
Abstract
The realization of human embryonic stem cells (hESC) as a model for human developmental hematopoiesis and in potential cell replacement strategies relies on an improved understanding of the extrinsic and intrinsic factors regulating hematopoietic-specific hESC differentiation. Human mesenchymal stem cells (hMSCs) are multipotent cells of mesodermal origin that form a part of hematopoietic stem cell niches and have an important role in the regulation of hematopoiesis through production of secreted factors and/or cell-to-cell interactions. We have previously shown that hESCs may be successfully maintained feeder free using hMSC-conditioned media (MSC-CM). Here, we hypothesized that hESCs maintained in MSC-CM may be more prone to differentiation toward hematopoietic lineage than hESCs grown in standard human foreskin fibroblast-conditioned media. We report that specification into hemogenic progenitors and subsequent hematopoietic differentiation and clonogenic progenitor capacity is robustly enhanced in hESC lines maintained in MSC-CM. Interestingly, co-culture of hESCs on hMSCs fully abrogates hematopoietic specification of hESCs, thus suggesting that the improved hematopoietic differentiation is mediated by MSC-secreted factors rather than by MSC-hESC physical interactions. To investigate the molecular mechanism involved in this process, we analyzed global (LINE-1) methylation and genome-wide promoter DNA methylation. hESCs grown in MSC-CM showed a decrease of 17% in global DNA methylation and a promoter DNA methylation signature consisting of 45 genes commonly hypomethylated and 102 genes frequently hypermethylated. Our data indicate that maintenance of hESCs in MSC-CM robustly augments hematopoietic specification and that the process seems mediated by MSC-secreted factors conferring a DNA methylation signature to undifferentiated hESCs which may influence further predisposition toward hematopoietic specification.
Introduction
H
Human mesenchymal stem cells (hMSCs) are multipotent cells of mesodermal origin that form a part of hematopoietic stem cell (HSC) niches. They have a key role in the regulation of the hematopoiesis, as they produce hematopoietic cytokines, support hematopoietic progenitors in vitro, and possess potent immunosuppressive properties [17,18]. In fact, co-transplantation of MSCs has also been shown to enhance the hematopoietic engraftment after human leukocyte antigen (HLA)-identical HSC transplantation [19 –23].
In an attempt to facilitate hESC maintenance and to reduce the use of xeno-components, both chemically defined commercial media and feeder-free culture systems based on the use of media conditioned by human feeders are evolving as the most commonly extended culture conditions for hESC maintenance [24 –26]. Despite the fact that human foreskin fibroblasts (HFFs) are the most commonly used human feeders for hESC co-culture [27,28], we and others have recently shown successful hESC maintenance in hMSCs or hMSC-conditioned media (MSC-CM) [26,29,30].
Here, we hypothesized that hESCs maintained in MSC-CM may be more prone to differentiation toward hematopoietic lineage than hESCs grown in standard HFF-CM. The rationale behind this hypothesis is 3-fold: (i) the existence of an embryonic population of mesodermal prehematopoietic precursors that give rise to both blood/vasculature and MSCs have long been suggested [31 –33], (ii) hMSCs produce hematopoietic cytokines which support hematopoietic progenitors in vitro, and (iii) a number of MSC-produced soluble factors with novel stem cell expansion activities were recently identified as being associated with mesodermal induction [34]. Our data indicate that maintenance of several hESC lines in MSC-CM robustly augments hematopoietic specification. This process seems mediated by MSC-secreted factors conferring a DNA methylation signature to undifferentiated hESCs that may influence further predisposition toward hematopoietic specification.
Materials and Methods
hESC culture
The hESCs lines AND1, H9, H13C, and SHEF2 were cultured in Matrigel (BD Biosciences)-coated T25 flasks in either HFF-CM or MSC-CM supplemented with 8 ng/mL basic fibroblast growth factor (bFGF) (Miltenyi). Media were changed daily, and the cells were split weekly by dissociation with 200 U/mL of collagenase IV (Invitrogen). hESC cultures were visualized daily by phase-contrast microscopy. The basal media used to prepare the CM as well as the procedure to produce both HFF-CM and MSC-CM have been extensively reported elsewhere [26,30]. Approval from the Spanish National Embryo Ethical Committee was obtained to work with hESCs.
