
Abstract
The identification of intrinsic factors required for propagation of self-renewing embryonic stem (ES) cells is important to improve the efficiency of expansion of ES cells for therapeutic purposes. Here, we report a novel role for natriuretic peptide receptor-C (NPR-C) in the survival of murine ES cells. We found that NPR-C was highly expressed in ES cells and was downregulated during ES cell differentiation. Knockdown of NPR-C in ES cells by using a small-interfering RNA resulted in apoptotic cell death, and the induction of p53 protein expression. Conversely, chemical inhibition of p53 by α-pifithrin significantly reduced apoptosis in NPR-C-deficient cells. cANF(4–23), a selective NPR-C agonist, protected ES cells against oxidative stress-induced apoptosis, and blocked activation of p53 and Nanog suppression in the presence of DNA-damaging agents. Thus, NPR-C is required to control DNA damage-induced p53 levels to maintain ES cell self-renewal.
Introduction
E
Recently, we showed that brain natriuretic peptide (BNP) and natriuretic peptide receptor A (NPR-A) are required for self-renewal and pluripotency of ES cells [8,9]. Currently, there are no data available concerning the role of natriuretic peptide receptor C (NPR-C) in ES cells. NPR-C is a receptor for natriuretic peptides [10,11] and was originally considered a clearance receptor without any physiological roles [10]. Several studies have shown that NPR-C is coupled to the adenylyl cyclase/cAMP signal transduction system and performs several physiological functions [11 –13] including protecting astrocytes from oxidative stresses [14].
It remains unclear as to how ES cells respond to the low levels of DNA damage that are normally introduced into ES cells. Here, we provide evidence demonstrating that NPR-C is expressed in undifferentiated murine ES cells, and that it is critical for murine ES cell viability by regulating p53 levels.
Materials and Methods
ES cell culture
Murine ES cells (E14TG2a) (CRL-1821; American Type Culture Collection) were maintained in DMEM/F-12 medium (Sigma) that was supplemented with 1,000 U/mL leukemia inhibitory factor (LIF) (Chemicon), 11% FBS, 2 mM glutamine (Nacalai Tesque), 1 mM sodium pyruvate (Sigma), 1% MEM nonessential amino acids (Gibco), 0.1 mM 2-mercaptoethanol (Sigma), and 1% penicillin–streptomycin. ES cells for small-interfering RNA (siRNA) transfection were cultured in the same medium, except that FBS was replaced with 15% knockout serum replacement (Gibco). For differentiation, embryoid bodies (EBs) were grown in bacteriologic dishes without LIF. cANF(4–23) (American Peptide) and pifithrin-α (Sigma) were added, to the cultured ES cells, whereas the control cells received the vehicle alone.
Immunofluorescence
ES cells grown on glass coverslips or 35 mm dishes were briefly rinsed with phosphate-buffered saline (PBS) and fixed for 20 min in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Blastocysts were collected from ICR mice at 3.5 dpc and fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). All experimental procedures and protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the Shiga University of Medical Science, Otsu, Japan, and conformed to the NIH Guide for the Care and Use of Laboratory Animals.
The ES cells and blastocysts were permeabilized for 10 min with 0.1% Triton X-100 in PBS, and blocked for 40 min with 4% BSA in PBS at room temperature. They were then incubated at 4°C overnight with the following antibodies: anti-NPR-C (1:1,000; Abcam) and anti-Oct-4 (1:200, sc-5279; Santa Cruz Biotechnology). This was followed by incubation with the following secondary antibodies: Alexa Fluor 488-labeled anti-rabbit IgG and Alexa Fluor 555-labeled anti-mouse IgG (1:500; Molecular Probes). Nuclei were counterstained with Hoechst 33342 (1 μg/mL) (Invitrogen). The slides were examined by confocal lazer microscopy (C1si; Nikon), and the images were processed by using the Nikon EZ-C1 viewer software.
