
Abstract
In contrast to hematological malignancies, meaningful improvements in survival statistics for patients with malignant brain tumors have not been realized in >40 years of clinical research. Clearly, a new medical approach to brain cancers is needed. Recent research has led to a new concept that needs to destroy all cancer subclones to control the cancer progression. However, this new concept fails to distinguish the difference between dominating subclones and dormant subclones. Here, we address the issue of clonal switch and emphasize that there may be one or more than one dominant clones within the tumor mass at any time. Destructing one dominant clone triggers activating other dormant subclones to become dominating subclones, causing cancer progress and post-treatment cancer recurrence. We postulate the concept of subclonal switchboard signaling and the pathway that involved in this process. In the context of stem cell and development, there is a parallel with the concept of quiescent/dormant cancer stem cells (CSC) and their progeny, the differentiated cancer cells; these 2 populations communicate and co-exist. The mechanism with which determines to extend self-renewal and expansion of CSC is needed to elucidate. We suggest eliminating the “dominating subclonal switchboard signals” that shift the dormant subclones to dominating subclones as a new strategy.
Background
T
The Hypotheses
Burgess' conclusion was based on 2 Nature articles on genetic complexity and heterogeneity of cancer. Anderson and colleagues found that the classic model of the linear clonal evolution could not explain their data because multiple subpopulations (also known as subclones) co-exist [4]. Burgess proposed a hypothesis, “genetic heterogeneity and the branching evolutionary trajectories,” to explain the Darwinian perspective of evolving these leukemia-initiating cells [3]. Surprisingly, Notta and colleague reported that these co-existed subclones of many leukemia patient samples co-evolve during disease progression and post-treatment relapse by shifting subclonal dominance [5]. Most importantly, this shifting subclonal dominance can reproduce by using transplantation assays.
Here, we provide a new hypothesis that this shifting subclonal dominance is controlled by the subclonal SSS (Fig. 1). Using experimental models [3,5], we can decipher these SSS, so we can specifically block their signal transduction and stop the subclonal switchboard function. However, we must be ready to co-exist with the cancer cells in our body. These cancer stem cells (CSC) may be not detrimental as long as we can keep them in surveillance.