Hematopoietic differentiation from hESCs through embryoid body formation
Undifferentiated hESCs at confluence were treated with collagenase IV and scraped off of the Matrigel attachments. To allow human embryoid body (hEB) formation, hESC clumps were transferred to low-attachment plates (Corning) and incubated overnight in differentiation medium [DM; Knock-out-Dulbecco's modified Eagle's medium (KO-DMEM) supplemented with 20% non-heat-inactivated fetal bovine serum (FBS), 1% nonessential amino acids (NEAA), 1 mmol/L l-glutamine, and 0.1 mmol/L β-mercaptoethanol]. The medium was changed the next day (day 1) with the same DM supplemented with hematopoietic cytokines: 300 ng/mL stem cell factor (SCF), 300 ng/mL Flt3L, 10 ng/mL interleukin (IL)-3, 10 ng/mL IL-6, and 50 ng/mL granulocyte-colony stimulating factor (G-CSF) and 25 ng/mL bone morphogenetic protein 4 (BMP-4) [7,8]. hEBs were dissociated using collagenase B (Roche Diagnostic) for 2 h at 37°C followed by 10 min incubation at 37°C with enzyme-free Cell Dissociation Buffer (Invitrogen) at day 10, 15, and 22 of development. A single-cell suspension was obtained by gentle pipetting and passage through a 70-μm cell strainer (Becton Dickinson), and the dissociated cells were stained with anti-CD34-FITC, anti-CD31-PE, and anti-CD45-APC (all from Miltenyi) antibodies and 7-actinomycin D (7AAD). Live cells identified by 7AAD exclusion were analyzed using a FACSCanto II flow cytometer equipped with FACS Diva software (Becton Dickinson).
hESC-OP9 co-cultures
hESC-OP9 co-cultures were performed as previously described [11]. Briefly, OP9 stroma was prepared by plating OP9 cells in gelatin-coated 10-cm dishes in alpha-minimum essential media (MEM) basal medium supplemented with 20% non-heat-inactivated FBS. The hESC lines AND1 and H9 grown in Matrigel-coated flasks were prepared as a suspension of small aggregates using Collagenase IV treatment followed by gentle scraping in DM (alpha-MEM basal medium, 10% non-heat-inactivated FBS, 100 μM monothioglycerol, and 50 μg/mL ascorbic acid). One-fifth of this suspension was plated on top of the 8-day overgrown OP9 stroma in 10 mL of DM. Next day, media were replaced by 20 mL of DM to remove unattached cells. From day 4 of co-culture, a half-volume media change was performed every other day. Hematopoietic differentiation was assessed by flow cytometry (at day 6, 8, 10, and 15 of co-culture) and colony forming unit (CFU) assays (at day 8 of co-culture). Cells were stained with anti-mouse CD29-FITC and anti-human CD31-PE, CD34-PE-Cy7, and CD45-APC. The proportion of hemogenic progenitors (CD31+ CD45−), primitive blood cells (CD34+ CD45+), and total blood cells (CD45+) was analyzed within the hESC-derived cell population identified as CD29 negative.
hESC-hMSC co-cultures
hMSCs were obtained from postnatal adipose tissue or bone marrow from healthy donors on informed consent as previously described [35 –37]. hMSC were fully characterized and showed typical fibroblast-like morphology, MSC immunophenotype, and in vitro differentiation potential into osteoblasts, adipocytes, and chondrocytes [17,32,38]. Established MSC cultures were trypsinized, resuspended in culture medium (Advanced DMEM, 10% FBS, 1% Glutamax), and mitotically inactivated by ionizing radiation (42 Gy). Irradiated hMSCs were then plated in 6-well plates (105cells/well) and allowed to attach overnight. Next day, media were changed to DM (KO-DMEM, 20% FBS, 1% Glutamax, 1% NEAA, and 0.2% ß-mercaptoethanol). The hESCs lines AND1 and H9 grown in Matrigel-coated flasks were prepared as a suspension of small aggregates using Collagenase IV treatment followed by gentle scraping in DM. One-fifth of this suspension was plated on top of irradiated hMSCs. Next day, the media were replaced by DM with or without cytokines (25 ng/mL BMP4, 300 ng/mL SCF, 10 ng/mL IL3, 10 ng/mL IL6, 300 ng/mL FLT3L, and 50 ng/mL G-CSF) to remove unattached cells. Every 3 days, a half-volume media change was performed. Hematopoietic differentiation was assessed by flow cytometry (at day 8, 15, 21, and 28 of co-culture) and CFU assays (at day 8 of co-culture). We stained the cells with anti-human CD31-FITC, CD73-PE, CD34-PE-Cy7, and CD45-APC. The proportion of hemogenic progenitors (CD31+ CD45−), primitive blood cells (CD34+ CD45+), and total blood cells (CD45+) was analyzed within the CD73- fraction (non-hMSCs).
CFU assays
CFU assays were performed by plating 35,000 cells from either day 15 hEBs or day 8 hESC-OP9/hESC-hMSC co-culture into methylcellulose H4230 (Stem Cell Technologies) supplemented with the recombinant human growth factors SCF (50 ng/mL), erythropoietin (EPO) (3 U/mL), granulocyte macrophage-colony stimulating factor (GM-CSF) (10 ng/mL), and IL-3 (10 ng/mL). Cells were incubated at 37°C in a 5% CO2-humidified atmosphere, and colonies were counted after 14 days using standard morphological criteria [8,39].