Reverse transcription–polymerase chain reaction
Experiments were performed as described [9]. A detailed description is given in Supplementary Data (available online at
RNA interference
RNA interference in murine ES cells was carried out according to the manufacturer's protocol by using Lipofectamine RNAiMAX (Invitrogen). Two pairs of siRNAs (Invitrogen) were designed for NPR-C (BC055897) by using the BLOCK-iT RNAi Designer software. NPR-C siRNA2 gave a higher level of NPR-C knockdown and was, therefore, used for most of the experiments. The appropriate siRNA negative control Duplex (Cat. No. 12935–300; Invitrogen) was selected based on the percentage G/C. The NPR-C siRNA and control siRNA were transfected at a final concentration of 40 nM for 24 h in triplicate for each treatment. The sequences of the NPR-C siRNAs were as follows:
NPR-C siRNA1- sense, 5′- CAAAUGUCCUGUUCCAAGUCUGCUG -3′; antisense, 5′-CAGCAGACUUGGAACAGGACAUUUG-3′; NPR-C siRNA2- sense, 5′-AACAGUUCCUCUCGAGUUUGUCGUC -3′; antisense, 5′-GACGACAAACUCGAGAGGAACUGUU -3′.
Western blotting
Western blotting was performed by using standard methods as described [9]. Proteins were detected by using antibodies against NPR-C (Abcam) [15], p53 (Santa Cruz Biotechnology), Oct4 (sc-5279; Santa Cruz Biotechnology), Nanog (Bethyl Laboratories), NPR-A (Santa Cruz Biotechnology), NPR-B (Santa Cruz Biotechnology) [16], BNP (Chemicon), and β-actin (sc-47778; Santa Cruz Biotechnology). The secondary antibodies were peroxidase-conjugated IgG (Jackson ImmunoResearch Lab, Inc.). The blots were developed by using SuperSignal West Pico Chemiluminescent substrate (Pierce) and visualized by using an LAS-3000 FujiFilm Lumino-Image Analyzer (FujiFilm).
In vitro differentiation
Undifferentiated ES cells were trypsinized and resuspended in ES cell culture medium without LIF. Differentiation was induced by hanging drop method as a standard method of EB formation, and treatment with retinoic acid (RA) for 3 days. EBs were further cultured for 7 days in differentiation medium.
Apoptosis assay
Annexin V staining was performed by using flow cytometry according to the manufacturer's guidelines (MBL). For fluorescence microscopy, adherent ES cells were stained with Annexin V, and then, the cells were washed and fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4).
Statistical analysis
The results are expressed as mean±SD, as indicated in the figure legends. Statistical significance was assessed by 2-tailed Student's t-tests. Values of P<0.05 were considered significant.
Results and Discussion
Expression of NPR-C in ES cells and preimplantation embryos
To examine NPR-C expression in ES cells, we measured NPR-C mRNA and protein levels in murine ES cells before and after differentiation. Under the conditions that promote differentiation (without LIF), the mouse ES cells lost their pluripotent stem cell markers, such as Oct4. Reverse transcription–polymerase chain reaction (RT-PCR) (Fig. 1A), western blot (Fig. 1B), and double-immunofluorescence (Fig. 1C) analyses showed that NPR-C is highly expressed in pluripotent ES (Oct-4-positive) cells that were cultured in the presence of normal ES media containing+LIF. Expression of NPR-C is downregulated on differentiation induced by culturing ES cells without LIF (−LIF) (Fig. 1). The differentiated cells, which are negative for Oct-4 expression, are also negative for NPR-C expression (Fig. 1C). These observations indicate that NPR-C expression is reduced during differentiation.
