
Abstract
The discovery that mesenchymal stem cells (MSCs) secrete SOD3 may help explain studies in which MSCs have direct antioxidant activities both in vivo and in vitro. SOD3 is an antioxidant enzyme that dismutes toxic free radicals produced during inflammatory processes. Therefore, MSC production and secretion of active and therapeutically significant levels of SOD3 would further support the use of MSCs as a cellular based antioxidant therapy. The aim of this study was therefore to investigate in vitro if MSC differentiation down the adipogenic, chondrogenic, and osteogenic lineages influences the expression of the antioxidant molecule SOD3. Human bone marrow MSCs and their differentiated progeny were cultured under standard conditions and both the SOD3 gene and protein expression examined. Following adipogenesis, cultures demonstrated that both SOD3 protein and gene expression are significantly increased, and conversely, following chondrogenesis SOD3 protein and gene expression is significantly decreased. Following osteogenesis there were no significant changes in SOD3 protein or gene expression. This in vitro study describes the initial characterization of SOD3 expression and secretion by differentiated MSCs. This should help guide further in vivo work establishing the therapeutic and antioxidative potential of MSC and their differentiated progeny.
Introduction
H
Overproduction of reactive oxygen species (ROS) induces toxic cellular events including oxidative damage to DNA, lipids, and proteins, which can lead to cellular mutations, death, and tissue damage. hMSCs are considered to have direct antioxidant effects in vivo [10] and recent reports have suggested that secretion of the extracellular antioxidant molecule superoxide dismutase-3 (SOD3) by bone marrow-derived hMSCs may, at least in part, be responsible for their antioxidant activity [7,8]. SOD3 is the only antioxidant enzyme that scavenges the ROS superoxide in the extracellular compartment and catalyses the dismutation of superoxides, which are generated during the inflammatory cascade, by conversion into hydrogen peroxide, which is subsequently detoxified into water and oxygen by enzymes including catalase and glutathione peroxidase [13]. SOD3 additionally competes with nitric oxide for superoxide anions, thus reducing the production of peroxynitrite, which is a major effector of tissue injury [14].
It has been demonstrated in a variety of studies that SOD3 attenuates tissue damage and inflammation [15 –21]. Oxidative stress has been implicated as a causative factor in numerous chronic and degenerative conditions, and in particular, numerous pathologies involving cells of mesenchymal origin, for instance osteoarthritis [22]. Interestingly SOD enzymes (SOD1, SOD2, and SOD3) in human cartilage are down-regulated during the progression and end stages of osteoarthritis [23]. Furthermore, adipose tissue of obese subjects has been shown to have increased levels of oxidative stress, which is considered to contribute to insulin resistance, type 2 diabetes, and atherosclerosis [24]. Oxidative modification of low-density lipoproteins has also long been associated with atherosclerosis [25,26] and has recently been shown to be prevented by addition of extracellular SOD3 or SOD3 overexpression [27,28].
Utilizing the antioxidant potential of pharmaceuticals or cellular therapies for conditions in which oxidative stress is a major etiological component is a topic of much interest. Although preclinical evidence suggests a benefit of antioxidant therapies, antioxidant use in clinical practice is limited and systematic reviews report mixed outcomes of antioxidants. Specifically, conditions investigated by systematic reviews include various cancers [29 –32], diabetes [33,34], cardiovascular disease [35 –37], and neurodegenerative conditions [38 –40]. Thus future studies are required to provide robust evidence for the potential of specific pharmaceuticals and cellular therapies, such as hMSCs, as alternative sources of antioxidants to promote antioxidant use in clinical practice.
In order to comprehensively assess the antioxidative potential of bone marrow-derived hMSCs it is important to consider whether antioxidant secretion is a property inherent to hMSCs or a commonality across hMSC cells and their more differentiated progeny. Here we utilize hMSC cultures differentiated down adipogenic, chondrogenic, and osteogenic cell lineages to investigate if hMSC differentiation in vitro affects SOD3 production and secretion. This is particularly important if hMSC transplants are exposed to in vivo environments potentially conducive to differentiation.
Materials and Methods
Establishment of human mesenchymal cultures
Bone marrow samples were obtained by an orthopedic surgeon at Southmead Hospital, Bristol, United Kingdom with informed written consent and hospital ethics committee approval. Bone marrow was harvested at the time of total hip replacement surgery from the femoral shaft and placed into a sterile 50-mL tube containing 1,000 IU of heparin. Patients with a history of malignancy, immune disorders, or rheumatoid arthritis were excluded from the study. Femoral shaft bone marrow donors were healthy apart from osteoarthritis and were not receiving drugs known to be associated with myelosuppression or bone marrow failure.
Femoral shaft marrow samples were broken up with a scalpel and washed with Dulbecco's Modified Eagles Medium (DMEM) (Sigma-Aldrich) until remaining material (bone) looked white at the bottom of the 50-mL tube. All washings were pipetted into a new 50-mL tube and kept for centrifugation. The suspension was centrifuged and re-suspended in DMEM and overlaid onto an equal volume of Lymphoprep™ (Axis-Shield; density 1.077±0.001 g/mL) and centrifuged at 600 g for 35 min at room temperature to separate the mononuclear cells (MNC) from neutrophils and red cells. The MNC layer was harvested and washed twice in DMEM.