Real-time reverse transcriptase–polymerase chain reaction
Total RNA was extracted from hEBs using Total RNA Purification Kit (Norgen Biotek). First-strand cDNA synthesis was performed using the First-Strand cDNA Synthesis Kit (Amersham). The resulting cDNA was analyzed for differential gene expression by using Platinum SYBR Green qPCR Super Mix-UDG (Invitrogen) on Mx3005P Q-PCR System (Stratagene). Real-time polymerase chain reaction (PCR) conditions were as follows: 10 min at 95°C; 40 cycles of 30 s at 95°C followed by 60 s at 60°C; 30 s at 72°C. GAPDH was used as housekeeping gene. Primer sequences are listed in Supplementary Table S1 (Supplementary Data are available online at
Enzyme-linked immunosorbent assays
MSCs and HFFs were grown in DMEM. At confluence, MSC-CM and HFF-CM were harvested for prostaglandin 2 (PGE2), leptin, and adiponectin detection. The concentration of these factors secreted into the media was measured by enzyme-linked immunosorbent assay (ELISA) using human ELISA Kits from Cayman Chemicals, following the manufacturer's guidelines.
Pyrosequencing assays
Sodium bisulphite modification of 0.5 μg of genomic DNA isolated from the hESCs lines H9, H13C, and SHEF2 grown in either MSC-CM or HFF-CM was carried out with the EZ DNA Methylation Kit (Zymo Research) following the manufacturer's protocol [40]. Primers for PCR amplification and sequencing were designed using the software PyroMark Assay Design (version 2.0.01.15; Qiagen). Primer sequences were designed to hybridize with CpG-free sites to ensure methylation-independent amplification (Supplementary Table S2). PCR was performed using primers biotinylated to convert the PCR product to single-strand DNA templates. We used the Vacuum Prep Tool (Biotage) to prepare single-strand PCR products according to the manufacturer's instructions. Pyrosequencing reactions and methylation quantification were performed in a PyroMark Q24 System (version 2.0.6; Qiagen). LINE-1 methylation levels were analyzed as an indicator of global DNA methylation. Two differentially methylated genes detected in the array, LEFTY1 and TEK, were validated by pyrosequencing. Graphs of methylation values show bars identifying CpG sites with values ranging from 0% to 100%.
Promoter DNA methylation profiling using bead arrays
Microarray-based DNA methylation profiling was performed in the hESCs lines H9, H13C, and SHEF2 grown in either MSC-CM or HFF-CM with the HumanMethylation27 DNA Analysis BeadChip (Illumina). The Infinium Methylation Assay combines bisulphite conversion of genomic DNA and whole-genome amplification sample preparation with direct, array-based capture and enzymatic scoring of the CpG loci. Bisulphite conversion of DNA was performed using the EZ DNA Methylation Kit (Zymo Research) according to the manufacturer's procedures. Processed DNA samples were hybridized to the BeadChip (Illumina). The assay interrogates the chemically different loci using 2 site-specific probes, 1 designed for the methylated locus (M bead type) and another for the unmethylated locus (U bead type). Single-base extension of the probes incorporates a labeled ddNTP, which is subsequently stained with a fluorescence reagent. The methylation level for the interrogated locus is determined by calculating the ratio of the fluorescent signals from the methylated versus unmethylated sites. The ratio of fluorescent signals is then computed from the 2 alleles according to the following formula: Beta=Max(M,0)/Max(U,0)+Max(M,0)+100. This beta value (β) is a quantitative measure of DNA methylation levels of specific CpG sites and ranges from 0 for completely unmethylated to 1 for completely methylated [41,42]. To analyze methylation changes in hESCs cultured in different conditions, only probes with methylation differences (β) over 0.2 (more than 20% difference in methylation level) were selected. Taking cells cultured in HFF-CM as reference, a CpG site was considered as hypermethylated or hypomethylated in cells cultured in MSC-CM when an increase or decrease of the β by 20% was detected, respectively. Before analyzing the methylation data, we excluded possible sources of technical biases that could alter the results. Every β in the array platform is accompanied by a detection P value. We based the filtering criteria on these P values reported by the assays. All probes with detection P values>0.01 were removed. The clustering heatmaps and bar plots using methylation data were performed with BeadStudio software (Illumina) and Microsoft Excel. Methylation profiling data have been deposited and are available at GEO (GSE30456;
Statistical analysis
All data are expressed as mean±standard error of the mean. Statistical comparisons were performed with a paired Student's t-test. Values were considered statistically significant at P<0.05.
Results and Discussion
Maintenance of hESC in MSC-CM enhances hematopoietic specification
We have previously reported successful long-term feeder-free maintenance of hESC lines in both HFF-CM and MSC-CM [26,43]. Here, we initially confirmed that the hESC lines AND1, H9, and H13C maintained either in HFF-CM or MSC-CM retained typical hESC morphology (Supplementary Fig. S1A); similar levels of the ESC-associated surface markers SSEA-3, SSEA-4, Tra-1–60, Tra-1–81, and OCT-4 (Supplementary Fig. S1B); and expressed identical or slightly higher levels of the ESC-transcription factors OCT4, SOX2, NANOG, and REX1 (Supplementary Fig. S1C).