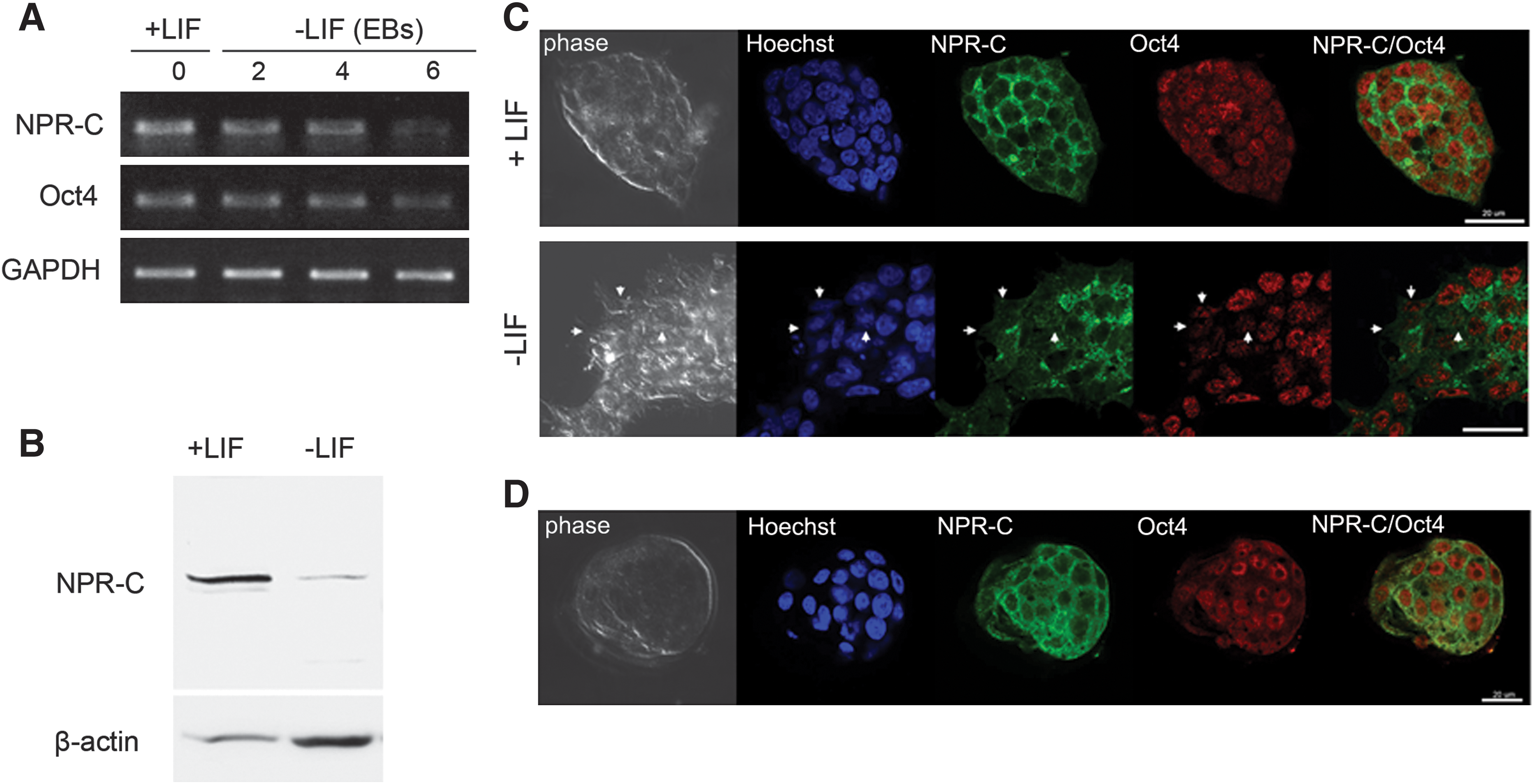
Expression of NPR-C in murine ES cells and preimplantation embryos.
One potential approach to support these results would be to examine the inner cell mass of the blastocyst, which is the in vivo compartment from which ES cells originate. Consistent with their presence in the undifferentiated ES cells, NPR-C is coexpressed with Oct4 in the 3.5-day-old murine blastocysts (Fig. 1D). The linkage between NPR-C expression in ES cells and in blastocysts strongly suggests that signaling through this receptor plays a role in maintaining ES cell functions.