MSC culture
Isolated MSCs from 3 independent patient samples were centrifuged and re-suspended in MSC medium consisting of DMEM with 10% fetal calf serum (FCS) which was selected for the growth of MSCs (StemCell Technologies), and 1% penicillin and streptomycin (Sigma-Aldrich). Vented flasks (25 cm2) containing 5 mL of MSC medium were seeded with 1×107 cells for primary culture. Flasks were incubated at 37°C in a humidified atmosphere containing 5% CO2 and fed every week with MSC medium by half medium exchange to remove non-adherent hematopoietic cells until the adherent fibroblast-like MSCs reached approximately 70% confluence.
On reaching confluence the adherent cells were re-suspended using 0.25% trypsin (Sigma-Aldrich) and re-seeded at 2.25×105 cells per (75 cm2) flask into first passage. Cultures were then incubated, fed every week with MSC medium by half medium exchange, and again trypsinised, a cell count taken, and re-seeded at 2.25×105 cells per flask (75cm2).
MSC immunophenotype
MSCs harvested from femoral shaft bone marrow were cultured and characterized according to previous published reports from our laboratory [41] and the position statement of the International Society for Cellular Therapy [1]. Cells harvested from femoral shaft marrows displayed all the typical characteristics of MSC in culture. Briefly, MSCs had characteristic spindle-like morphology, were plastic adherent, and demonstrated adipogenic, osteogenic, and chondrogenic differentiation potential. To ensure a homogenous population of MSC had been cultured, immunophenotyping of surface markers was carried out using flow cytometry. Cells were examined at third passage using anti-CD105, anti-CD45 (eBioscience), anti-CD166, and anti-CD44 (Serotec). hMSC used within this study were uniformly positive for the mesenchymal markers CD105, CD166, CD44, but negative for the hematopoieticmarker CD45, which is consistent with the known MSC phenotype and excludes contamination of cultures with hematopoietic cells.
Human MSC differentiation
Bone marrow-derived hMSCs were seeded onto 6-well and 24-well plates as a control sample at 3.1×103 per cm2 and prior to induction adipogenesis at 2.1×104 per cm2 and osteogenesis at 3.1×103 per cm2. Prior to chondrogenesis, bone marrow-derived hMSCs were seeded at 2.5×105 per 15m-polypropylene culture tube. Differentiation was induced using Adipogenic, Osteogenic, and Chondrogenic Differentiation Media Bulletkits (Lonza), respectively, according to manufactures instructions. Cells were cultured for specific time periods outlined in the manufactures instructions. Human MSC differentiation along adipogenic, chondrogenic, and osteogenic lineages was characterized by histocytochemistry using Oil Red O (Sigma Aldrich) with Hematoxylin (Surgipath) counter stain, Alcian Blue (Sigma Aldrich) with Nuclear Fast Red counter stain (Sigma Aldrich), and Alizarin Red (Fulka) stains, respectively. Briefly, chondrocyte pellets were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned at 5 μM and deparaffinized. Human MSC, adipocyte, and osteocytes were cultured on coverslips in 24-well plates, fixed in 4% paraformaldehyde, and washed with phosphate-buffered saline (PBS). Stains were applied and then samples were dehydrated and mounted using Clearium (Surgipath).
Immunocytochemisty
Immunocytochemistry was used to identify SOD3 expression of bone marrow-derived hMSCs and hMSC differentiated adipocytes, chondrocytes, and osteocytes. Samples were prepared as described in the human MSC differentiation section. Primary antibodies were added following permeabilization of cells with 100% methanol for 10 min at −20°C. The primary antibody used for all cultures was mouse and human anti-SOD3 [Abcam anti-superoxide dismutase 3 antibody (1H12) (ab28442), 2μg/mL]. Primary antibody staining was visualized using species specific (1:500) Alexa Fluor® 488 and 555 conjugated secondary antibodies (Invitrogen). DAPI (4′, 6-diamidino-2-phenylindole dihydrochloride) Vectasheid™ was used for nuclear identification.
Real-time PCR
Human MSC and hMSC differentiated adipocytes, chondrocytes, and osteocytes were washed in PBS at 37°C. RNA was extracted and cDNA produced using the Taqman gene expression cells-Ct-kit (Applied Biosystems) according to the manufacturer's instructions. Cells were added to lysis solution plus DNase (5 min) before the addition of stop solution (2 min). All RNA samples were quantified using a Qubit® Fluorometer and Quant-iT™ RNA assay kit (Invitrogen) according to manufacturers' instructions to ensure equal loading of RNA samples. All samples were stored at −80°C prior to use. To synthesize cDNA, 50 ng of extracted RNA was added to the reverse transcription buffer and RT enzyme mix, placed in a thermal cycler, and incubated at 37°C for 1 h and 95°C for 5 min. Real-time polymerase chain reaction (RT-PCR) was performed using the StepOnePlus™ Real-Time PCR System (Applied Biosystems) with Assay-on-Demand Gene Expression Products for SOD3 (Taqman MGB probe, FAM dye-labeled, Applied Biosystems, Assay ID_Hs00162090_m1) or 18S rRNA (Taqman MGB probe, VIC™ dye-labeled, Applied Biosystems) using 10 ng of cDNA in a total volume of 20ul. Reactions were run at 50°C for 2 min; 95°C for 10 min; and 40 cycles of 95°C for 15 seconds and 60°C for 1 min. All samples were analyzed in triplicate. The relative gene expression (RQ value) of SOD3 was calculated using the 2-ΔΔCt method, and the geometric mean taken for each group; 18S rRNA was used as the reference housekeeping gene.
