
Abstract
We describe a new, efficient protocol that involves the serial addition of noggin, basic fibroblast growth factor (bFGF), retinoic acid, and sonic hedgehog (Shh) for the differentiation of human induced pluripotent stem cells (hiPSC) to retinal pigmented epithelium (RPE) in a serum- and feeder-free adherent condition. hiPSC-RPE cells exhibited RPE morphology and specific molecular markers. Additionally, several hiPSC lines were generated from retinal-specific patients with Leber's congenital amaurosis, Usher syndrome, two patients with retinitis pigmentosa, and a patient with Leber's hereditary optic neuropathy. The RPE cells generated from these disease-specific hiPSCs expressed specific markers by the same RPE lineage-directed differentiation protocol. These findings indicate a new short-term, simple, and efficient protocol for differentiation of hiPSCs to RPE cells. Such specific retinal disease-specific hiPSCs offer an unprecedented opportunity to recapitulate normal and pathologic formation of human retinal cells in vitro, thereby enabling pharmaceutical screening, and potentially autologous cell replacement therapies for retinal diseases.
Introduction
T
Recent groundbreaking developments regarding induced pluripotent stem cells (iPSCs) produced from somatic cells [3] have been proposed as providing a nearly inexhaustible source of cells (for review, see references 4 and 5] quite similar to their respective embryo-derived embryonic stem cell (ESC) counterparts [6,7]. Human embryonic stem cells (hESCs) can differentiate into RPE cells [8 –23]. Similar techniques can be applied to iPSCs to generate RPE cells in vitro [10,16,17,19,24]. These hESC-RPE and human induced pluripotent stem cells (hiPSC)-RPE have been demonstrated to be similar to native RPE in many ways, including expression of markers, transcriptome [10,21], function, and rescue of visual function in the dystrophic rat [13 –15,24,25]. These reports describe the differentiation of RPE from hESCs or hiPSCs by several approaches (for review, see references 26,27). In one approach, hESCs and hiPSCs were allowed to overgrow and become multilayered, and spontaneous differentiation was induced by the withdrawal of basic fibroblast growth factor (bFGF) from the expansion medium [9,10,13,15,16,25]. Timescales vary between these reports, but approximately 1 to 8 weeks after growth factor removal, brown pigmented spots were observed. Approximately 6 to 14 weeks later, the pigmented spots were of sufficient size for mechanical excision and removal to new plates for their expansion [16,25]. In another method for RPE differentiation from hESCs and hiPSCs, cells were co-cultured with PA6 stromal cells [12,18,23,28]. The third approach for differentiation of hESCs and hiPSCs to RPE included embryoid body (EB) formation [8,14,17,19 –22,24,25]. The resultant EBs were maintained in suspension for a varying amount of time, from approximately 1 to 3 weeks [8,14,17,19 –22,24] or up to 9 months [24,25]. Consequently, the aggregates were replated in tissue culture plates and further differentiated. The first RPE areas were visualized after 4 to 8 weeks from the start of the protocol. Several different growth factors and chemicals have been examined for their effects on the induction of RPE differentiation in these approaches. Such supplements included WNT antagonists (eg, Dickkopf-1, Dkk-1) combined with a bone morphogenetic protein (BMP) antagonist (eg, noggin), NODAL antagonists [eg, lefty-A, a transforming growth factor beta (TGF-β) ligand], insulin-like growth factor (IGF), activin, and small molecules such as dorsomorphin, XAV939, nicotinamide (vitamin B3), SB431542, N2 or B27 supplement, and heparin [19 –22,24,29].
Overall, despite numerous attempts, the differentiation process of hESCs and hiPSCs to RPE is not well defined and most often necessitates a long-term differentiation time with low efficiency; the ideal supplements for the induction of directed differentiation are still unclear.
Importantly, there are limited reports regarding the generation of hiPSCs from patients with retinal disease [19,30], an approach that may provide appropriate cellular sources for biomedical applications. Therefore, this study describes and illustrates a new, simple, efficient, and fast protocol for direct differentiation of hiPSCs toward RPE generation, as well as the generation and characterization of hiPSC lines in serum- and feeder-free culture conditions from patients diagnosed with retinal disease. These hiPSCs are indistinguishable from hESCs with respect to colony morphology, passaging, surface and pluripotency markers, normal karyotype, and differentiation potential. We have also differentiated these cell lines into RPE in vitro.