To determine the propensity of several hESC lines grown for several weeks on either HFF-CM or MSC-CM to differentiate toward the hematopoietic lineage, the differentiation was induced through the formation of hEBs in the presence of BMP-4 and hematopoietic cytokines. We then analyzed the emergence of hemogenic precursors (CD45-CD31+), primitive (CD45+ CD34+), and mature (CD45+ CD34−) blood cells as well as CFU potential at different time points during hEB differentiation (day 10, 15 and 22) (Fig. 1A and Supplementary Fig. S2). hESC lines maintained in MSC-CM displayed a highly augmented hematopoietic differentiation at any time point analyzed (Supplementary Fig. S2) as compared with those hESCs maintained in HFF-CM. The emergence of hematopoietic cells from hESC lines maintained in MSC-CM was significantly higher than from hESC lines maintained in HFF-CM: hemogenic precursors (∼3–5-fold) (Fig. 1B), primitive blood cells (∼10-fold) (Fig. 1C), mature blood cells (∼8–10-fold) (Fig. 1D), and CFUs (∼3–4-fold) (Fig. 1E). Importantly, scoring of CFUs revealed no significant differences in the CFU types (granulocyte, macrophage, granulo-monocyte, and erythroid) obtained from hESCs grown in HFF-CM versus MSC-CM (Fig. 1A and Supplementary Fig. S3). This suggests that the maintenance of hESCs in MSC-CM enhances the in vitro clonogenic potential without impairing normal developmental stem cell fate.
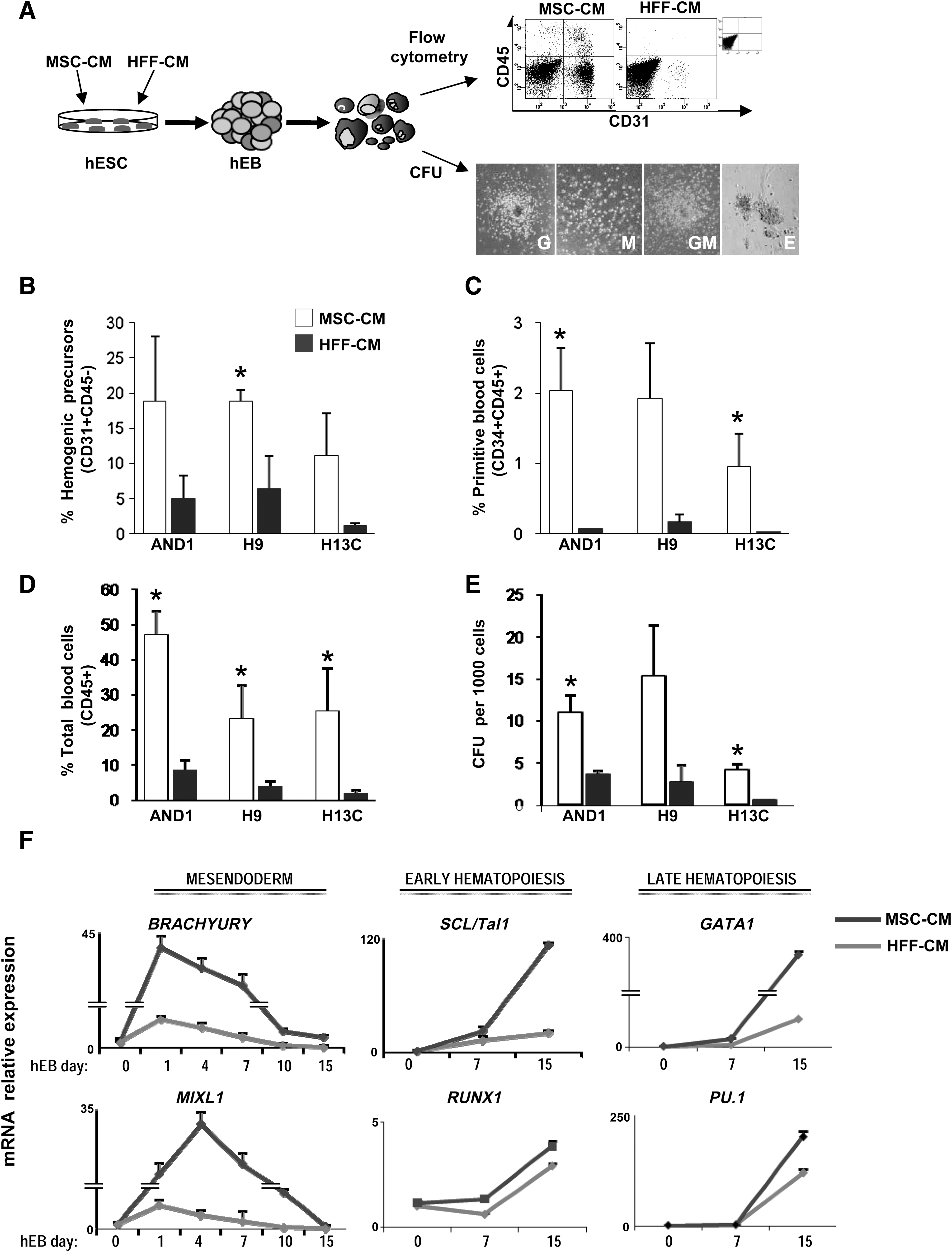
Maintenance of hESC in MSC-CM enhances hematopoietic differentiation.