NPR-C is required for ES cell viability
The functional significance of NPR-C expression was assessed by examining whether it is essential for the maintenance of ES cell self-renewal. A siRNA-based technique using 2 independent siRNAs that target different regions of the NPR-C mRNA were used to specifically knockdown the NPR-C gene in ES cells maintained in a feeder-free culture. The knockdown efficiency was analyzed at 48 h after transfection of the siRNA. RT-PCR and western blotting revealed a marked reduction in the level of NPR-C mRNA and protein at 48 h post-transfection in the ES cells that were transfected with NPR-C-targeting siRNA (NPR-C siRNA1 and NPR-C siRNA2), compared with the ES cells transfected with a nontargeting siRNA (control siRNA) (Fig. 2A–C). Recently, we have reported that other natriuretic peptides (NPR-A, NPR-B, and BNP) are also expressed in murine ES cells [8,9]. Western blot analysis showed that there were no significant changes in the expression levels of NPR-A, NPR-B, and BNP after NPR-C knockdown (Fig. 2D), thus suggesting that the effect of NPR-C knockdown on ES cells is due to a specific knockdown of endogenous NPR-C.

Knockdown of NPR-C induces apoptosis in murine ES cells.
To determine the effects of knockdown of NPR-C on ES cell viability, cell apoptosis was examined in siRNA-treated ES cells by using flow cytometry and immunofluorescence for Annexin V-positive cells and western blotting for activated caspase-3. Flow cytometry analysis and immunofluorescence microscopy revealed a significant increase in Annexin V-positive cells in both siRNAs targeting NPR-C (Fig. 2E–G). In addition, loss of NPR-C resulted in an increase in the level of activated caspase-3 (Fig. 2H). Further, NPR-C siRNA-treated cells exhibited a reduction in colony and cell numbers in comparison to those treated with control siRNA (Fig. 2I and Supplementary Fig. S1A, B). These findings suggest that NPR-C has a functional role in ES cell viability.
A previous study reported that activation of caspsase 3 is increased during ES cell differentiation [17]. However, in the current study, although caspase 3 was activated in response to NPR-C knockdown, the ES cells were maintained undifferentiated. In support to our results, a recent study has shown that loss of nucleostemin in ES cells leads to apoptosis with a dramatic increase in the levels of activated caspase 3 without affecting ES cell pluripotency [18]. Taken together, these findings suggest that loss of NPR-C may cause a low level of caspase 3 activation, which is not sufficient to induce differentiation.
To further confirm that endogenous NPR-C protects murine ES cells from apoptosis, we treated ES cells with various doses of cANF(4–23) (a specific NPR-C agonist) [13], and examined its effects on ES cell viability under conditions of H2O2-induced oxidative stress. Pretreatment of ES cells with cANF(4–23) increases the resistance of ES cells to H2O2-induced apoptosis in a concentration-dependent manner (Fig. 3). Western blot analysis showed that cANF(4–23) causes a concentration-dependent reduction in caspase-3 activity (Fig. 3A, B), thus suggesting a protective effect of cANF(4–23). Further, apoptosis analysis using flow cytometry showed a significant reduction in Annexin V-positive cells in a concentration-dependent manner (Fig. 3C, D). These results show that NPR-C activation by cANF(4–23) is able to attenuate H2O2-induced apoptosis, thus suggesting an anti-apoptotic role for NPR-C in murine ES cells.

NPR-C agonist (cANF4-23) protects murine ES cells from H2O2-induced apoptosis.