Materials and Methods
Establishment and culture of hiPSCs from patients' fibroblasts
Human skin biopsies were obtained in accordance with the Declaration of Helsinki, following approval from the Royan Institute Review Board and Ethical committee and after obtaining informed consent from Iranian patients diagnosed with eye disease (Table 1;
hiPSCs were generated by transduction with retroviral vectors that contained Oct4, Sox2, c-Myc, and Klf4, as described earlier [31]. The transduced cells were passaged and expanded on Matrigel (1:30, Sigma-Aldrich) under feeder-free culture conditions as previously described (31) in hESC medium that contained Dulbecco's modified Eagle's medium (DMEM)/F12 medium (Gibco) supplemented with 20% knockout serum replacement (KOSR, Gibco); 2 mM
Additionally, a hiPSC line (hiPSC1, passage 48) [31] from a normal individual and a hESC line (Royan H6, passage 35) [34] were used in this study.
Immunofluorescence staining
The hiPSCs were fixed in 4% paraformaldehyde for 30 min, permeabilized with 0.2% Triton X-100 for 20 min (except for surface markers), washed in phosphate-buffered saline (PBS), and blocked in PBS that contained 1% BSA and 10% normal goat serum for 1 h at 37°C.
Cells were incubated with primary antibody for 1 h at 37°C, then washed and incubated with the following fluorescein isothiocyanate (FITC)-conjugated secondary antibodies: anti-mouse IgM (1:100, Sigma-Aldrich); anti-rat IgM (1:200, eBioscience); anti-mouse IgG (1:200, Sigma- Aldrich); and goat anti-rabbit IgG-Texas red conjugated secondary antibody (1:100, Santa Cruz), as appropriate, for 1 h at 37°C.
The following primary antibodies were used for pluripotency determination: anti-TRA-1-60 (1:100) and TRA-1-81 (1:100, gifts from Dr. P. Andrews, Sheffield, UK); Oct4 (1:100, Santa Cruz Biotechnology) and SSEA-3 (1:100, Chemicon). In addition, anti-MITF (1:100, Abcam,); OTX2 (1:100, Abcam); PAX6 (1:100, Santa Cruz); ZO-1 (1:30, Abcam); RPE65 (1:250, Abcam6); CRALBP (1:250, abcam); and bestrophin (1:250, Abcam) were used for RPE lineage determination. Nuclei were counterstained with DAPI (Sigma) and cells analyzed under the fluorescence microscope (Olympus).
Alkaline phosphatase staining and karyotype analysis
Alkaline phosphatase staining was conducted based on the manufacturer's recommendations (Sigma). Karyotype analysis was performed as previously described [32].
RNA isolation and quantitative reverse transcription-polymerase chain reaction
Total RNA was isolated by TRI reagent and treated with DNase I (Takara) to remove genomic DNA contamination. Two micrograms of total RNA was used for reverse transcription polymerase chain reaction (qRT-PCR)with the RevertAid™ H Minus First Strand cDNA Synthesis Kit (Fermentas) and random hexamer primers (Fermentas), according to the manufacturer's instructions. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) reactions were set up in duplicate with the Power SYBR Green Master Mix (Applied Biosystems). Relative gene expression was analyzed by comparative Ct method, 2-ΔΔCt [35]. Target genes were normalized by the reference gene GAPDH and calibrated for each sample against control cells (hiPSCs or hESCs, as appropriate from the same cell lines). Each experiment included 3 replicates. Thermal conditions for all genes were the same and the annealing temperature was 60°C.