To determine whether the observed cellular differences in hematopoietic differentiation from hESCs grown in HFF-CM versus MSC-CM are reflected at the molecular level, we evaluated the gene expression kinetics of mesendoderm (BRACHYURY and MIXL1), early (SCL and RUNX1), and late (GATA1 and PU1) hematopoietic transcription factors (Fig. 1F) in differentiating hEBs. All hESC lines regulated both BRACHYUTY and MIXL1 at early time points (days 1–4 of hEB development), regardless of the culture conditions (Fig. 1F). In differentiating hEBs formed from hESCs grown in MSC-CM, the expression of these early mesendodermal markers emerged rapidly during hEB differentiation, peaked by day 1–4 of hEBs, and then switched off rapidly to become almost undetectable around day 15 of hEB differentiation. This suggests that differentiating hEBs from hESC maintained in MSC-CM display an early gene expression pattern reminiscent of cells predisposed to mesoderm specification. In contrast, the regulation of BRACHYURY and MIXL1 is less pronounced in differentiating hEBs from hESCs maintained in HFF-CM, in line with their less efficient hematopoietic differentiation (Fig. 1F). Additionally, the expression of the early hematopoietic transcription factors SCL and RUNX1 emerged more rapidly in differentiating hEBs formed from hESC grown in MSC-CM as compared with hEBs from hESC grown in HFF-CM (Fig. 1F). Similarly, the expression of the late hematopoietic transcription factors GATA1 and PU1 emerged around day 7 of hEB differentiation, and their expression was more (∼2–8-fold) up-regulated in hEBs formed from hESC grown in MSC-CM as compared with hEBs from hESC grown in HFF-CM (Fig. 1F). Thus, the specification into hemogenic progenitors and subsequent differentiation into primitive blood cells and mature blood cells is significantly enhanced in hESC cultured with MSC-CM, whereas the temporal expression pattern of hematopoietic-related genes supports an enhanced hematopoietic commitment within differentiating hEBs from hESC grown in MSC-CM.
We next asked whether the enhanced hematopoietic specification in response to the maintenance of hESCs in MSC-CM could be equally achieved by differentiating hEBs in the presence of such MSC-CM (in the absence of bFGF). Interestingly, no hematopoietic differentiation could be observed by flow cytometry or CFU assay even after 22 days of hEB differentiation (data not shown), thus indicating that secreted factors present in the MSC-CM enhance hematopoietic specification when acting on undifferentiated hESCs but not on differentiating hEBs. Due to the rather impermeable physical architecture of the hEBs [44], it cannot be ruled out that the potential hematopoietic-promoting MSC-secreted factors may encounter physical barriers to diffuse into the hEB structures where they are expected to exert its hematopoietic-promoting function, thus explaining the differential functional impact of MSC-CM when used in undifferentiated hESCs versus differentiating hEBs.
Co-culture of hESCs on hMSCs impedes hematopoietic specification of hESCs
We next wanted to determine whether the mechanisms by which hMSCs augment hematopoietic differentiation of hESCs rely on the MSC secretome or MSC-hESC physical interactions. Co-culture of hESCs with the stromal cell line OP9 has widely been shown to successfully promote in vitro hematopoietic differentiation of hESC [2,6,10,11]. Thus, the hESC lines AND1 and H9 were allowed to differentiate in either the presence or absence of hematopoietic cytokines on OP9 (positive control) or hMSCs co-cultures (Fig. 2A). As previously described [10,11], hESCs grown on OP9 gave rise to hemogenic precursors (4%±0.5%), CD45+blood cells (7%±2%), and CFU potential, thus indicating that hESC-OP9 co-cultures resulted in efficient hematopoietic differentiation (Fig. 2B, C). In contrast, the same hESC lines cultured on hMSCs were unable to differentiate into either hemogenic precursors or CD45+blood cells (Fig. 2B). Similarly, CFU assays revealed a complete absence of hematopoietic progenitor function from differentiating hESCs co-cultured on hMSCs (Fig. 2C).
