
Abstract
To grow more robust cardiac tissue for implantation in vivo, strategies to improve survival of implanted stem cells are required. Here we report the protective effects of hypoxic preconditioning (HPC) and identify mechanisms for improving survival of adipose-derived stem cells (ASC) in vitro. Human ASC were preconditioned for 24 h with hypoxia and then exposed to simulated ischemia for a further 24 h. HPC significantly increased ASC viability, and reduced cell injury and apoptosis compared with non-preconditioned cells under ischemic conditions, as shown by 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), lactate dehydrogenase-release, and caspase activity assays. Preconditioned ASC increased levels of hypoxia-inducible factor-1 alpha and secreted significantly more of the downstream target vascular endothelial growth factor (VEGF-A; 13-fold) compared with control during the 24 h. Exogenous VEGF (50 ng/mL) increased phosphorylation of Akt without affecting ERK1/2, JNK, or p38 MAPK protein levels. Phospho-Akt was also increased in preconditioned ASC compared with non-preconditioned ASC, an effect that may be mediated via VEGF-A. Importantly, the protective effects of HPC were abolished by a neutralizing antibody against VEGF-A and the phosphoinositol 3-kinase inhibitor LY294002, demonstrating the importance of VEGF-A and Akt in hypoxia-induced ASC survival. Importantly, we showed that media derived from hypoxic preconditioned ASC support endothelial cell survival and endothelial tube formation in vitro. Our in vitro findings indicate that HPC may be a promising strategy to improve survival of ASC and promote angiogenesis in ischemic environments.
Introduction
R
Preconditioning occurs when cells are able to survive a lethal insult due to prior activation of survival mechanisms by a sub-lethal stimulus, and this is one method shown to improve stem cell survival in harsh environments. Multiple studies have demonstrated that both pharmacological preconditioning and genetic manipulation improves the survival of bone marrow-derived mesenchymal stem cells (BM-MSC) after implantation into the infarcted heart [4,5]. Preconditioning with hypoxia is another clinically viable way in which cells can be protected [6,7].
Hypoxia is involved in both physiological and pathophysiological processes. Although severe hypoxia can lead to cell death in certain cell types such as endothelial cells, exposure to brief hypoxia (i.e., preconditioning) has been shown to confer cytoprotective benefits [6 –9]. These include promoting cell survival and angiogenesis, both of which are fundamental for the success of cell therapies. Hypoxia exerts these protective effects through stabilization and accumulation of hypoxia inducible factor-1 alpha (HIF-1α), which in turn leads to activation of downstream mediators and signaling molecules involved in the regulation of glucose and energy metabolism, angiogenesis, and cell survival, such as vascular endothelial growth factor (VEGF-A) and protein kinase B (Akt) [8,10]. In stem cells, hypoxia has been shown to increase expression and secretion of growth factors, including VEGF-A, basic fibroblast growth factor (bFGF), insulin-like growth factor 1 (IGF-1), hepatocyte growth factor (HGF), and stromal-derived factor 1 (SDF-1), all of which have known cytoprotective actions [6,7,11 –13]. Although the pro-survival effect of hypoxic preconditioning (HPC) has been widely studied in BM-MSC and neural progenitor cells [6,7,14,15], whether HPC is also beneficial for adipose-derived stem cells (ASC) is currently unknown and is the aim of the present study.
ASC are an easily accessible source of stem cells. They are rapidly expandable and can be obtained in large numbers through minimally invasive, routine liposuction procedures, in contrast to their bone marrow counterparts [16,17]. ASC are phenotypically similar to BM-MSC and give rise to adipocytes, chrondrocytes, and osteoblasts [18]. Interestingly, we have recently shown that ASC are able to differentiate into cardiomyocytes in vitro and in vivo, suggesting that they may be a promising source of cells for cardiac repair [19,20].
The present study demonstrates for the first time that preconditioning ASC with hypoxia increases cell survival via a HIF-1α/VEGF-A/phosphoinositide 3-kinase (PI3K)-Akt-mediated pathway. Further to this, we found that HPC of ASC increases their paracrine effects in promoting endothelial cell survival and proangiogenic responses in vitro, both of which are important elements of efficacy for stem cell-based regenerative therapies.
Materials and Methods
Isolation and culture of primary human ASC
ASC were isolated from freshly excised human female abdominal adipose tissue (donor age 45–63) according to a previously described method [21]. Approval for experimentation was obtained from the St. Vincent's Health Human Research Ethics Committee. Briefly, tissue was minced and washed with equal volumes of phosphate-buffered saline (PBS) before incubation at 37°C for 60 min in 0.1% collagenase type I (Worthington Biochemical). Cells were then centrifuged at 1,200 rpm for 10 min to remove adipocytes. The pellet was resuspended in 0.16 M NH4Cl and incubated for 5 min at room temperature to lyse red blood cells. Cells were collected by centrifugation at 1,200 rpm for 5 min and filtered through a 100 μm nylon mesh to remove debris. Finally, cells were incubated in complete DMEM-low glucose (DMEM-LG containing 10% fetal calf serum (FCS) and 1% antibiotic/antimycotic; Invitrogen) in culture flasks at 37°C in a humidified CO2/O2 incubator overnight. The next day, cells were washed extensively to remove nonadherent cells. The mesenchymal population (ASC) was selected based on their adherence to plastic and nonadherent cells were removed by regular medium changes. The cells were previously shown by flow cytometry to be positive for MSC markers (CD73, CD90, and CD105) and negative for CD34 and CD45 [19]. At confluence, cultures were harvested with 0.25% trypsin/EDTA and passaged at a ratio of 1:3. Passage 4–6 ASC were used in this study. ASC were plated at 5,000 cells/cm2 for 16 h at 37°C in a humidified CO2/O2 incubator before all experiments.
HPC and simulated ischemia in ASC
All experiments were conducted in serum-free DMEM-LG media. Ischemia was simulated under hypoxic conditions using serum- and glucose-free DMEM. Hypoxia was induced in a sealed, hypoxia GENBox Jar (BioMérieux) at 37°C, in which oxygen is depleted by an enzymatic reaction. Final gas compositions in the chamber were <0.1% O2 and 15% CO2.