Immunoblotting for SOD3
Human MSCs and hMSC differentiated adipocytes and osteocytes, were lysed using Beadlyte cell signaling universal lysis buffer (Upstate™). The Qubit® Fluorometer and Quant-iT™ Protein assay kit (Invitrogen) was then used to quantify the concentration of total protein within each cell lysate sample according to manufacturers' instructions to ensure equal loading of cell lysates. Lysates were heated to 95°C for 5 min with Laemmli 2×sample buffer (Invitrogen) and run on Tris-HCl 10–20% ready gels (Bio-Rad). After transfer to nitrocellulose membrane (Bio-Rad) and blocking in Tris-buffered saline/5% bovine serum albumin (BSA), membranes were incubated overnight in primary antibody at 4°C (in Tris-buffered saline/5% BSA). Antibodies used were mouse anti-SOD3 [Abcam anti-superoxide dismutase 3 antibody (1H12) (ab28442), 0.067μg/mL] and anti-GAPDH (Abcam Anti-GAPDH antibody (ab9484), 0.10μg/mL]. Immunoreactivity was detected using secondary anti-rabbit horseradish peroxidase conjugated antibodies (Abcam) (in Tris-buffered saline/5% BSA) and specific protein expression patterns were visualized by chemiluminescence using an Amersham ECL Plus™ Western Blotting Detection System (Amersham).
Reactive oxygen species/SOD3 activity assay
hMSCs and hMSC differentiated adipocytes, chondrocytes, and osteocytes were cultured for 24 h in minimal base media, which consisted of DMEM supplemented with insulin-free Sato (containing 100 μg/mL BSA, 100 μg/mL transferrin, 0.06 μg/mL progesterone, 16 μg/mL putrescine, 0.04 μg/mL selenite, 0.04 μg/mL thyroxine, and 0.04 μg/mL triiodothryonine) (Sigma-Aldrich). The conditioned medium was then removed and stored at −80°C and defrosted prior to use in SOD activity assays. SOD activity within the conditioned medium was directly measured using the Superoxide Dismutase Assay Kit (Cayman Chemical Company, US item number 706002) according to manufacturer's instructions. As an indirect measure of SOD activity within the conditioned medium, 0.5 mL of conditioned medium, 0.5 mL minimal based medium, and 0.5mL minimal medium supplemented with 40U Cu/Zn SOD derived from human erythrocytes (Sigma-Aldrich) were exposed to DETANONOate (0.15 mM; Enzo Life Sciences) at 37°C for 60 min; 60 μL of media was then removed and stored at −80°C. Levels of hydrogen peroxide in all samples before and after DETANONOate exposure was determined using Colorimetic Hydrogen Peroxide Kit (Enzo Life Sciences) according to manufacturer's instructions.
Statistical analysis
Statistical comparisons of parametric data were analyzed using paired t-tests for comparison between non-differentiated and differentiated cell types. Values are expressed as the mean±SEM from at least 3 independent experiments, taking P<0.05 to represent statistical significance.
Results
Bone marrow-derived human MSCs differentiate down the adipogenic, chondrogenic, and osteogenic lineages
Bone marrow-derived hMSCs from 3 independent patient samples were cultured with adipogenic, chondrogenic, and osteogenic induction media to initiate differentiation down respective lineages. Cell cultures were differentiated for the period as outlined in manufacturer's instructions. Following this, cultures were subjected to histochemical staining and morphological observation to successfully confirm cell differentiation (Fig. 1).