The sequences of primers for OCT4 and NANOG, transgenes, and other embryonic germ layer genes were previously presented [31]. The primer sequences of RPE-related genes are presented in Supplementary Table S1 (Supplementary Data available online at
Differentiation, enrichment, and culture of pigmented cells
The differentiation procedure is outlined in Fig. 1A. hiPSCs were induced to neuroectoderm (NE) in induction medium that contained DMEM/F12 medium, 5% KOSR, N2 (2%, Invitrogen; which included recombinant insulin, human transferrin, sodium selenite, putrescine, and progesterone), 1% NEAAs, 2 mM
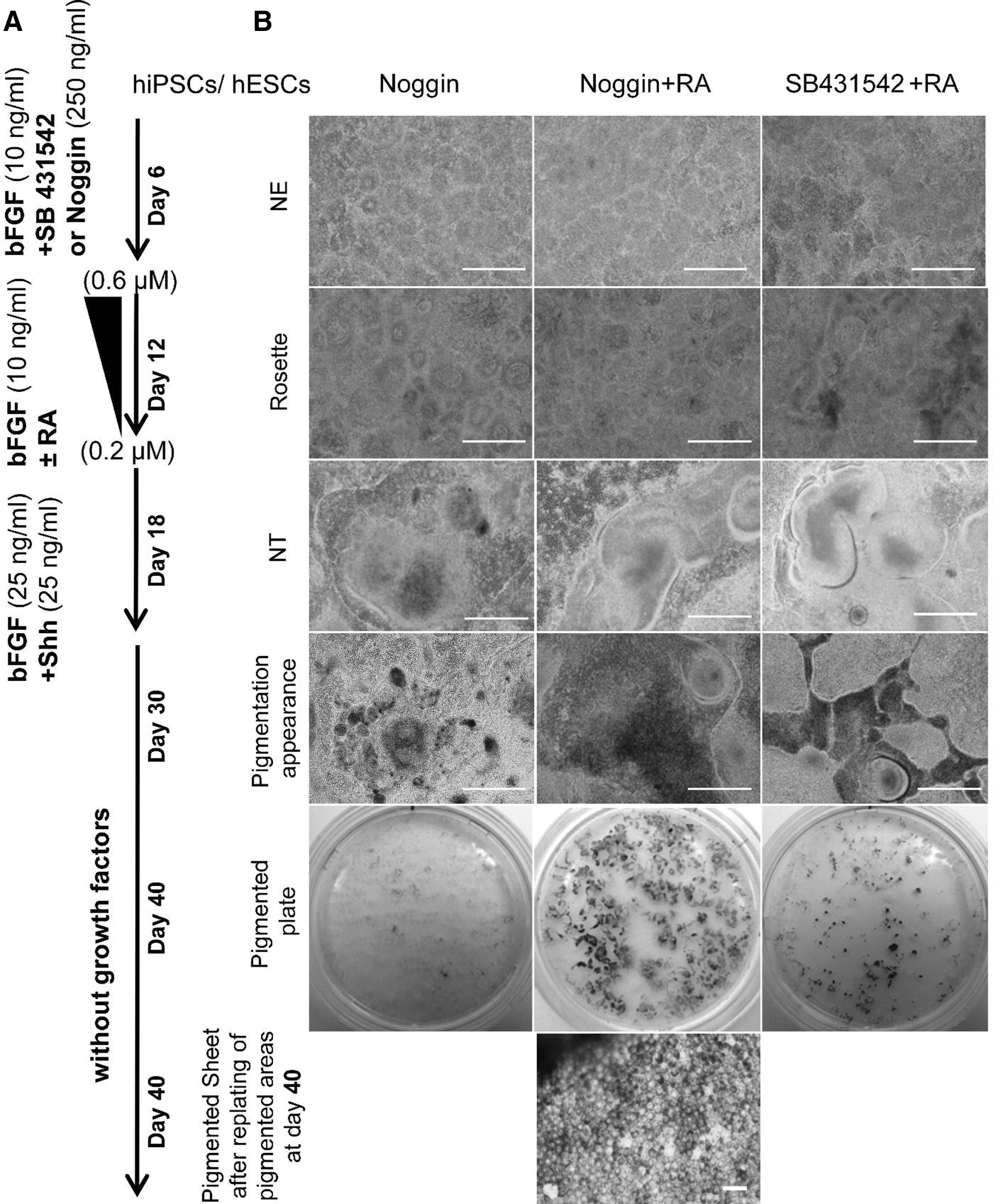
The differentiation of induced pluripotent stem cells (hiPSCs) into retinal pigmented epithelium (RPE).