To assess the effects and mechanisms of HPC, ASC were assigned to 1 of 8 experimental groups as follows (Fig. 1A): (i) control (48 h normoxia); (ii) HPC (24 h hypoxia+24 h normoxia); (iii) LY294002 (a PI3K inhibitor, 10 μM); (iv) VEGF neutralizing antibody (VEGF-Ab, 0.6 μg/mL); (v) simulated ischemia; (vi) HPC+simulated ischemia (24 h HPC+24 h ischemia); (vii) HPC+LY294002+simulated ischemia; and (viii) HPC+VEGF-Ab+simulated ischemia. LY294002 and VEGF-Ab were given during the 24 h of HPC.
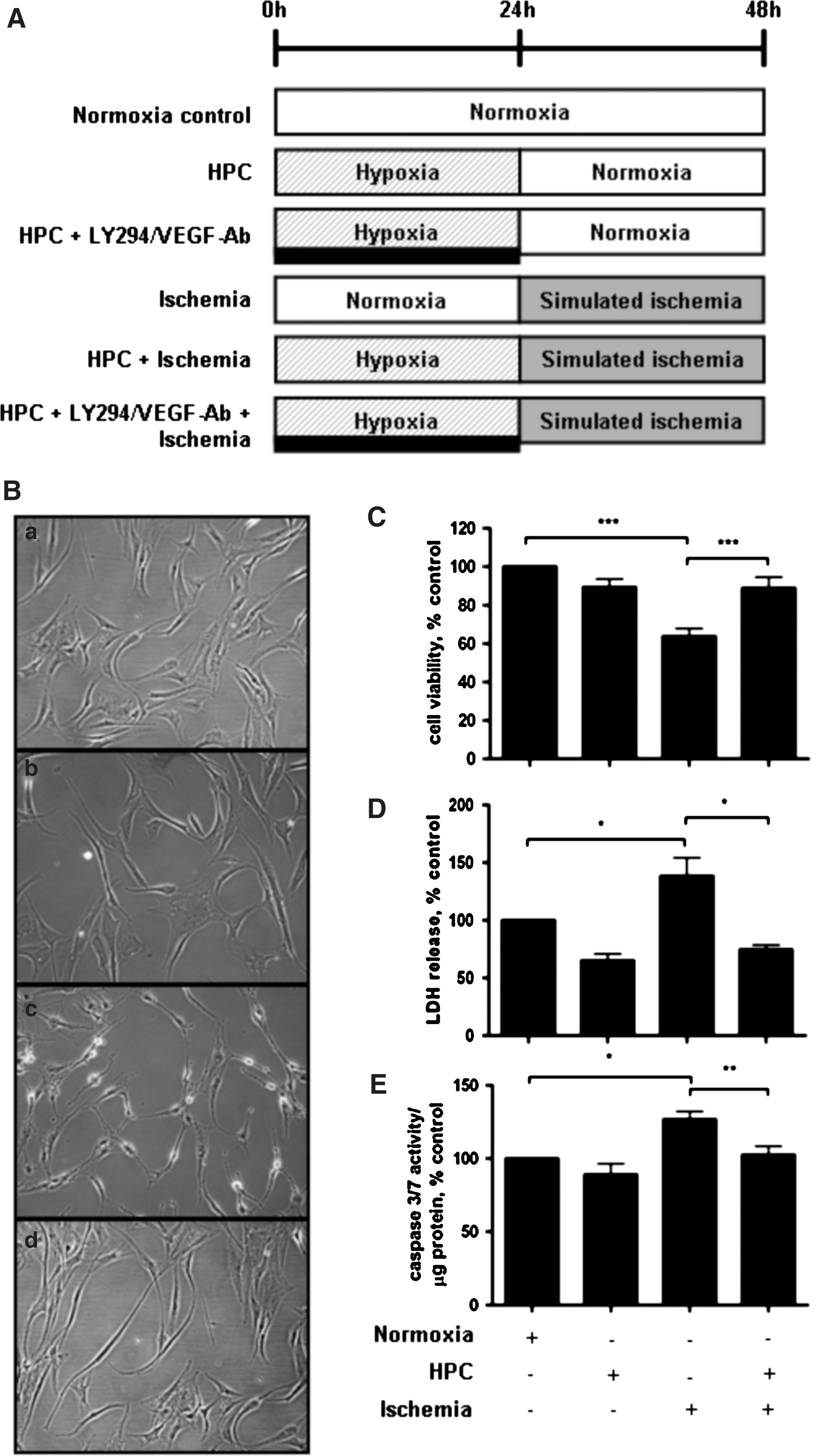
ASC were preconditioned with hypoxia for 24 h before simulated ischemia
Measurement of cell viability
Cell viability was measured by the MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay (Sigma-Aldrich). Briefly, a 5 mg/mL MTT salt solution was prepared and added to cells after 24 h simulated ischemia to give a final concentration of 2.5 mg/mL MTT. Cells were then incubated at 37°C for 1 h. The final formazan product was dissolved in dimethyl sulfoxide and absorbance measured at 550 nm. The amount of formazan was directly proportional to the number of live cells.
Measurement of lactate dehydrogenase release
Cellular injury was evaluated after exposure to ischemia for 24 h using a lactate dehydrogenase (LDH) release assay. Cell supernatant was analyzed using an LDH assay kit (Sigma-Aldrich) as per the manufacturer's instructions at 490 nm.
Measurement of caspase 3/7 activity
Caspase 3/7 activity was measured using a commercially available assay (Caspase-Glo® 3/7; Promega). Briefly, 50 μL caspase 3/7 reagent was added to 20 μL cell lysate and incubated for 5 min at room temperature to ensure complete cell lysis. Caspase 3/7 assays were conducted in duplicate in a 96-well plate and luminescence measured using a POLARstar OPTIMA (BMG Labtech) at 25°C. Caspase 3/7 activity was standardized to protein content and expressed as caspase 3/7 activity/μg protein.