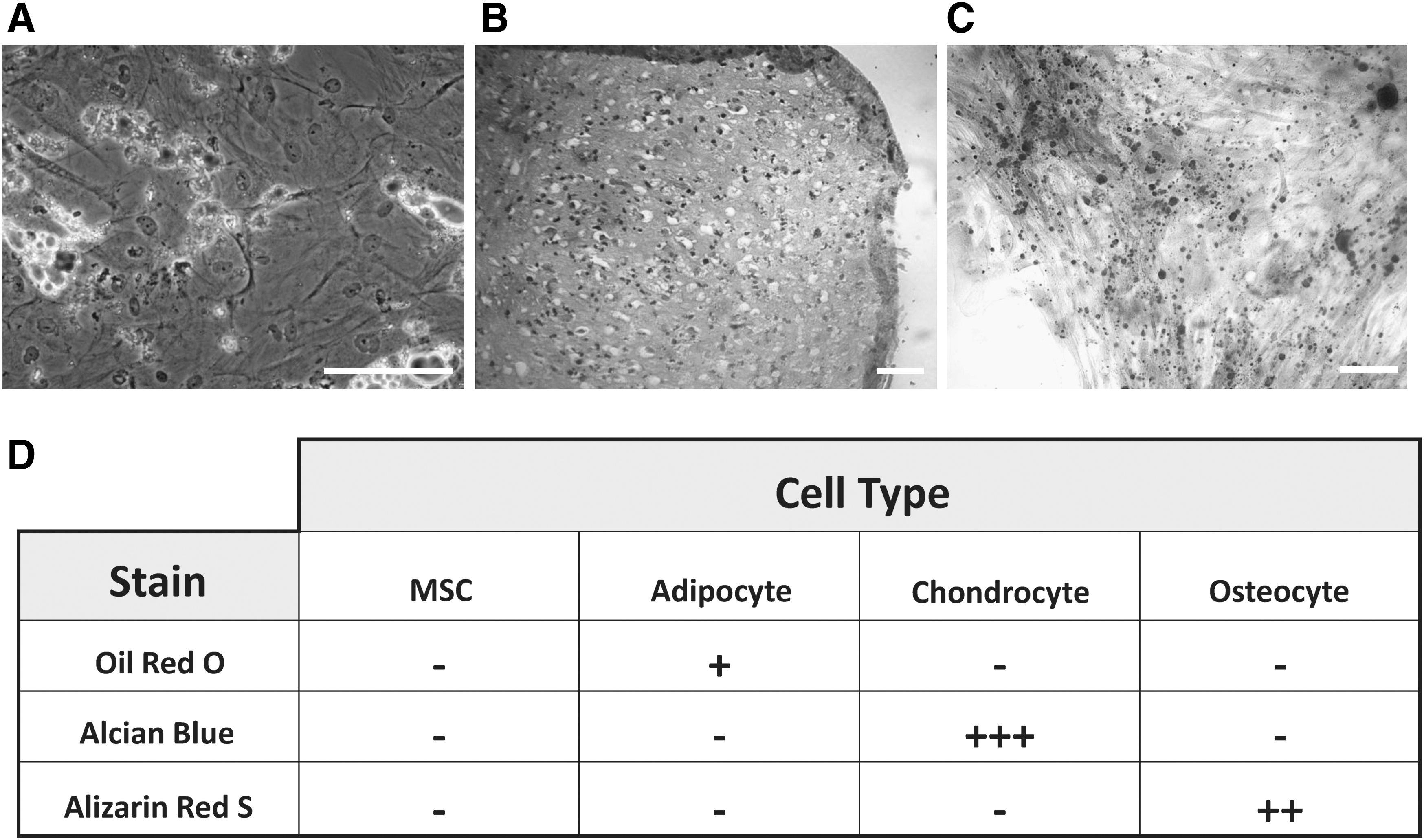
Histological analysis of human mesenchymal stem cells (MSC) differentiation.
Mesenchymal stem cell-derived osteocyte, adipocyte, and chondrocyte cells differentially express SOD3
To investigate SOD3 expression following differentiation of bone marrow hMSCs we initially determined the presence of SOD3-positive cells in the hMSC cultures. Cell cultures were differentiated for the period as outlined in manufacturer's instruction. Following this, hMSC cultures differentiated along adipogenic, osteogenic, and chondrogenic lineages were fixed and immunocytochemically labeled for SOD3. All cultures contained SOD3-positive cells. The percentage of SOD3-positive cells was significantly lower in hMSC-derived adipocyte, chondrocytes, and osteocytes cell cultures compared with hMSC cell culture (P<0.05; Fig. 2).
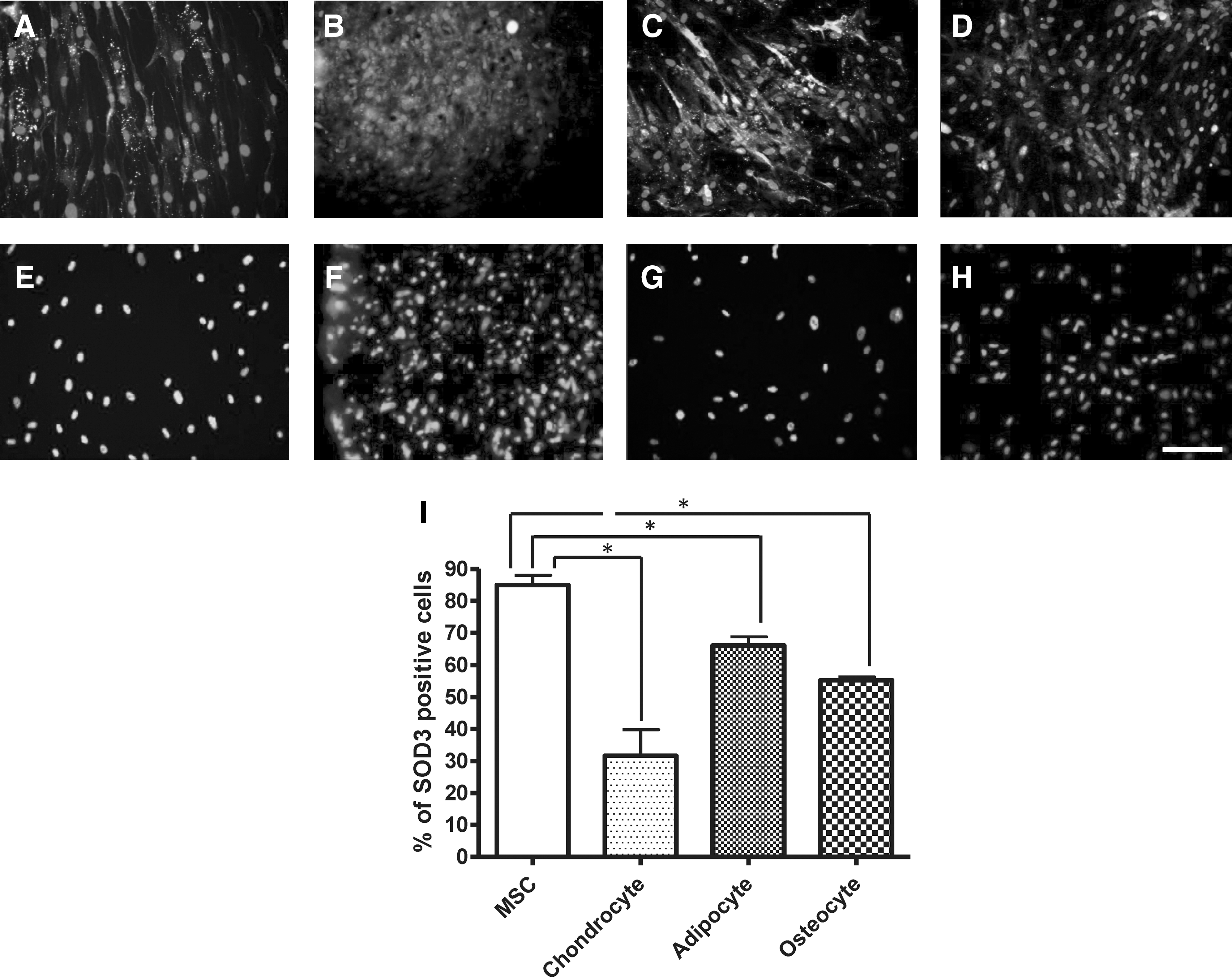
Human MSC-derived cultures immunocytochemically labeled for SOD3 (superoxide dismutase-3;) and DAPI (4′, 6-diamidino-2-phenylindole dihydrochloride) nuclear stain.
To assess if the observed changes in percentage of SOD3-positive cells reflected levels of SOD3 gene expression, RT-PCR was used to determine mRNA levels. We found levels of SOD3 mRNA were significantly higher following induction of adipogenesis and significantly lower following