The cells were subsequently grown without noggin for another 6 days in the same medium in the absence or presence of all-trans-retinoic acid (RA, Sigma-Aldrich) at a decreasing concentration from 0.6 to 0.2 μM over 6 days to promote additional rosette formation. The medium was renewed every other day at these stages. Then, cells were exposed to 25 ng/mL bFGF along with 25 ng/mL sonic hedgehog (Shh, R&D) in the absence of RA for an additional 6 days giving rise to what appeared to be neural tube (NT)-like structure-organized rosettes that included lumen. On day 18, the culture was maintained in DMEM-F12 medium supplemented with 5% KOSR, 1% NEAAs, and 2 mM
Flow cytometric analysis
Initially, cells were washed in PBS supplemented with 2% fetal calf serum (FCS) followed by dissociation in 0.05% trypsin-ethylenediaminetetraacetic acid (Sigma-Aldrich). Consequently, all staining reactions were performed in washing buffer that consisted of 0.1% bovine serum albumin (BSA) in PBS. After determination of cellular viability by trypan blue exclusion, cells were fixed in 4% paraformaldehyde for 15 min or in –20°C ethanol/acetone (3:7) for 10 min at 4°C. After washing, cells were permeabilized with Triton X-100 (0.1%) for 10 min at room temperature. Non-specific antibody binding was blocked with 10% heat-inactivated goat serum in washing buffer. A total of 1–1.5×105 cells were used per sample. Subsequently, the cell suspension was incubated overnight with the appropriate diluted primary antibodies at 4°C. After washing, cells were incubated for 60 min at 4°C with goat anti-mouse FITC-conjugated secondary antibody (1:100, Santa Cruz). The control sample contained only the secondary antibody. All experiments were replicated 3 times with a BD-FACS Calibur flow cytometer and the acquired data was analyzed with WinMDI2.9 software.
Statistical analysis
All experiments were conducted in at least 3 independent cultures. Data from qRT-PCR and flow cytometry were expressed as mean±standard error and analyzed by one-way analysis of variance followed by the post hoc least significant difference (LSD) test for multiple comparisons.
P values less than 0.05 were considered significant.
Results
Development of hiPSC-derived pigmented cells
Given the reported roles of noggin [36,37], RA [38], and a selective inhibitor of the TGF-β superfamily type I receptors (SB) ([37] in cell differentiation into neural cells, we sought to study their influence on the differentiation of hiPSCs to RPE. Initially, a population of NT-like structures was established without EB formation through feeder-free, adherent, and defined conditions. hiPSCs were induced to produce neural rosettes by the combination of bFGF along with noggin, noggin+RA, or SB+RA. Neural ectoderm was noted by the appearance of rosettes and NT (Fig. 1B). Consequently, the rosettes and NTs were exposed to Shh, a RPE inducer [39 –41], from day 12 to day18.
In the presence of noggin+RA, pigmented areas began to appear within the differentiating clusters after 4 weeks; most hiPSC clusters had pigmented cells by day 35 of differentiation (Fig. 1B). Morphologically, differentiated dark cells seemed to be putative RPE cells because they contained pigment granules and were organized with a cobblestone appearance.
In the noggin only group, few pigmented areas were observed. In the SB+RA group, more pigmented areas of the hiPSC clusters developed after 6 weeks, but were much less than those seen in the noggin+RA group (Fig. 1B). Noggin+RA promoted the appearance of more pigmented areas within the differentiating hiPSC clusters.
Pigmented foci were mechanically dissected out of the culture and placed in new Matrigel-coated plates at day 30 after which they developed into an enriched and nearly uniform population of a single layer RPE phenotype by day 40 (Fig. 1B). Given these results, we further studied the influence of noggin+RA on RPE differentiation.
The effect of noggin+RA on differentiation toward pigmented cells was confirmed in an additional hESC line (Royan H6; Supplementary Fig. S1).