Real-time reverse transcriptase–polymerase chain reaction
RNA was extracted from cells using the RNAqueous® Kit (Ambion) according to the manufacturer's instructions. Real-time reverse transcriptase–polymerase chain reaction (PCR) of human-specific glucose transporter 1 (GLUT1), bFGF, HGF, IGF-1, SDF-1, VEGF-A, VEGF receptor (VEGFR1 and VEGFR2), and 18S genes was performed using TaqMan technology. Reactions were carried out in duplicate in 96-well plates with the ABI Prism 7300 Real-Time PCR System (Applied Biosystems) and analyzed using the ΔCt method. Relative fold changes were compared to control (normoxia), which were normalized to 1, and all results were expressed as relative fold-change in candidate gene expression for each treatment group.
Western immunoblotting
Cells cultured in 100 mm dishes were washed with cold PBS and lysed with 100 μL of cell lysis buffer (pH 7.5) containing 100 mM NaCl, 1% Triton X-100, 10 mM Tris, 2 mM EDTA, and a protease inhibitor cocktail (Roche). Cell lysates were centrifuged at 13,000 rpm at 4°C for 15 min and the supernatants mixed with (6×) Laemmli buffer and boiled for 5 min. Equal amounts of protein were separated through SDS-PAGE gels by electrophoresis and transferred onto a Hybond nitrocellulose membrane (Amersham) by semi-dry electrophoretic transfer. The membrane was then blocked with 5% nonfat milk in TBS (pH 7.5) and hybridized overnight at 4°C with primary antibodies against pAkt (Cell Signalling; 1:1,000), Akt (Cell Signalling; 1:2,000), HIF-1α (Santa Cruz; 1:200), pERK (Cell Signalling; 1:1,000), ERK (Cell Signalling; 1:2,000), p-p38 MAPK (Cell Signalling; 1:1,000), p38 MAPK (Cell Signalling; 1:2,000), pJNK (Cell Signalling; 1:2,000), JNK (Cell Signalling; 1:1,000), and β-actin (Sigma-Aldrich; 1:5,000). Proteins were detected using enhanced chemiluminescence (ECL) with horseradish peroxidise-conjugated secondary antibodies (Amersham).
ELISA
Secreted VEGF-A, SDF-1, bFGF, and IGF-1 were quantified using a human Quantikine ELISA kit (R&D systems) according to the manufacturer's instructions. Results were compared with a standard curve and each assay was carried out in duplicate. Absorbance was measured at 450 nm using a microplate reader (Molecular Devices).
Activity of conditioned media on human endothelial cells
Endothelial cell survival and tube formation was assessed in the presence of ASC-conditioned media. Confluent ASC were washed extensively with PBS, serum-free DMEM-LG was replaced, and cells were subjected to normoxia or hypoxia (0.1% O2) for 48 h. At the end of the experimental time period, the conditioned media (Norm-ASC-CM and Hyp-ASC-CM, respectively) were collected and centrifuged at 300 g for 5 min to remove any cellular debris. Samples were frozen at −80°C until further use.
Human dermal microvascular endothelial cells (HMEC-1; a gift from Prof. Philip Hogg, University of New South Wales, Sydney, Australia) were cultured in endothelial growth media (EGM, BulletKit; Cambrex Corporation) containing 2% FCS, bovine brain extract, human epidermal growth factor, hydrocortisone, gentamicin, and amphotericin B at 37°C in a humidified CO2/O2 incubator. For survival and apoptosis assays, HMEC-1 were seeded at 10,000 cells/cm2 in EGM and incubated at 37°C in a humidified chamber overnight. The next day, media were removed and cells were washed with PBS. The medium was then replaced with serum-free DMEM-LG (control), Norm-ASC-CM, or Hyp-ASC-CM, and cells were incubated under hypoxia (0.1% O2) for 48 h. After hypoxia, the effects of ASC-CM on hypoxia-induced endothelial cell death were measured by MTT survival assay as described above.
To measure the angiogenic effects of ASC-CM (normoxic and hypoxic) on endothelial cells, 25,000 HMEC-1 (in control media or ASC-CM) were seeded onto growth factor-reduced Matrigel™ in a 48-well plate in the presence of Norm-ASC-CM, Hyp-ASC-CM, or Norm-ASC-CM supplemented with 50 ng/mL of VEGF. Tube formation was measured at 12 h.
Statistical analysis
All experiments were performed at least 3 times and data are expressed as mean±SEM. Statistical significance was assessed by analysis of variance followed by Bonferroni post-hoc testing or unpaired t-test using Prism 5 software (GraphPad). P values<0.05 were considered statistically significant.
Results
Protective effects of HPC
Hypoxia alone for 24 h did not affect ASC morphology (Fig. 1B). After 24 h of simulated ischemia, ASC morphology was changed from a regular spindle-shape as seen in control cells cultured under normoxic conditions to cells with shrunken cytoplasm, which were beginning to lift off the tissue culture plastic surface (Fig. 1B). HPC was able to prevent these morphological changes (Fig. 1B).
Cell viability, as assessed by MTT assay, was significantly reduced by 24 h simulated ischemia. Compared to non-preconditioned ASC, cells subjected to HPC had significantly higher cell viability when challenged with ischemia (Fig. 1C). Similarly, an LDH release assay revealed that ASC were protected against ischemia-induced cellular injury by HPC (Fig. 1D). Apoptosis, as assessed by caspase 3/7 activity, was also significantly decreased in the HPC group compared with the non-preconditioned ischemic group (Fig. 1E). Importantly, HPC alone did not affect ASC viability compared with control.