chondrogenesis when compared with SOD3 mRNA expression in undifferentiated hMSC cultures (P<0.05; Fig. 3A). SOD3 mRNA was expressed after induction of osteogenesis, but there were no significant changes in SOD3 mRNA expression when compared with SOD3 mRNA expression in undifferentiated hMSC cultures (Fig. 3A).
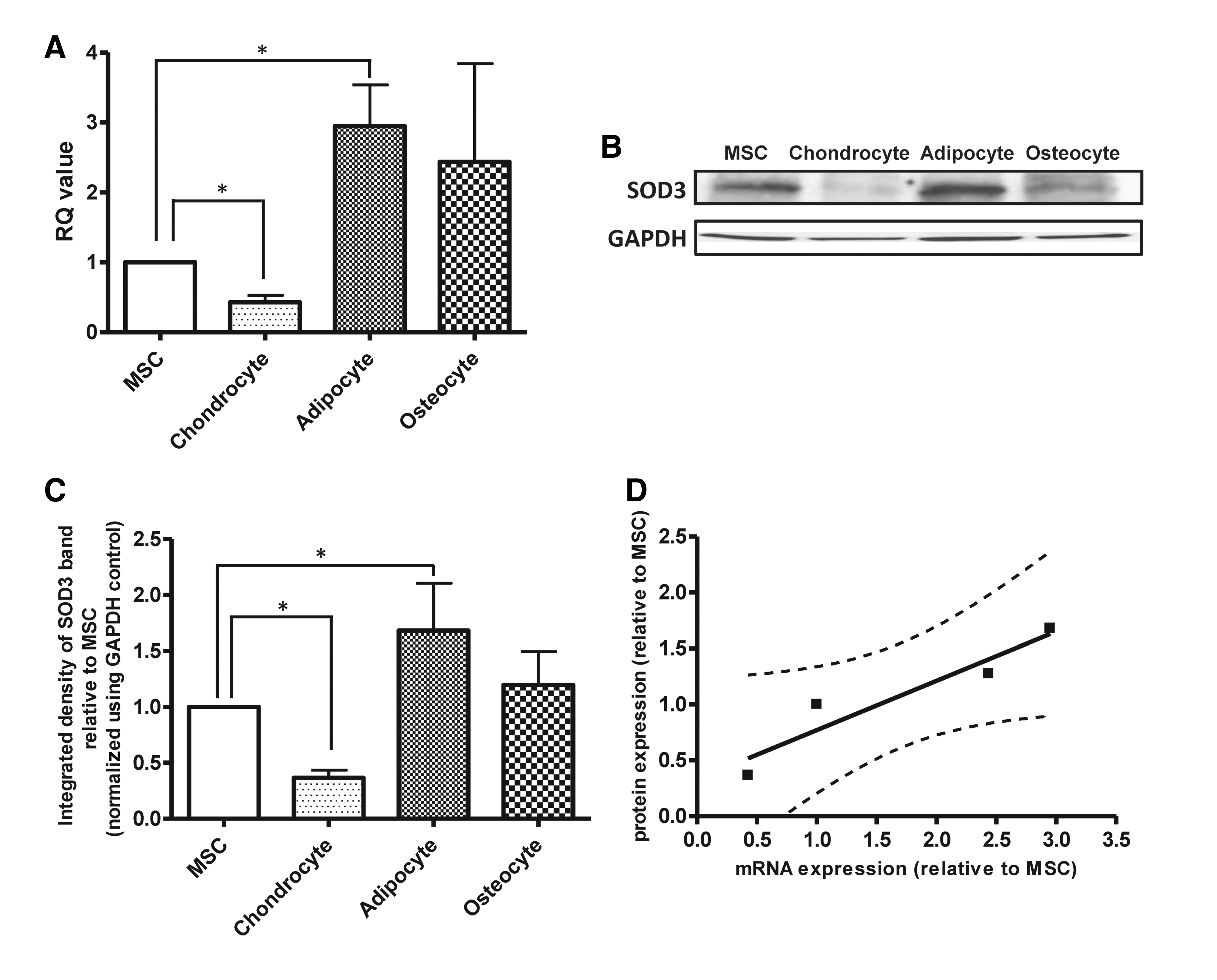
SOD3 mRNA and protein are differentially expressed by human MSC-differentiated chondrocyte and adipocyte cell cultures.
Next we determined if changes in BM-hMSC SOD3 gene expression following differentiation reflected levels of SOD3 protein expression. BM-hMSC–derived adipocyte, chondrocyte, and osteocyte cultures, cells were lysed and immunoblotting was performed for SOD3 (Fig. 3B). A positive band was found at a weight of 35 kDa, corresponding to the predicted band size of SOD3 and with previous published reports. The integrated density of the SOD3 bands are expressed as a percentage of GAPDH control. SOD3 protein expression was detected at significantly higher levels in hMSC-derived adipocytes and at significantly lower levels in hMSC-derived chondrocytes compared with the hMSC control (P<0.05; Fig. 3C). No significant changes in SOD3 protein expression were seen after induction of osteogenesis (Fig. 3C). Linear regression was then used to demonstrate the correlation