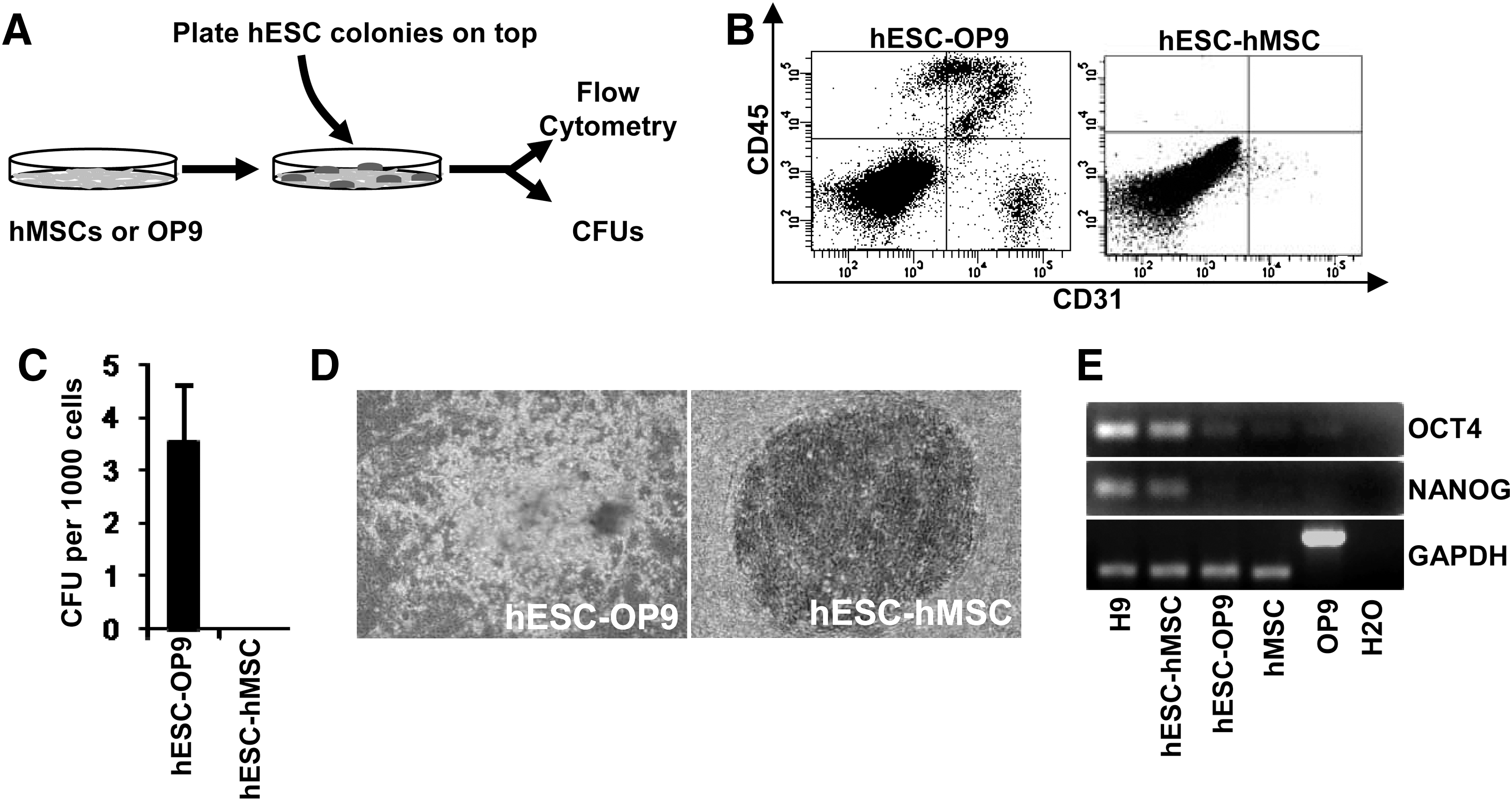
Coculture on hMSCs does not support hematopoietic differentiation from hESC.
Interestingly, phase-contrast examination of hESC-OP9 and hESC-hMSC co-cultures revealed that hESCs cultured on OP9 for 15 days display many clusters of differentiation, whereas hESCs cultured on hMSCs for up to 28 days retain the typical undifferentiated ESC morphology (Fig. 2D). Reverse transcriptase–PCR analysis of the pluripotency-associated transcription factors OCT4 and NANOG confirmed that hESCs cultured on hMSC retain expression of both OCT4 and NANOG, whereas hESCs cultured on OP9 for 15 days mostly lost the expression of both OCT4 and NANOG (Fig. 2E). These data suggest that hMSCs-based co-cultures prevent hESC differentiation rather than inducing hematopoietic differentiation. Since hESCs maintained in MSC-CM are more prone to differentiation toward hematopoietic lineage than hESCs grown in HFF-CM, but the co-culture of hESCs on hMSCs fully abrogates hematopoietic specification of hESCs, it is tempting to speculate that MSC-hESC physical interactions may be responsible for inhibiting hematopoietic differentiation of hESCs.