HPC induces HIF-1α and VEGF-A synthesis
To determine the mechanisms responsible for the observed protection afforded to ASC by HPC, we evaluated gene and protein expression levels of several cytoprotective molecules. HIF-1α protein expression was up-regulated in ASC after HPC (Fig. 2A). Furthermore, preconditioned ASC expressed significantly increased mRNA levels of the HIF-1α downstream target genes VEGF-A (21.3±3.5-fold at 24 h, Fig. 2B) and SDF-1α (1.5±0.04-fold at 12 h, data not shown). Other growth factors (bFGF and IGF-1) were not affected, but GLUT1, an important regulator of glucose metabolism, was significantly up-regulated during the period of HPC (data not shown). Similar upregulation of basal VEGF-A mRNA by HPC was found in mesenchymal stem cells derived from bone marrow (see online Supplementary Fig. S1; Supplementary Data are available online at
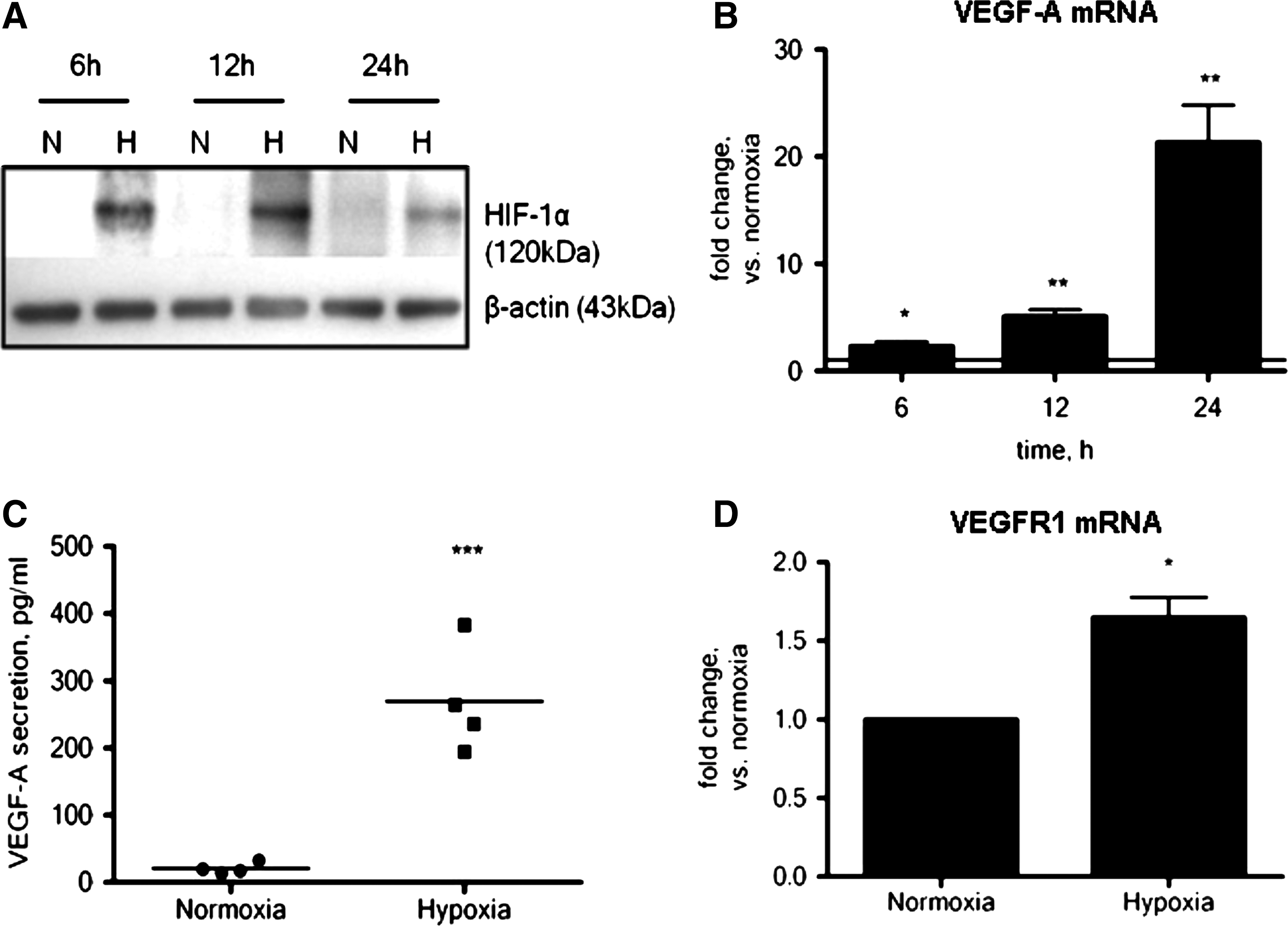
HIF-1α protein and its downstream target genes are regulated by hypoxia. HIF-1α was stabilized during the period of hypoxia (n=3,
Expression and secretion of growth factors were confirmed by ELISA. Basal VEGF-A secretion (20.9±4.1 pg/mL) was significantly increased after the 24 h period of HPC (270±41 pg/mL), representing a 13-fold increase (Fig. 2C). The observed increase in SDF-1α gene expression under hypoxic conditions did not, however, translate to an increase in SDF-1α protein secretion (data not shown). Similarly, HPC did not increase bFGF or IGF-1 secretion (data not shown), suggesting that under these conditions, the protective effects of HPC on ASC is at least in part related to the production of VEGF-A and not SDF-1α, IGF-1, or bFGF. It is important to note that ASC do express mRNA for the VEGF receptor, VEGFR1, allowing for the possibility of an autocrine mechanism. In fact, HPC caused a small but significant increase in VEGFR1 gene expression (1.6-fold compared with normoxia; Fig. 2D). VEGFR2 was undetectable in ASC, and HPC did not increase VEGFR2 gene expression (data not shown).
Exogenous VEGF-A activates Akt in ASC
VEGF-A, which is increased by HPC in the present study, can activate various downstream signaling pathways such as the PI3K/Akt pro-survival pathway. To investigate the involvement of this pathway in ASC, we evaluated the phosphorylation state of Akt after VEGF stimulation. Figure 3A shows that VEGF (50 ng/mL) increased pAkt in ASC. ERK, which is known to promote proliferation, was not activated after VEGF treatment, nor were the stress-activated kinases p38 MAPK and JNK (Fig. 3B–D). Time-course studies confirmed that VEGF-A-mediated activation of Akt is decreased in the presence of LY294002 and a VEGF-A neutralizing antibody (Fig. 3E).