between the mean SOD3 mRNA and protein levels by hMSC and hMSC-derived adipocyte, chondrocyte, and osteocyte cells (Fig. 3D). Linear regression showed a significant correlation between SOD3 mRNA expression and SOD3 protein expression for the cells tested (P<0.05, R 2=0.84).
MSC-derived chondrocytes, adipocytes, and osteocytes secrete an active and functional SOD3 enzyme
In order to demonstrate the activity and functional capability of the secreted SOD3 enzyme we cultured bone marrow-derived hMSC and bone marrow-derived hMSC differentiated chondrocytes, adipocytes, and osteocytes in minimal base media for 24 h to produce a cell conditioned medium. Conditioned media from cell cultures was then subjected to a Superoxide Dismutase Assay according to manufacturer's instructions to determine secreted SOD3 levels. We demonstrated that all media conditioned by hMSC cultures and hMSC cultures differentiated along adipogenic, osteogenic, and chondrogenic lineages secreted SOD3. The levels of SOD3 in the conditioned media from the hMSC-derived adipocyte cultures were significantly increased compared with hMSCs cultures grown under standard conditions (P<0.05). There were no significant changes in SOD3 levels in the conditions media from hMSC-derived osteocyte or chondrocytes cultures compared with hMSC cultures (Fig. 4A).