To further analyze differentiating hiPSC-RPE in vitro, we performed qRT-PCR, immunostaining, and FACS for the expression of markers of undifferentiated hiPSCs, as well as retinal and RPE development during differentiation. During initiation of early neural differentiation, expression of markers of undifferentiated hiPSCs (OCT4 and NANOG) was down-regulated (Fig. 2A). During this process, hiPSCs acquired expression of transcription factors associated with eye field specification (PAX6, RX, SIX3) [22], and CHX10, which regulates the proliferation of neural retinal progenitors [17]. qRT-PCR analysis further demonstrated that expression of RX, SIX3 and CHX10 were up-regulated, whereas PAX6 was down-regulated from days 15 through 60 of differentiation. The expression of RPE markers, microphthalmia-associated transcription factor (MITF), and RPE-specific protein 65 kDa (RPE65), was strongly increased. RPE65 is involved in the conversion of all-trans retinol to 11-cis retinol and plays a principal role in visual pigment regeneration. The expression of cellular retinaldehyde-binding protein (CRALBP; also known as RLBP1), involved in vitamin A metabolism, was also augmented. Other more mature RPE cell markers, such as pigment epithelium derived factor (PEDF), DCT, and tyrosinase (TYR) pigmentation synthesis enzymes, were expressed relatively early during the differentiation process from day 15; DCT was further up-regulated with additional culture. Additionally, we observed the expression of other genes known to be expressed in the RPE, including orthodenticle 2 isoform b homeobox protein (OTX2), mer tyrosine kinase (MERTK), membrane-associated protein bestrophin (BEST), and endothelial growth factor (VEGF) by RT-PCR (data not shown).