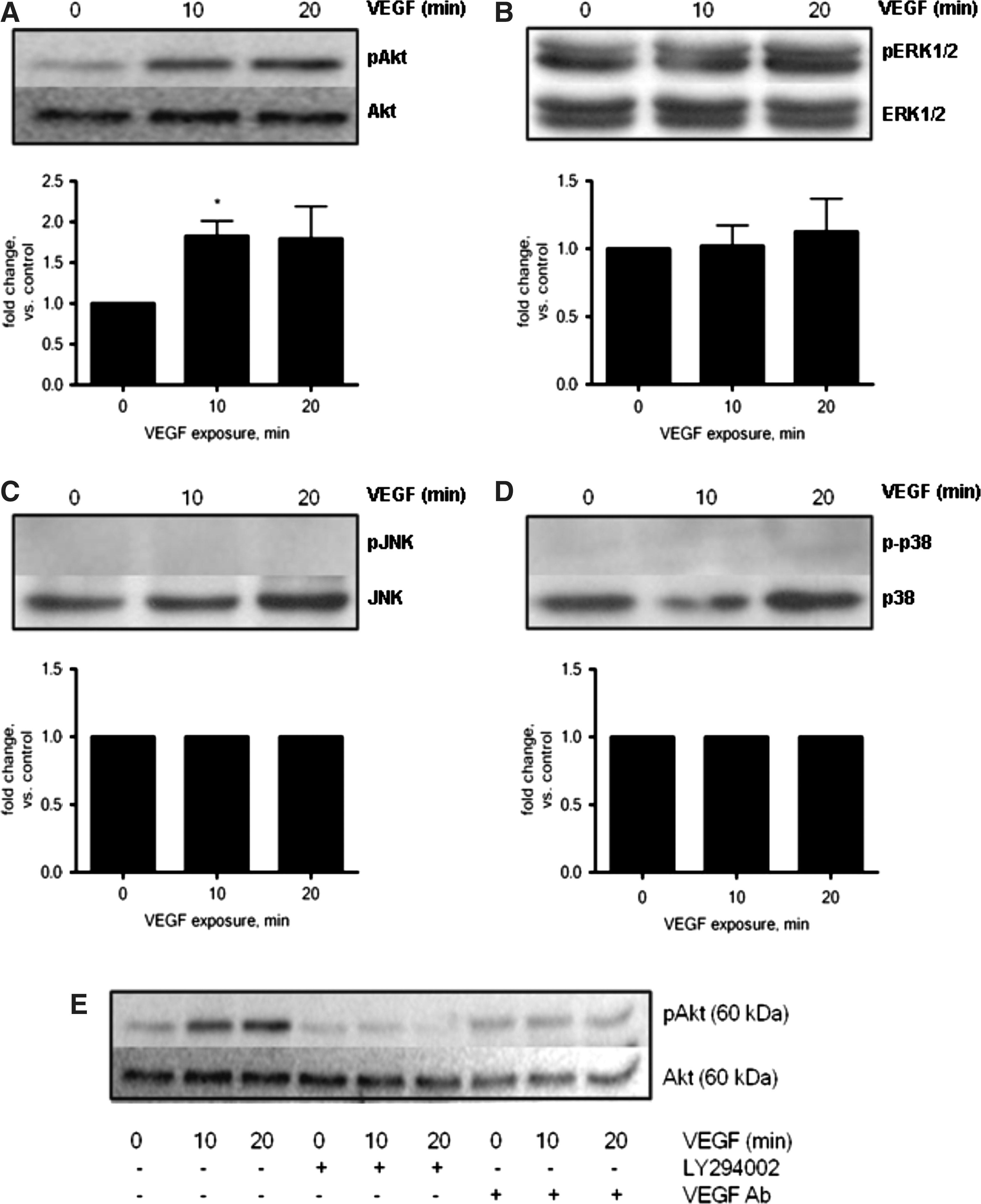
VEGF-mediated activation of the pro-survival signaling molecule Akt. ASC were treated with VEGF (50 ng/mL) for 10 or 20 min. VEGF activated Akt in ASC (n=3, P<0.05,
HPC improves ASC survival through VEGF and Akt pathways
Given the degree by which VEGF-A was elevated over the period of HPC, we investigated the importance of VEGF in HPC-induced ASC protection under ischemic conditions. The protective effect of HPC was abolished in the presence of a VEGF-A neutralizing antibody (Fig. 4A), suggesting that secreted VEGF-A (Fig. 2C) may act in an autocrine fashion to promote ASC survival. The neutralizing antibody against VEGF-A alone did not affect ASC viability.
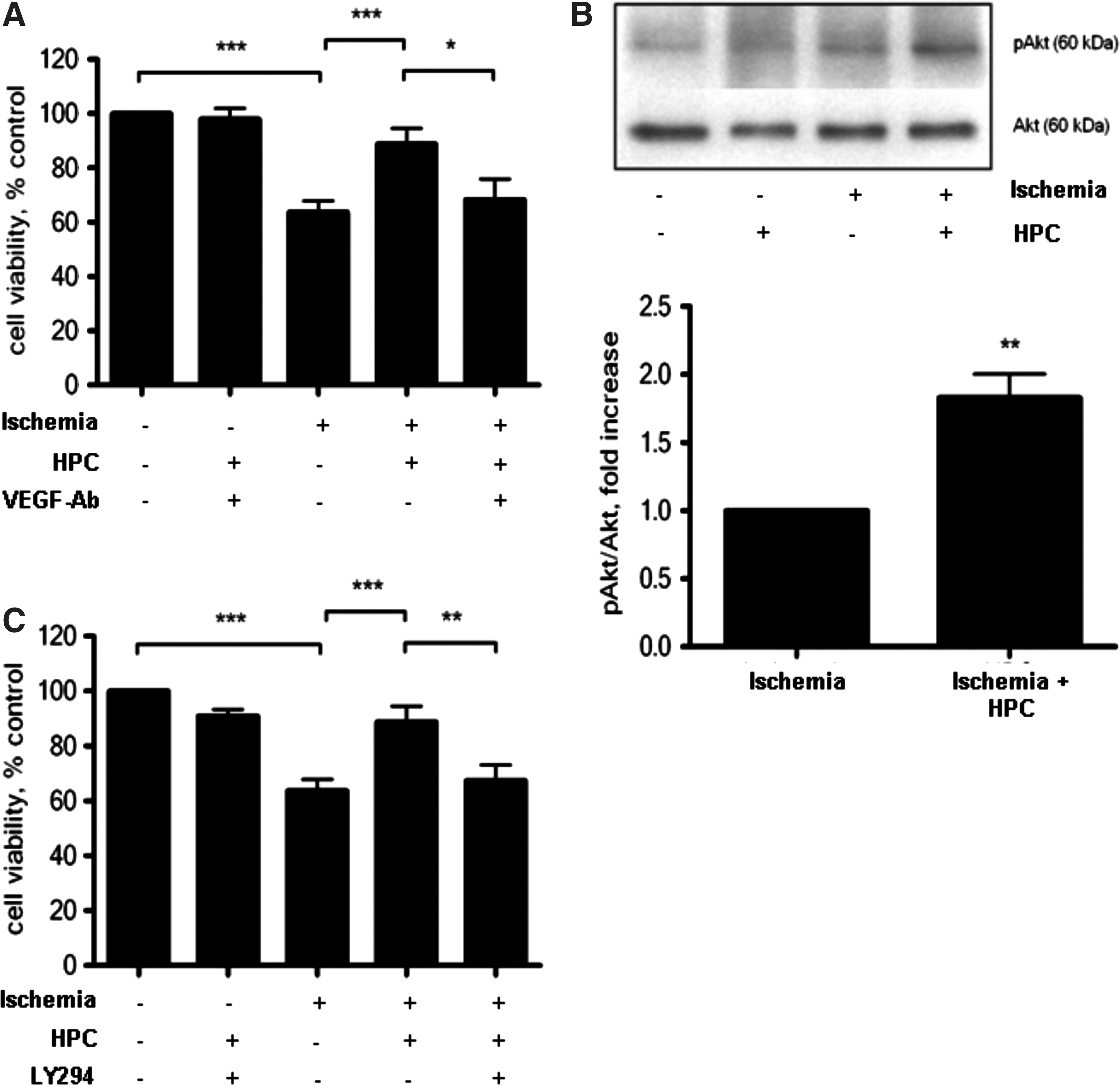
HPC protects ASC from ischemia in VEGF- and Akt-dependent manner. The protective effects of HPC were abolished in the presence of a VEGF-A neutralizing antibody (
HPC also significantly increased Akt phosphorylation (1.8-fold, Fig. 4B) in ASC compared to their non-preconditioned ischemic counterparts. The protective effect of HPC was abrogated in the presence of the PI3K