in vivo [119], suggesting an essential role of BMP signals in DPC functions. These results also imply the complex epithelial–mesenchymal crosstalk in follicle regeneration.
Bone Marrow-Derived Cells in Wound Healing
Previous studies suggest that the bone marrow (BM)-derived cells may play a role in cutaneous repair/regeneration [120]. BM-derived cells have been found in the epidermis, hair follicles, sebaceous glands, and dermis of the adult skin [121]. Our recent studies and others' suggest that BM-derived mesenchymal stem cells (BM-MSCs) may be involved in cutaneous regeneration [120,122]. When grafted in a matrix gel into excisional wounds, glandular appendage-like structures formed by cells derived from transplanted BM-MSC were found, which resembled developing sweat or sebaceous glands; however, these structures were short-lived and disappeared when wounds completely closed [122]. Consistent with our findings, epithelial cells derived from endogenous BM cells were found to appear in the cutaneous wound transiently during wound healing [123]. These results suggest that BM-MSC may not provide long-term self-renewal stem cells for keratinocytes and appendages. Instead, BM-MSC may contribute to cutaneous regeneration through a paracrine mechanism. Previous studies suggest that certain molecules essential for the integrity of the skin appear to be exclusively released by BM-derived cells. BM-MSCs are capable of releasing some of these molecules [120,124]. For example, BM transplant treatment of adult collagen XVII (Col17)-deficient mice induced donor-derived Col17 expression associated with the recovery of hemidesmosomal structures, and BM-MSCs have the potential to produce Col17 [125]. In an ex vivo study, BM-MSCs in a collagen matrix induced keratinocytes on the top of the matrix gel to migrate downward into the matrix and form glandular structures, whereas subcutaneous preadipocytes or dermal fibroblasts did not [126]. In consistence with these findings, BM-MSCs have been shown to secrete a large variety of growth factors, cytokines, and extracellular matrix molecules, which are distinctively different in expression levels from those released by dermal fibroblasts [127]. Some of the highly expressed cytokines have been known to be involved in tissue regeneration of other organs [128 –130]; however, their roles in cutaneous regeneration have not been examined.
Conclusion
The embryonic hair follicle development and adult hair follicle regeneration are highly related processes; both rely on populations of stem cells to undergo a highly coordinated and stepwise program of differentiation to produce the completed structure. Convincing evidence indicates that there are cutaneous stem cells in the adult skin, which are capable of forming de novo hair follicles and other structures of the epidermis. However, in humans and most mammals, the injured skin is repaired by formation of scar tissue rather than restored to its original structure. Recent studies have suggested that the absence of regeneration to the wounded skin is not due to malfunction of cutaneous stem cells, but due to a lack of proper inductive signals for the neogenesis of hair follicles and other appendage structures. Because of diverse gene KO mouse models developed in the past decades, tremendous progress has been made in understanding of signals that regulate hair follicle development. Though signals that regulate follicle regeneration have far less been understood, recent studies suggest that similar molecular signals to follicle development are likely to be used (Table 1). With a better understanding of signals for hair follicle development and the identification and synthesis of key inductive signaling molecules, novel molecular therapies for cutaneous regeneration will be developed.
BMP, bone morphogenetic protein; DPCs, dermal papilla cells; EDA, ectodysplasin A; EDAR, EDA receptor; HED, hypohidrotic (anhidrotic) ectodermal dysplasia; DKK, Dickkopf; SHH, sonic hedgehog; EGF, epidermal growth factor; EGFR, EGF receptor; KGF, keratinocyte growth factor; FGF, fibroblast growth factor.
Footnotes
Acknowledgments
The authors thank H.A. Shankowsky for her excellent assistance with editing and secretarial assistance. This work was supported by grants from Natural Science Foundation of China (No. 30971496, U1032003) and from Shenzhen (JC201005280597A to Y.W.).
Author Disclosure Statement
No competing financial interests exist.