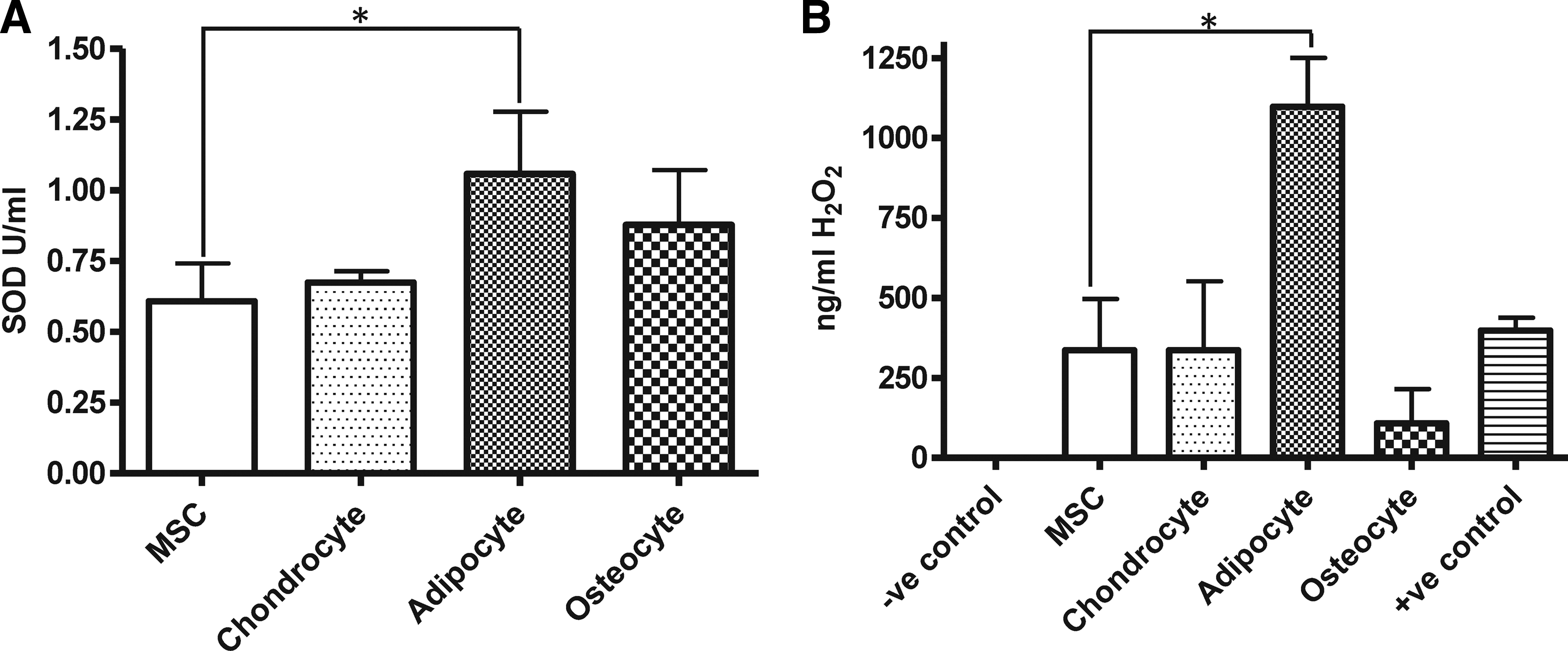
Human bone marrow-derived MSC and MSC-differentiated chondrocyte, adipocyte, and osteocyte cell cultures secrete active and functional superoxide dismuting enzymes.
To determine the functional capacity of SOD3 secreted by the cell cultures, conditioned medium was exposed to 0.15 mM DETANONOate, which was used as a nitric oxide donor. Nitric oxide spontaneously forms superoxide with water, and levels of hydrogen peroxide within the conditioned media were measured as an assay of the dismutation of superoxide by SOD3. We demonstrated that all media condition by hMSC cultures and hMSC cultures differentiated along adipogenic, osteogenic, and chondrogenic lineages secrete a functional form of SOD3 capable of dismuting superoxide to produce hydrogen peroxide in vitro (Fig. 4B). Prior to the addition of nitric oxide, all conditioned medium and minimal base medium controls contained no detectable hydrogen peroxide (results not shown). The amount of hydrogen peroxide within the conditioned medium obtained from hMSC-derived adipocyte cultures was significantly higher than that of the conditioned medium obtained from hMSC cultures when grown under standard culture conditions (P<0.05). No significant differences in the levels of hydrogen peroxide within the conditioned media were evident between hMSC and hMSC-derived chondrocyte and osteocyte cultures.
Discussion
Human mesenchymal stem cells have been shown to have antioxidative properties. Recent evidence from our laboratory and from others has demonstrated that bone marrow-derived hMSCs specifically secrete the extracellular antioxidant molecule SOD3, which directly promotes cellular survival following oxidative stress by reducing cellular toxic ROS [7,8,10]. Therefore, SOD3 production and secretion by transplanted MSC offers a potential future therapeutic antioxidant option. Here we report that SOD3 expression is dependent on the differentiation state of bone marrow-derived hMSC cultures down adipocyte, chondrocyte, and osteocyte cell lineages.
Our results in this study demonstrate a significant increase in the expression of SOD3 mRNA and protein following adipogenic differentiation of hMSC cultures. Furthermore, there was an evident increase in SOD secretion, and hence SOD activity present within the conditioned medium derived from cells differentiated down the adipogenic lineage. Similarly, a recent report has demonstrated up-regulation of the SOD3 gene expression during the early adipogenesis of mouse 3T3-L1 pre-adipocyte cells [43]. Transcription factors controlling adipogenesis include CCAAT/enhancer-binding protein alpha (C/EBP-α) and beta (C/EBP-ß) and peroxisome proliferator-activated receptors gamma (PPAR-γ) [44]. Interestingly, increases in SOD3 gene expression have been found to correlate well with changes in both PPAR-γ and C/EBP-α expression during adipogenesis [43]. Furthermore, the C/EBP-ß enhancer (prolactin) has been shown to induce SOD3 mRNA and protein expression in cultured fibroblast cell lines; however in contrast, PPAR-γ ligands (thiazolidinediones) are reported to show no effect on SOD3 expression in these culture systems [45].