inhibitor, LY294002, demonstrating the importance of Akt in mediating HPC-induced ASC survival (Fig. 4C). LY294002 alone did not affect ASC viability. We therefore propose that in this model, HPC stabilized HIF-1α and enhanced the production of the downstream target, VEGF-A. Binding of VEGF-A to VEGFR1 activates the PI3K pathway, leading to downstream phosphorylation of Akt and subsequently increased survival of ASC under ischemic conditions (Fig. 5).
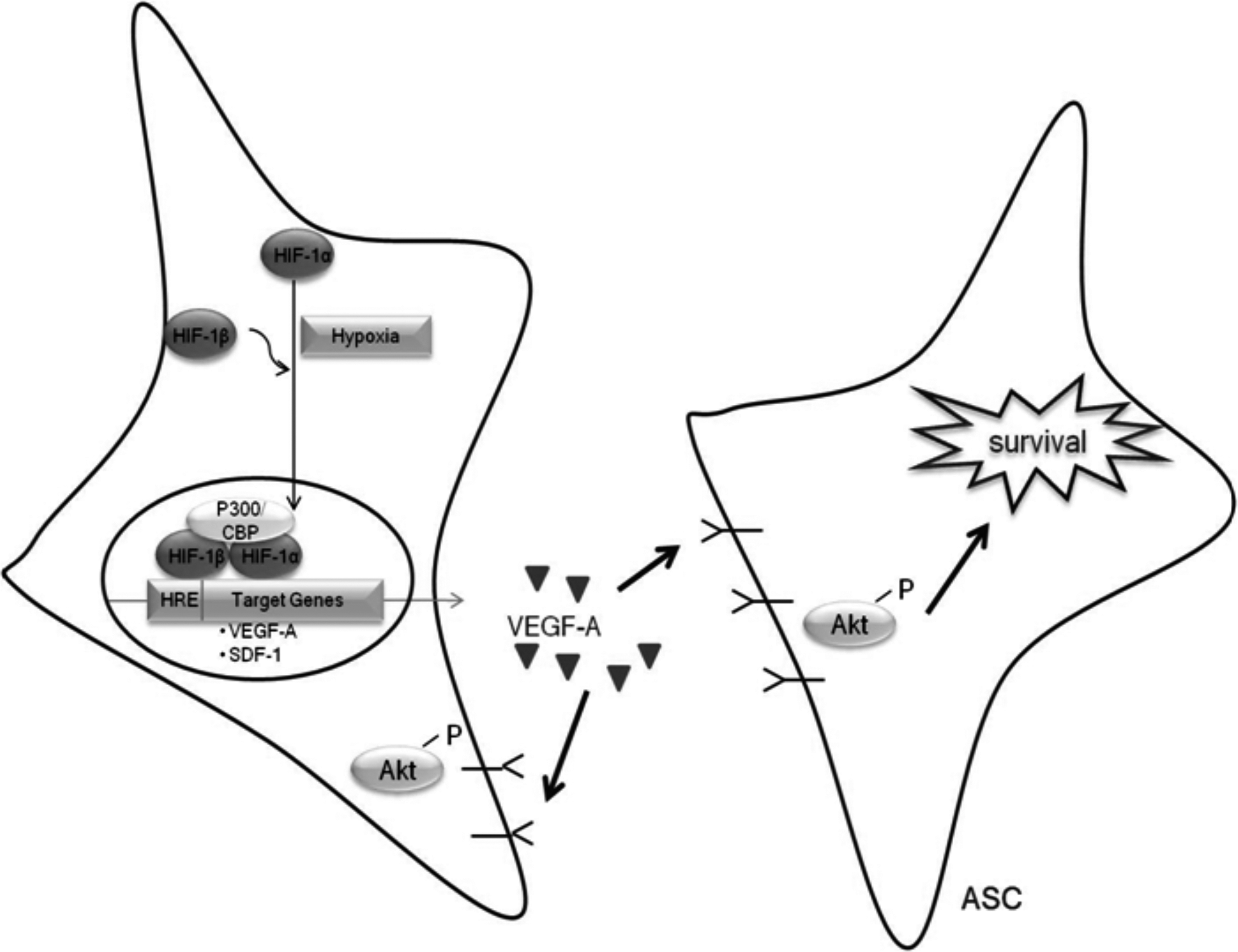
ASC survival response to HPC. Preconditioning ASC with hypoxia induced the stabilization of HIF-1α. Stabilized HIF-1α dimerises with the constitutively active HIF-1β and the HIF-1 dimer translocates to the nucleus, where it interacts with the hypoxia-responsive element on target genes, including VEGF-A. Hypoxia increases VEGF-A transcription and protein secretion from ASC. Secreted VEGF-A is then able to interact with its receptor, VEGFR1, which is present in ASC. Secretion of VEGF-A also leads to activation of its downstream target Akt, which is a key regulator of HPC-mediated survival of ASC under ischemic conditions.