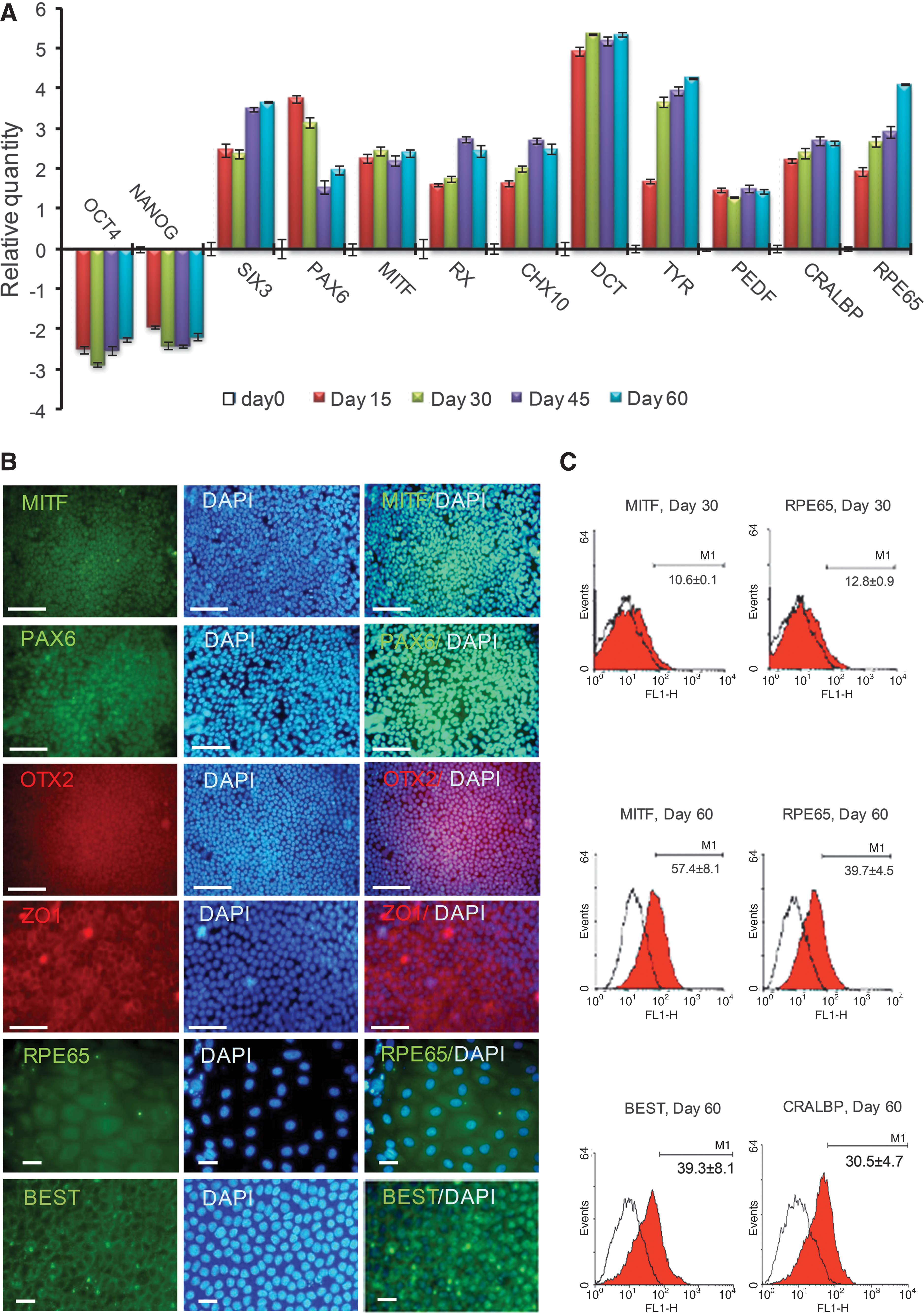
Characteristics of hiPSC1-RPE.
To confirm that the isolated cells expressed proteins typical of RPE phenotype, the expression and localization of OTX2, PAX6, MITF, RPE65, and ZO1 were analyzed by immunostaining (Fig. 2B). MITF, PAX6, and OTX2 localized to the nuclei and RPE65 was observed in the cytoplasm. ZO1 localized to the tight junctions on the cell membrane.
FACS analysis after 30 and 60 days of initiation of differentiation in the presence of noggin+RA demonstrated that 10.6%±0.1% and 57.4%±8.1% of the cells expressed MITF and 12.8%±0.9% and 39.7%±4.5% of the cells expressed RPE65, respectively (Fig. 2C). Additionally, 30.5%±4.7% of the total cells at day 60 expressed CRALBP, whereas 39.3%±8.1% of the total cells at day 60 expressed BEST (Fig. 2C).
Therefore, we concluded that hiPSCs differentiated into cells with RPE characteristics in a noggin+RA culture.
Production of retinal disease-related hiPSCs
To develop the potential for studying eye disease-specific RPE, we generated retinal disease-specific hiPSCs and examined their potential for RPE differentiation. Human dermal fibroblasts (HDF) were reprogrammed into hiPSCs under serum- and feeder-free culture conditions as described above. Three to twelve colonies per patient were picked from transduced HDFs and expanded (Table 1). One hiPSC line per patient was further expanded for analysis (Table 1). All selected hiPSC lines showed hESC-like morphology: compact colonies whose cells demonstrated high nucleus-to-cytoplasm ratios as well as prominent nucleoli (Fig. 3). These lines were expanded in a continuous culture for several months by weekly passaging with a split ratio of 1:3 to 1:6.