The reduction in ES cell survival caused by abrogation of NPR-C signaling had no effect on the undifferentiated status of the ES cells, as determined by evaluation of colony morphology and measurements of alkaline phosphatase (AP) activity (Supplementary Fig. S1A, C). Identical levels of AP staining were observed in the control siRNA-treated and NPR-C siRNA-treated cells (Supplementary Fig. S1C). In addition, alterations in the expression of the pluripotency markers Oct4 and Nanog were not observed after NPR-C knockdown (Supplementary Fig. S1D, E). Further, RT-PCR analysis of the early differentiation markers 48 h after siRNA transfection showed no significant difference between control siRNA-treated and NPR-C siRNA-treated cells (Supplementary Fig. S1F). Based on these observations, we propose that NPR-C knockdown in ES cells does not affect ES cell pluripotency under normal conditions.
We examined whether NPR-C knockdown has any effect on ES cell proliferation. Cell cycle analysis examined 48 h after siRNA transfection showed no clear difference between control siRNA and NPR-C siRNA-treated cells (Supplementary Fig. S2A). This indicates that the reduction in ES cell number observed with NPR-C knockdown is not due to the suppression of mitogenic activity but is rather due to an increase in the rate of cell death.
We next examined the differentiation potential of NPR-C-deficient ES cells by EB formation in the presence of RA. ES cells are commonly differentiated in vitro by spontaneously self-assembling in hanging drop culture to form EBs, which model many of the hallmarks of early embryonic development. Analysis of specific markers (GATA4, GATA6, and AFP for endoderm, Brachyury for mesoderm, nestin for ectoderm, and Eomes for trophectoderm) showed that NPR-C-deficient ES cells were capable of differentiation to all germ layers (Supplementary Fig. S2C). Morphologically, NPR-C siRNA-derived EBs were similar to those derived from control siRNA-treated cells (Supplementary Fig. S2D). Taken together, these findings indicate that the role of NPR-C is to protect ES cells from accumulating damage which induces apoptosis without affecting ES cell pluripotency.
NPR-C protects ES cells from apoptosis by regulating p53