Hypoxic preconditioned ASC promote endothelial cell survival and angiogenesis
To evaluate the capacity of these VEGF-producing cells to support therapeutic angiogenesis in a paracrine manner, the effect of ASC-conditioned media on endothelial cell survival and the formation of a functional vascular network was assessed. Here we showed that endothelial cells, in the absence of serum, are susceptible to hypoxia-induced death (Fig. 6A). Under these hypoxic conditions, endothelial cell viability was significantly improved by culture medium harvested from hypoxic preconditioned ASC (Hyp–ASC-CM) but not by media from non-preconditioned ASC (Norm-ASC-CM; Fig. 6A).
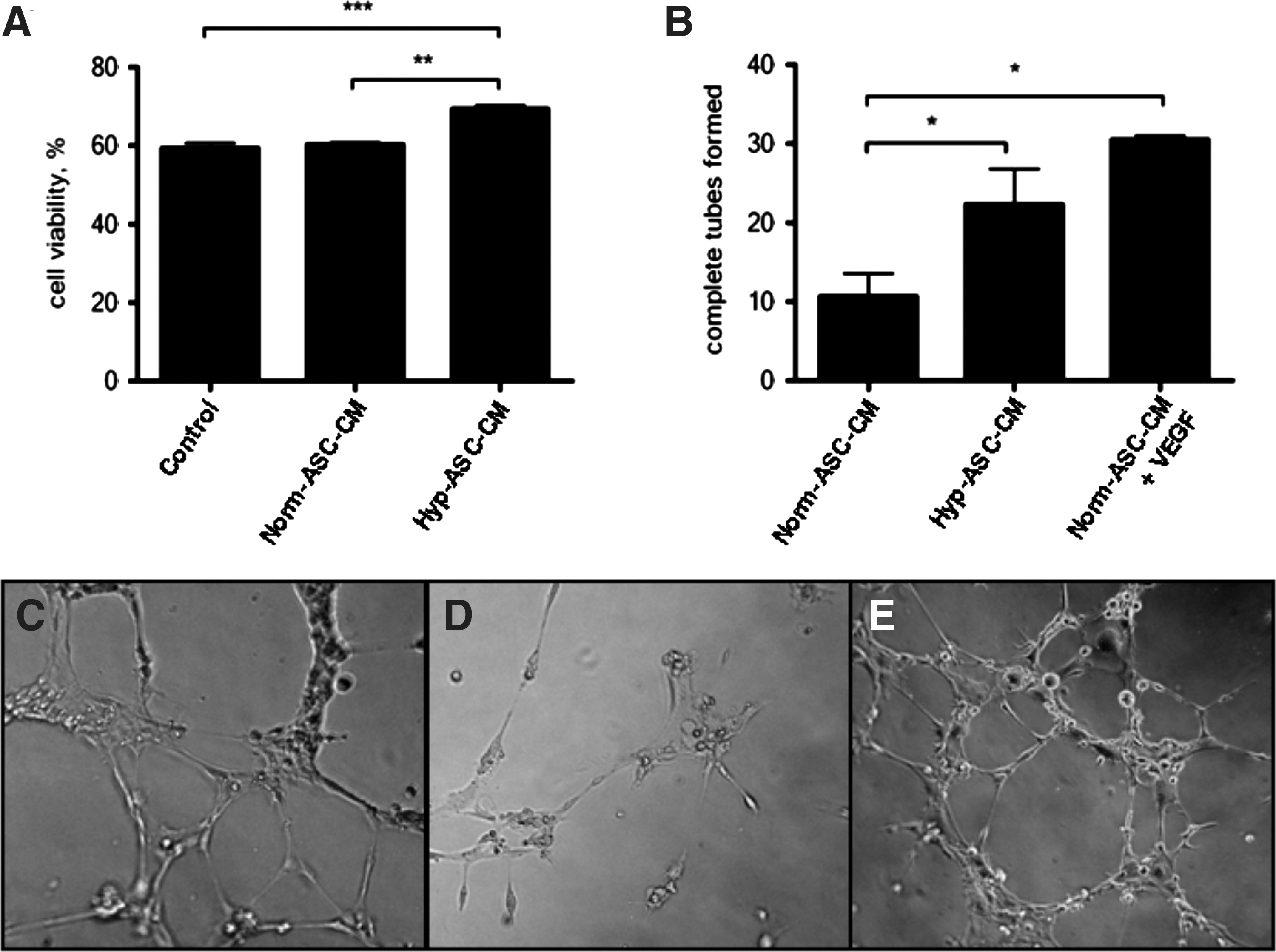
Hypoxic preconditioned ASC protect endothelial cells against hypoxia-induced cell death and improve endothelial tube formation. Conditioned medium from hypoxic preconditioned ASC (Hyp–ASC-CM) significantly increased the viability of endothelial cells compared with control unconditioned DMEM-LG medium (***P<0.001) and conditioned medium from non-preconditioned ASC (Norm-ASC-CM) (**P<0.01) (n=3,
Concomitant with the observed survival advantage, endothelial cell tube formation was significantly improved in cells cultured in Hyp-ASC-CM compared with Norm-ASC-CM (Fig. 6B–E). The angiogenic effect of Hyp-ASC-CM was comparable in magnitude to the effects of the known angiogenic factor, VEGF (Norm-ASC-CM+VEGF).