We next attempted to gain insights into which potential candidate hMSC-secreted factors might be responsible for such an effect (Supplementary Fig. S4). First, we carried out high-performance liquid chromatography and isobaric tag for relative and absolute quantitation (iTRAQ)-based proteomic assays (data not shown) with and without serum depletion with no success mainly due to 2 technical difficulties: (i) the secretome of hMSCs is extremely rich [45], thus making proteomic analyses very complex and barely informative and (ii) as hMSCs get confluent, age, and senescent, they differentiate by default into adipocytes loaded with vacuoles that make the MSC-CM very oily. We, therefore, analyzed the secreted levels of lipid molecules such as leptin and adiponectin. Neither MSCs nor HFFs produced leptin or adiponectin (Supplementary Fig. S4A). According to the literature, we further analyzed PGE2, a potent hematopoietic inducer [46,47]. Interestingly, the levels of PGE2 were robustly increased in MSC-CM as compared with HFF-CM (Supplementary Fig. S4A). However, despite hMSCs producing abundant levels of PGE2, functional assays based on supplementation of HFF-CM with exogenous human recombinant PGE2 did not improve/promote hematopoietic differentiation of hESCs, even though hESCs do express abundant levels of PGE2 canonical receptors (Supplementary Fig. S4B, C). This suggests that PGE2, at least on its own, is not the hMSC-secreted factor responsible for the improved hematopoietic differentiation (Supplementary Fig. S4C).
Genome-wide methylation analysis reveal a specific DNA methylation profile distinguishing hESCs cultured with MSC-CM from hESCs cultured in HFF-CM
Epigenetic marks are known to be established and modified in response to environmental factors and can influence gene expression and cellular differentiation. Genomic DNA methylation directly regulates gene expression, and alterations in this process are associated with pluripotency and differentiation [40]. To gain insights into the molecular mechanisms involved in the hematopoietic differentiation predisposition of hESCs cultured with MSC-CM, we analyzed global (LINE-1) methylation and genome-wide promoter DNA methylation in hESC cultures maintained in either MSC-CM or HFF-CM. We first looked at the global DNA methylation level by pyrosequencing analysis of LINE-1 element in H9, H13C, and SHEF2 hESC lines grown in either MSC-CM or HFF-CM (Fig. 3A). We found that hESCs grown in MSC-CM showed a consistent decrease of up to 17% in global DNA methylation as compared with the same hESC lines grown in HFF-CM (P value=0.021; Fig. 3A). It is tempting to speculate that this partial but consistent demethylation observed in hESCs maintained in MSC-CM may contribute to the expression of master genes involved in mesodermal and hematopoietic lineage specification [40].
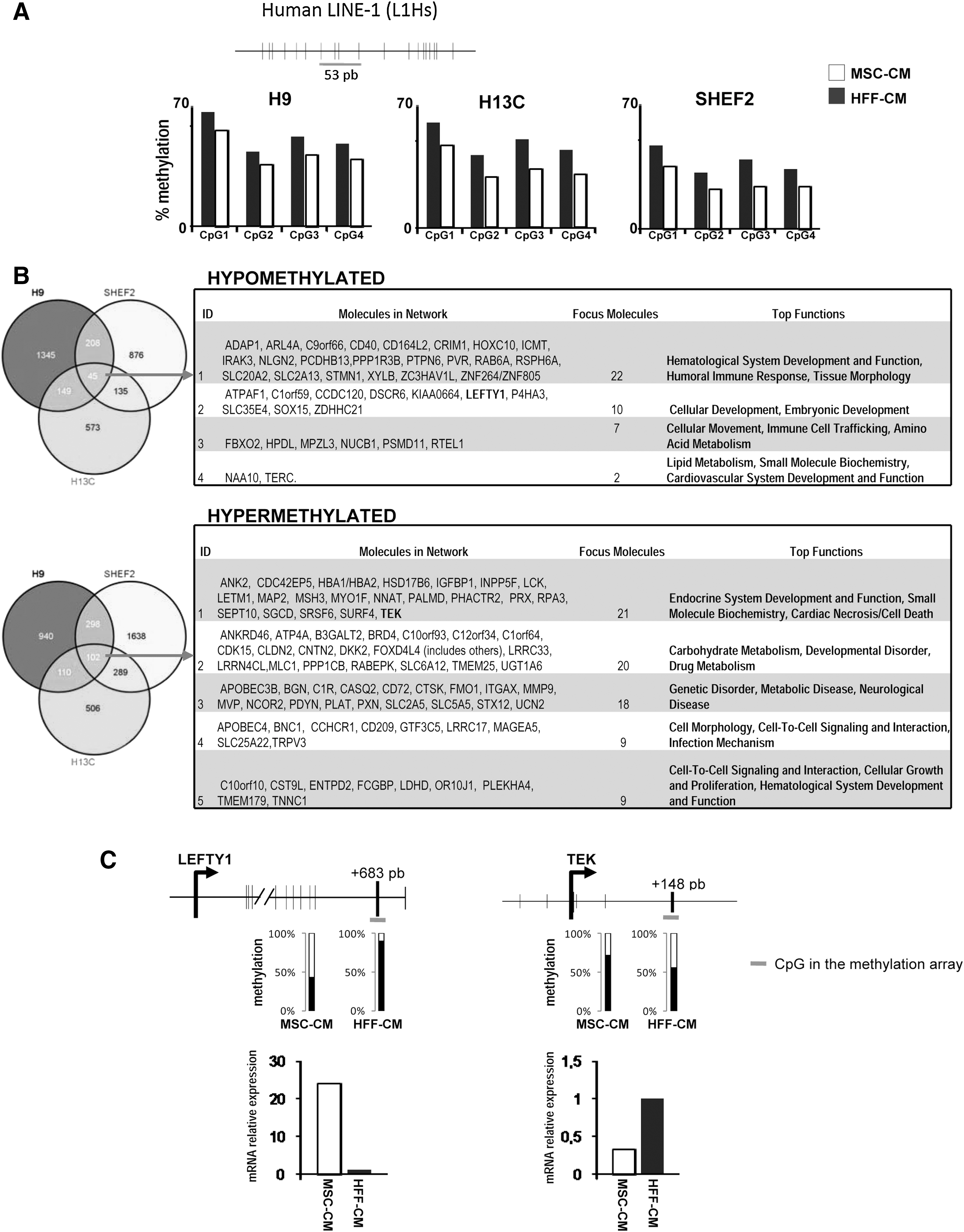
Specific DNA methylation profile distinguishes hESCs cultured with MSC-CM from hESCs cultured in HFF-CM.