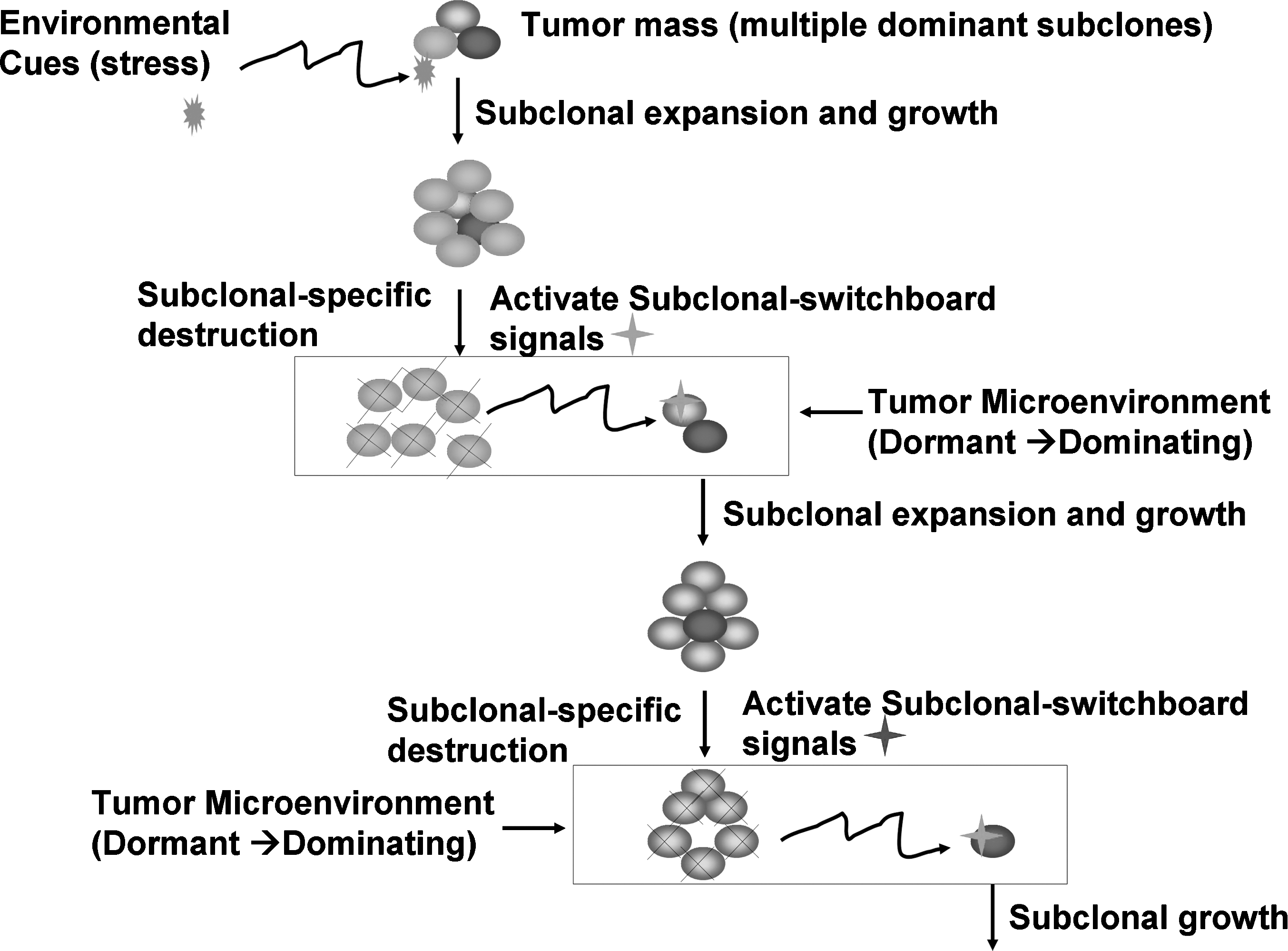
Subclonal switchboard signals (SSS) as mechanisms for leading to shift dominating subclones as triggered by environmental cues (stress) for cancer progression and post-treatment. A cancer subclone may gain a mutation that, in the appropriate environment cue, leads to dominating subclonal activation due to positive selection. Showed lettering and lines/arrows in black color is the current concept of treatment strategy for cancer-dominant subclonal cells (cancer stem cells) that may acquire a mutation, in the suitable environment, triggering to dominating subclonal expansion and growth. When this dominating subclone is specifically destroyed, it sends out dominating subclonal-SSS to a dormant/quiescent subclonal cell, which gets activated for dominating subclonal expansion and growth.
Emerging evidence supports the SSS concept. Cancer cells have been traditionally treated as invading aliens, which must be completely destroyed and removed. We may, however, argue for the need to view cancer differently from traditionally. We and others have found that similarities and overlapping mechanisms between induced cell plasticity and cancer formation shed new light on the emerging picture of p53 sitting at the crossroads between 2 intricate cellular potentials: stem cell versus cancer cell generation [6] and regulating the quiescence and self-renewal of hematopoietic stem cells [7]. We may over-react toward cancer cells, “the invading aliens,” which lead to over-treating and injure our own body by using aggressive multi-modalities (surgery, radiation, and chemotherapies) [8]. Perhaps, we ought to consider that cancer cells share similar citizenship, demanding to survive on the Earth because their survivorship is driven by their evolutional driving force [9]. Sustaining the biodiversity and heterogeneity may balance organisms or organs out of the hostile environment [10,11]. As such, managing tumor growth rather than eliminating it should be a new guideline for treating tumors. The eliminating-cancer treatments drive producing populations of drug-resistant tumor cells upon eliminating the drug-sensitive cells while managing tumor growth to treat tumors with minimum doses of drug so as to modulate the survival of some drug-sensitive cells [12,13]. This treatment paradigm may help the drug-sensitive cells out-compete the resistant ones upon completion of drug treatment, thereby keeping tumors alive but small and manageable [1,14].