Discussion
The use of stem cells for tissue repair and restoration continues to show promise, but may be hampered by early cell loss [1,2], in part due to oxygen or nutrient deprivation and loss of cell to cell contact [22]. It is therefore important to identify methods for improving the survival of cells in these environments so that functional outcomes can be improved. Preconditioning is a powerful tool through which cells can be protected and is one strategy that is easily adaptable to the clinic for improving cell survival in these environments.
In this study we focused on hypoxia as a strategy for preconditioning because HPC has been shown to exert powerful protective effects on multiple cell types (including stem cells) and in several organ systems [6,7,23 –25]. Furthermore, hypoxia provides a safe, clinically compliant alternative to other methods, such as genetic modification, which could be used to protect stem cells in harsh ischemic environments. Long-term hypoxia has been shown to affect the proliferation and differentiation of ASC [26 –28], but the cytoprotective effects of HPC have not been properly investigated. Ours is the first study to demonstrate clearly that HPC can protect ASC against ischemic injury in vitro, and here we evaluate the mechanisms involved.
In our model of HPC, we show that HIF-1α protein, which is degraded under normoxic conditions, is stabilized and accumulates in ASC. HIF-1α is known to trigger nearly all cellular responses to hypoxia through activation of its downstream targets, several of which promote cell survival [8]. VEGF is one such target gene [29,30]. Previous studies have shown that ASC cultured under hypoxic conditions increased VEGF gene expression [28,31], but we now show that relatively short-term exposure to hypoxia also significantly increases VEGF-A production in ASC and that VEGF-A contributes to the observed improvement in cell survival under simulated ischemic conditions in vitro. We also found similar VEGF-A mRNA responses to acute hypoxia in MSC derived from bone marrow, potentially broadening the application of this method. The protective actions of VEGF-A in ASC in this model of HPC are mediated via activation of VEGFR1, for RT-PCR revealed the presence of VEGFR1, but not VEGFR2, transcripts, and it is therefore unlikely that VEGFR2 is expressed in these cells. Furthermore, VEGFR1 mRNA is significantly up-regulated by hypoxia, in agreement with earlier studies which have shown that hypoxia regulates VEGFR1 expression in BM-MSC [32]. Others have reported that hypoxia increases the expression of other protective growth factors in stem cells, including HGF, bFGF, and IGF-1 [12,33], but this did not occur in ASC with our HPC protocol. Indeed, exclusive up-regulation of VEGF-A, which is an important angiogenic and pro-survival factor, suggests that ASC may have unique properties for targeted application in stem cell therapy. Interestingly, we also found that under normoxic culture conditions ASC secrete basal levels of VEGF-A and exhibit basal Akt activity, and both these levels were significantly increased by HPC. This may in part explain the survival advantage imparted to these ASC, in particular, under hypoxic conditions as compared to other stem cells [34,35].
To investigate further the downstream signaling pathway of VEGF-A in ASC, we examined the expression of important survival kinases in response to exogenous VEGF-A. We found that VEGF activates Akt in ASC and that this effect was abolished in the presence of a neutralizing antibody against VEGF and the PI3K inhibitor, LY294002. Akt has been shown to be an important mediator of survival in BM-MSC where BM-MSC over-expressing Akt alone enjoy a survival advantage and improve outcomes in the infarcted myocardium [36]. This finding led us to show the functional importance of Akt phosphorylation in mediating the pro-survival effect of HPC in ASC under simulated ischemic conditions, and this effect is mediated by VEGF-A. The important role of Akt activation in this scenario was confirmed by pharmacological inhibition of the PI3K/Akt pathway with LY294002.
A recent experimental study has shown that hypoxic preconditioned ASC enhance the survival of neural stem cells in co-culture both in vitro and in vivo [37]. Importantly, we also showed here that in addition to the protective autocrine effects of HPC on human ASC, conditioned media from preconditioned ASC also exert protective effects on endothelial cells subjected to in vitro hypoxic injury. Similar paracrine effects have been shown previously with BM-MSC, where conditioned media improved endothelial cell survival and tube formation [12,38] through an Akt-dependent pathway. Similarities in HPC-activated pathways between mesenchymal stem cell populations require further study but may provide an opportunity for pharmacological intervention. Although we have not probed the mechanisms of the observed paracrine effects in the present study, this anti-apoptotic effect is likely mediated by similar mechanisms as the pro-survival effects of HPC in ASC, specifically through increased secretion of VEGF-A by ASC. VEGF-A is not only important in endothelial cell survival but also promotes angiogenesis. Here we showed that the conditioned medium from hypoxic preconditioned ASC exerts a greater pro-angiogenic effect on endothelial cells than does the medium from non-preconditioned ASC, to a degree that is comparable to the known pro-angiogenic factor, VEGF-A.
Conclusion
We have demonstrated that HPC improves ASC survival under ischemic conditions in vitro. These survival benefits are mediated by the HIF-1α downstream target gene, VEGF-A, and the PI3K/Akt signaling pathway. Furthermore, the release of protective paracrine factor(s), particularly VEGF-A, by hypoxic preconditioned ASC improves endothelial cell survival and angiogenesis in a paracrine fashion. The current findings therefore suggest a new approach to improve ASC survival and enhancing their paracrine activities that would be more acceptable clinically than gene manipulation in regenerative therapies.
Footnotes
Acknowledgments
The authors would like to acknowledge support and Principal Research Fellowship (to G.J.D.) from the National Health and Medical Research Council of Australia (400303 and 509271) and the Australian Postgraduate Award (to S.S.). The O'Brien Institute acknowledges the support of JO and JR Wicking Trust and Victorian State Government's Department of Innovation, Industry and Regional Development's Operational Infrastructure Support Program. The authors also wish to acknowledge the technical advice of Caroline Taylor, Effie Keramidaris, and Catherine Chang.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.