To identify the mechanism by which NPR-C protects ES cells from apoptosis, we investigated the levels of p53 mRNA and protein in NPR-C-deficient ES cells. In ES cells, p53 is required to activate apoptosis [1,18]. We did not observe any increase in the levels of p53 mRNA (Supplementary Fig. S2B), but the level of p53 protein was greatly increased in NPR-C-deficient ES cells (Fig. 4A, B). These results indicate that the increased amount of p53 protein after NPR-C knockdown is regulated at the post-transcriptional level. Activation of p53 did not result in arrest of the ES cell cycle (Supplementary Fig. S2A), which is consistent with the lack of a G1 checkpoint in ES cells [19]. Although ES cells have high basal levels of p53, they do not undergo normal p53-mediated cell cycle arrest after DNA damage [1].
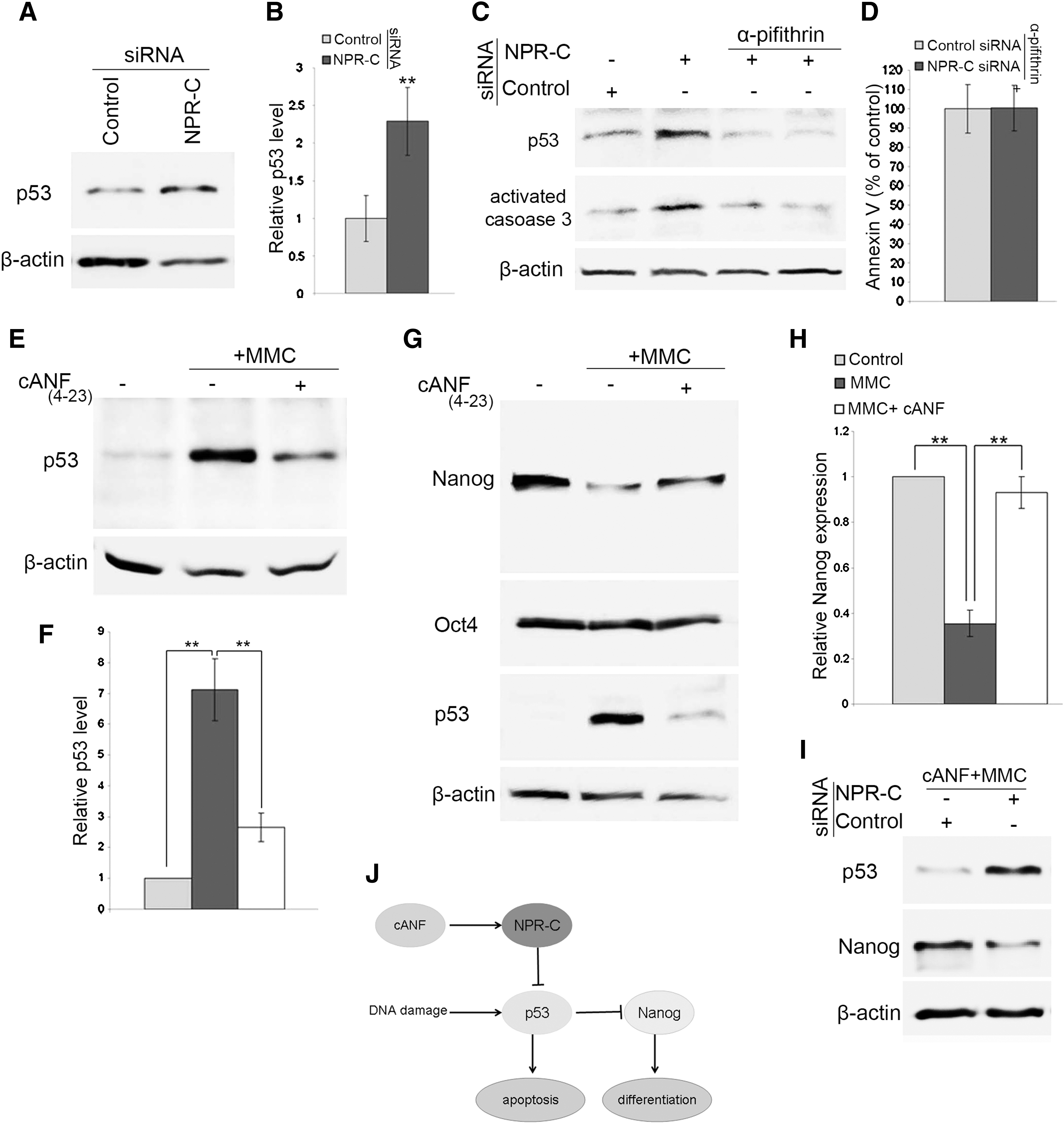
NPR-C protects ES cell from apoptosis by regulating p53 expression.
To investigate whether the effect of NPR-C on apoptosis is p53-dependent, we inhibited p53 activity in ES cells by treating them with 10 μM of pifithrin-α for 24 h [18,20]. Interestingly, inhibition of p53 in ES cells eliminated the difference in apoptosis observed between ES cells treated with control siRNA and those treated with NPR-C siRNA (Fig. 4C, D). This suggests that the cell death induced by NPR-C knockdown in ES cells may function in a p53-dependent manner.
Previous studies have reported that exposure of ES cells to DNA-damaging agents leads to activation of p53 and suppression of Nanog expression [4,21]. Therefore, to explore the relationship between NPR-C and p53, we treated ES cells with mitomycin C (MMC), an activator of the DNA damage p53/apoptosis pathway [22]. ES cells cultured in serum-free medium were treated for 6 h with MMC (5 μg/mL) alone or with MMC and cANF(4–23) (an NPR-C-specific agonist) together, and then we examined the expression of p53. As expected, MMC-treated cells showed a significant increase in the level of p53 protein. Pretreatment of ES cells with cANF(4–23) before MMC addition significantly inhibited p53 activation (Fig. 4E, F), thus indicating that activation of NPR-C can suppress p53 activation in the presence of DNA damage.