“Keeping tumors alive but small and manageable” sounds a reasonable strategy. However, how can we manage tumor growth rather than eliminate it? We argue that managing SSS may be an effective strategy.
Implications of the Hypothesis
Understanding the mechanisms for SSS will provide new insights to develop anti-cancer therapies. The SSS may be activated upon eliminating a drug-sensitive dominant subclonal population, and SSS may activate a neighboring dormant subclone within a microenvironment (tumor) or traffic out of the tumor microenvironment (Fig. 2). Upon characterization of SSS, we can better detect and control the outbreaks of SSS-driven cancers with defensive strategies for disruptions of SSS signal transduction during different stages of tumorigenesis and cancer progression.
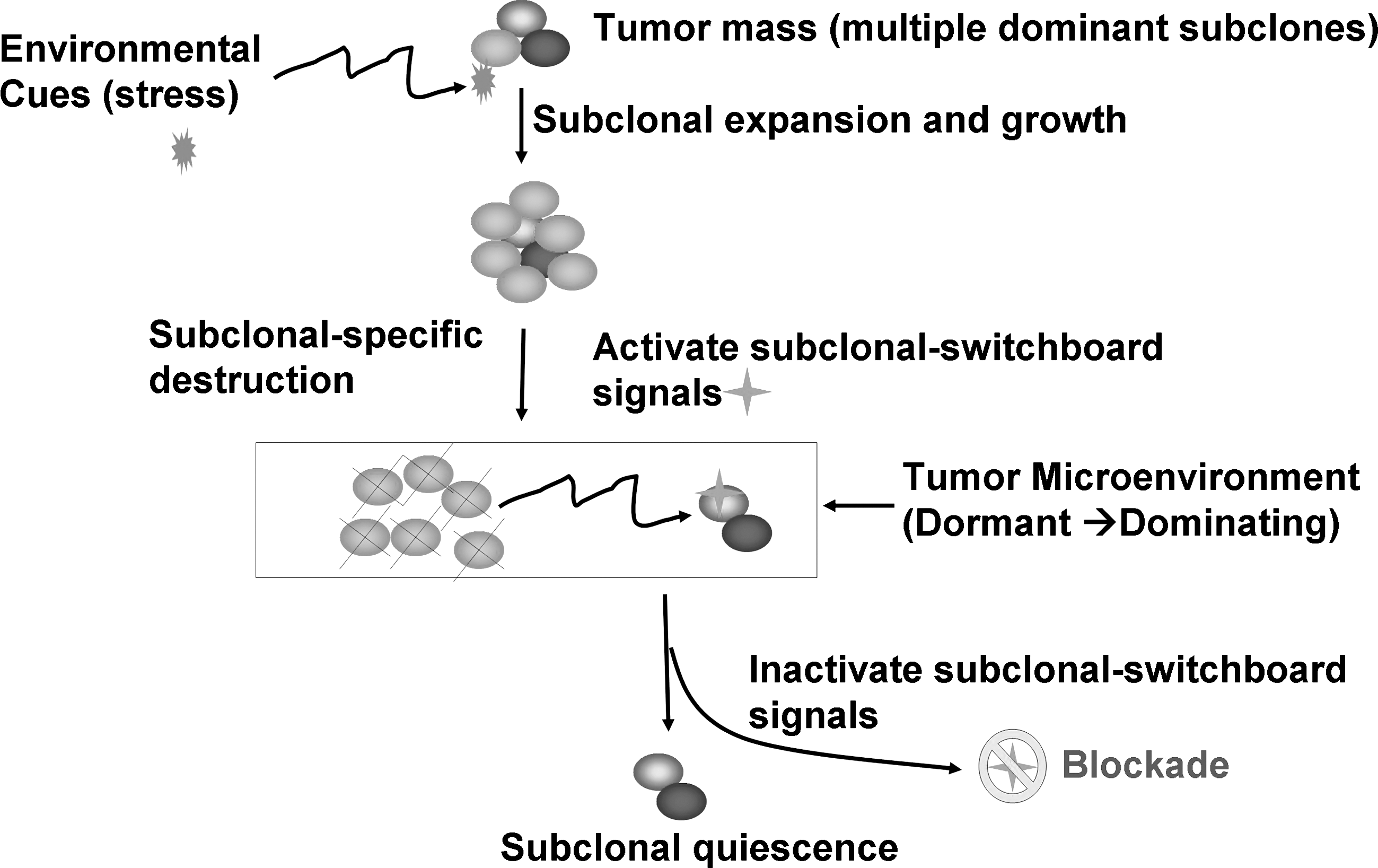
Blockade of the dominating subclonal SSS as a new therapeutic strategy to suppress the dominating subclone shift to control cancer progression and post-treatment cancer recurrence. Showed is the proposed new treatment paradigm that should target the subclonal-SSS. Blocking the dominating subclonal SSS leads to subclonal quiescence, so keeping tumors alive but small and manageable (dormant/quiescent subclone).