Oxidative stress is known to be an important etiological factor in numerous pathologies involving cells of mesenchymal origin. The adipose tissue of obese subjects has shown to exhibit high levels of oxidative stress, which is associated with insulin resistant diabetes, type 2 diabetes, and atherosclerosis [24]. Obese mice (ob/ob) compared with lean mice have been demonstrated to have increased SOD3 levels in the white adipose tissue, liver, and kidney but decreased in the lung. It is therefore suggested that increased SOD3 levels within white adipose tissue of these mice is a compensatory mechanism subsequent to increased levels of oxidative stress in obesity [46]. Oxidative stress has long been associated with atherosclerosis, in particular oxidized low-density lipoprotein (LDL) is considered as an important mediator of this disorder [25,26]. SOD3 is abundant in arterial interstitium [47] and has been suggested through in vitro studies as a potential anti-atherosclerotic factor [27,28,48]. Specifically, exogenous addition of SOD3 or SOD3 overexpression has been shown to reduce vascular endothelial cell-mediated LDL oxidation [27,28,49], yet subsequent in vivo studies of SOD3 in LDL receptor null mice report that SOD3 has no influence on atherogenesis [50,51]. Some caution is however needed when interpreting results from studies involving animal models with life-long deficiencies, as compensatory mechanism may have occurred, such as increases in alternative antioxidant mechanisms [52]. Indeed, the functional role of SOD3 is unclear [52]. Our findings help to further define the characteristics of SOD3 production and secretion by hMSC cultures and their progeny in vitro. However, further impetus for research in this area is required, particularly to establish if there is a functional role in vivo for the cellular expression of SOD3 following hMSC differentiation.
Our results in this study also demonstrate that SOD3 is expressed and secreted by hMSC-derived cultures differentiated down the chondrogenic lineage. However, it was evident that there was a significant decrease in both SOD3 protein and gene expression following chondrogenesis of bone marrow-derived hMSC cultures. It was noted in this study that the SOD activity within the conditioned medium (hence SOD3 secretion) did not correlate with the reduction of SOD3 protein expression after differentiation down the chondrogenic lineage. As SOD3 is secreted through the trans-Golgi network, these discrepancies may be due to differences in secretion rates between cell populations and thus differences in the amount of intracellular versus extracellular SOD3 levels. In relation to cartilage, oxidative stress is a process implemented in the pathogenesis of osteoarthritis [23,53]. SOD3 is normally highly expressed in human articular cartilage [54]. However, levels are described to be substantially decreased during progression and end stages of osteoarthritis [23,53]. Numerous studies have documented the use of antioxidants for arthritis [55] and SOD3 specifically has been shown to protect collagen-I from oxidative fragmentation [56]. In addition, ex vivo SOD3 gene transfer has been described to reduce severity of collagen-induced arthritis [57]. It is suggested that decreases in SOD enzymes levels in articular cartilage may lead to increased levels of oxidative stress contributing to the pathogenesis of osteoarthritis [58]. Our results also demonstrated hMSC cultures differentiated down the osteogenic lineage express SOD3; however, there were no significant differences in mRNA or protein SOD3 levels compared with hMSC cultures. Following bone fracture, large amounts of ROS are released [59]; this impairs fracture healing [60]. Estrogen is reported to promote fracture healing [61] by influencing endochondral ossification and chondrogenesis [62,63]. Interestingly SOD3 mRNA levels are down-regulated in the fracture callus and adjacent bone of ovariectomized female rats. However, estrogen supplementation in these rats subsequently increases SOD3 mRNA levels [63]. This suggests a cellular component of fracture callus or adjacent bone secretes SOD3, which may ameliorate oxidative stress following bone fracture.
In summary, we have identified that levels of SOD3 expression are changed following in vitro differentiation of bone marrow-derived hMSC cultures down the adipocyte, chondrocyte, and osteocyte cell lineages. Furthermore, we have shown a significant correlation between SOD3 mRNA expression and SOD3 protein expression in the cells tested. The functional role of differential changes in SOD3 levels during differentiation is unclear. Recently SOD3 expression has been suggested as a cellular differentiation marker [64], and it is possible that SOD3 may also be marker of MSC differentiation down adipogenic, chondrogenic, and osteogenic cell lineages. Site-specific adipogenic and chondrogenic differentiation have been reported following transplantation of bone marrow-derived hMSCs [65]. Therefore, our results may also have possible implications for the in vivo use of hMSC transplants exposed to environments conducive to adipogenesis or chondrogenesis. Indeed, this especially holds true if hMSCs and their mature progeny are going to be considered in transplantation studies as a therapeutic option for conditions in which oxidative stress is a major etiological component. The secretion of SOD3 by hMSCs at clinically significant levels would support hMSCs and hMSC lineage cells as a potentially viable therapeutic option for conditions amenable to treatment with antioxidants. However, further studies are required to comprehensively define the role of hMSC SOD3 production, secretion, and antioxidant activity in vivo.
Footnotes
Acknowledgments
This work was carried out using a project grant from The Burden Institute.
Author disclosure statement
The authors indicate no potential conflicts of interest.