We next used Infinium DNA methylation arrays to study the genome-wide role of promoter DNA methylation in 3 different hESCs maintained in MSC-CM versus HFF-CM (Fig. 3B). To exclude X chromosome inactivation-dependent DNA methylation, we selected autosomal probes [40]. To analyze methylation changes, only probes with methylation differences (β) higher or lower than 0.2 were selected. Taking cells cultured in HFF-CM as reference, a CpG site was considered as hypermethylated or hypomethylated in cells cultured in MSC-CM when an increase or decrease of the β by 20% was detected, respectively [40
–42]. Using this approach, we analyzed gene promoters differentially as well as commonly hypermethylated or hypomethylated in the 3 independent hESC lines in order to define a specific DNA methylation profile distinguishing hESCs cultured with MSC-CM from hESCs cultured in HFF-CM. Many promoters were found either differentially hypomethylated (between 573 and 1345) or hypermethylated (between 506 and 1638) in the individual hESC lines (Fig. 3B). Intriguingly, only 45 and 102 promoters were found commonly hypomethylated and hypermethylated, respectively, in MSC-CM as compared with HFF-CM (Fig. 3B and Supplementary Fig. S5). Ingenuity Pathway Analysis (IPA;
To validate the methylation array data, we selected 1 gene hypomethylated (LEFTY1) and 1 hypermethylated (TEK) gene [48] in hESC grown in MSC-CM and analyzed their methylation by pyrosequencing and expression by quantitative PCR (Fig. 3C). In both cases, bisulphite pyrosequecing data confirmed the array results, and the mRNA expression level correlated well with the methylation status (Fig. 3C). Importantly, the observation that LEFTY1 is hypomethylated and highly expressed (∼25-fold upregulated) in hESCs maintained in MSC-CM supports previous studies reporting that active Nodal/Lefty1 signaling predicts human pluripotent stem cell lines prone to differentiate toward the hematopoietic lineage [8]. These data reinforce the key role of the Nodal-Lefty-Activin signaling pathway as a potential positive regulator of embryonic hematopoiesis.
In summary, our data indicate that maintenance of hESCs in MSC-CM robustly augments hematopoietic specification and that the process seems mediated by MSC-secreted factors rather than by hESC-hMSC physical interactions. The culture of hESCs in MSC-CM seems to confer a global genome-wide partial demethylation and a specific DNA methylation signature to undifferentiated hESCs that may influence further predisposition toward hematopoietic specification. Further proteomic and metabolomic studies are required to gain insights into the nature of the MSC-secreted molecules/metabolites responsible for the improved hematopoietic differentiation.
Footnotes
Acknowledgments
P.M.'s group is funded by the Innovation and Science Department of the Junta de Andalucía (P08-CTS-3678), the FIS/FEDER (PI10/00449), the MICINN (PLE-2009-0111), and the Spanish Association Against Cancer (CI110023). F.M.'s group is funded by The Junta de Andalucía (P09-CTS-04532). C.B., P.J.R., P.A., and V.R.-M. are supported by the Instituto de Salud Carlos III (CP07/00059 to C.B, CP09/0063 to P.J.R. and CP09/00228 to P.A.) and the Marie Curie IIF (PIIF-GA-2009-236430) to V.R.-M., respectively. M.F.F. lab is supported by the CSIC (200820I172), the FIS/FEDER (PI061267 and PS09/02454), and the Community of Asturias (FICYT IB09-106). A.F.F. is supported by the IUOPA-Obra Social Cajastur.
Author Disclosure Statement
The authors report no potential conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.