A defining feature of SSS system is the presence of multiple structural elements specializing in distinct biological functions. As a general rule, these elements can stably maintain their identity over long periods despite fluctuations in their external physiological environment and internal regulatory networks [15]. Tumor–tissue barrier (physical boundary) may maintain the intra-tumor pressure during tumor development [16]. The quiescent/dormant CSC and their progeny, the differentiated cancer cells, may communicate and co-exist within the tumor microenvironment. Leakage of the tumor–tissue barrier via treatments (surgery, radiation, and chemotherapy) may lead to change the intra-tumor pressure that may act as a physical SSS signal to wake up a dormant subclone. How this is achieved at the molecular level is a central but poorly understood question. An interesting finding was that the matrix elasticity (physical signal) activates the stem cell differentiation signal pathway, directing the organ-specific stem cell lineage specification [17 –19]. The nature of the chemical-based SSS signals and the pathways that involved in the signaling process are not well understood. Some examples of the SSS signals may be cytokines [20,21], growth factors [22], angiogenesis factors [23,24], and the balance of their expression levels [25]. Indeed, irradiation of mouse bone marrow stromal cell lines induces release of significant levels of transforming growth factor (TGF)-beta into the tissue culture medium despite the lack of a detectable increase in TGF-beta mRNA [26]. TGF-beta regulates the coexistence and interconvertibility of CSCs that sustain tumor growth through their ability to self-renew and to generate differentiated progeny [27].
It is possible that SSS is maintained by modifying host gene products. This model predicts, for example, that many host gene products changes after the host is treated by an anti-tumor agent. This is in line with the recognition that there is a combined effect of 2 distinct inputs: positive and negative. The positive inputs from the SSS may be to activate biological events that favor the host, and the negative inputs may be detrimental to the host.
We can design a comprehensive strategy that allows the systematic identification of those inputs from the SSS. Specifically, we can fundamentally define the SSS by execution of the following experiments: 1. assess the time-course of dominant subclonal formation in vivo; 2. determine the biochemical nature of SSS molecules upon dominant subclonal destruction; 3. activate the dormant (quiescent) cancer subclone using SSS molecules.
The SSS hypothesis has broad impact on many areas of biology and medicine, including developmental biology, stem cell biology, cancer biology, aging, epigenetics, functional genomics, systems biology, regenerative medicine, molecular diagnostics, and drug discovery. For example, it implies that cancers and neurodegenerative disorders may be governed by the same SSS mechanism, which escapes the host surveillance system for their subclonal reproduction. The organizational structure of normal epithelium may be such a host surveillance system [12], and the tumor microenvironment may evolve in the SSS production and traffic [28]. Noninvasive monitoring of intra-tumor SSS behavior in vivo is potentially useful for evaluating the efficacy of individual treatment responses with prognostic value in the clinic [29]. However, understanding the SSS-evolved host surveillance for cancer suppression mechanisms is essential to defining the first steps of tumorigenesis and developing rational cancer prevention strategies.
Footnotes
Author Disclosure Statement
No competing financial interests exist.