p53 responds to DNA damage signals in ES cells by promoting differentiation via direct suppression of Nanog expression, which represents an alternative pathway to maintain genetic stability in these cells [3,4,21]. Analysis of Nanog expression found that it is markedly downregulated in ES cells treated with MMC concurrent with upregulation of p53. Pretreatment of ES cells with cANF(4–23) before MMC treatment prevented the downregulation of Nanog expression (Fig. 4G, H). Nanog is a master transcription factor that is crucial for the self-renewal of ES cells [6,23], and a 50% reduction in Nanog expression leads to spontaneous differentiation [7]. Taken together, these findings indicate that selective activation of NPR-C may be effective in counteracting the DNA damage effects on self-renewal of ES cells through preventing Nanog suppression.
The DNA damage-induced p53 accumulation had no direct effect on Oct4 expression (Fig. 4G). p53 directly suppresses Nanog expression in murine ES cells [4], but in human ES cells, p53 accumulation can suppress the expression of Oct4 and Nanog [3].
To confirm that cANF(4–23) protects ES cells from DNA damage through NPR-C, control siRNA-treated and NPR-C siRNA-treated cells were treated for 6 h with cANF(4–23) and MMC together, and then, the expressions of p53 and Nanog were measured. Although the pretreatment of ES cells with cANF(4–23) before MMC addition significantly inhibited p53 activation and Nanog downregulation in control siRNA-treated cells, cANF(4–23) could not prevent activation of p53 or Nanog downregulation in NPR-C siRNA-treated cells (Fig. 4I). These findings indicate that cANF(4–23) protects ES cells from DNA damage through NPR-C.
Our results showed that although p53 had been activated by NPR-C knockdown, Nanog expression was not changed. However, MMC treatment caused a dramatic reduction in Nanog levels due to the activation of p53 protein. One possible explanation for the difference between NPR-C knockdown and MMC treatment might be the severity of the DNA damage. Apoptosis assay showed that MMC treatment led to severe cell death and a marked activation of p53 protein (Supplementary Fig. S3A–C) compared with those caused by NPR-C knockdown (Fig. 2E-F). It is possible that the level of cell death and p53 activation caused by NPR-C knockdown may be not sufficient to cause downregulation of Nanog expression to induce ES cell differentiation. However, further studies are needed to elucidate the mechanisms of the ES cell response to the different levels of DNA damage.
It has been shown that some of the actions of atrial natriuretic peptide (ANP) and BNP are mediated through their binding to NPR-C [13,14]. Previously, we have shown that ANP and BNP are also expressed in ES cells and are involved in the self-renewal of ES cells through NPR-A and NPR-B [8,9]. It is not clear whether the survival-enhancing effect of NPR-C activation is mediated by ANP and BNP in ES cells. Further studies are required to elucidate the relationship between NPR-C and natriuretic peptides in ES cells.
A more comprehensive understanding of the molecular pathways controlling ES survival, in particular the signals controlling the p53 expression, would help generate clinically appropriate cell types from ES cells, and may also enhance the reprogramming of somatic cells into induced pluripotent stem (iPS) cells [25]. Recent studies of the generation of iPS cells have shown that the loss of p53 signaling promotes reprogramming [24,25], thus indicating that upstream signaling of p53 may be essential for reprogramming by maintaining cellular stress levels. Taken together with the results presented in this study, this suggests that activation of NPR-C may facilitate the reprogramming process.
In conclusion, we have demonstrated for the first time that NPR-C is expressed in undifferentiated ES cells, and its signaling is crucial for ES cell viability through inhibition of p53-dependent apoptosis. Knockdown of NPR-C in ES cells leads to apoptosis, associated with activation and accumulation of p53. Further, activation of NPR-C with its specific agonist protects ES cells against oxidative stress-induced apoptosis, and blocks activation of p53 and Nanog suppression in the presence of DNA damaging agents (Fig. 4J). These findings provide a novel signaling mechanism involved in maintaining ES cell viability.
Footnotes
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research (No. 21–09133) from the Japan Society for the Promotion of Science.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.