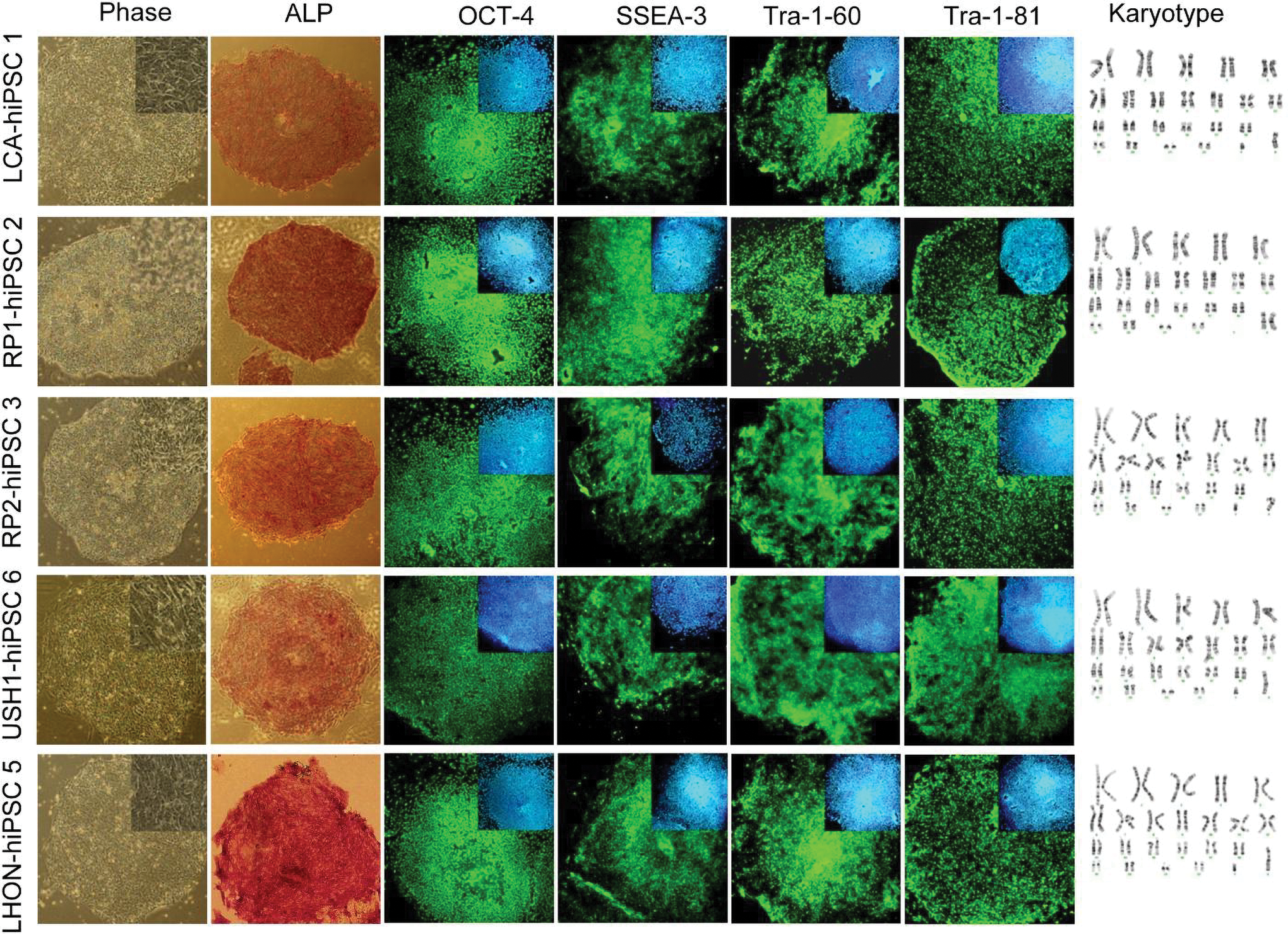
The characterization of established retinal and optic nerve disease-specific hiPSCs. Morphology, expression of different pluripotency and surface markers, and normal karyotypes are presented. The lines are characterized after at least 10 passages. Nuclei were stained with DAPI (blue). Color images available online at
Analysis of the colonies with hESC-like morphology showed that the clones demonstrated alkaline phosphatase (ALK) activity along with expression of OCT4, SSEA3, TRA-1-60, and TRA-1-81. Furthermore, all hiPSC lines also showed normal karyotype (Fig. 3).
qRT-PCR analysis using primers specific for OCT4, SOX2, c-MYC, and KLF4 transgenes showed that the hiPSC clones silenced expression of the retroviral transgenes (Fig. 4A). We also analyzed gene expression by qRT-PCR and noted that the expression of OCT4 and NANOG markedly increased over the respective fibroblast population and were comparable to that seen in hESCs (Fig. 4B). RT-PCR analysis of spontaneously differentiated cells showed gene expression of a number of lineage specific marker genes (Fig. 4C).
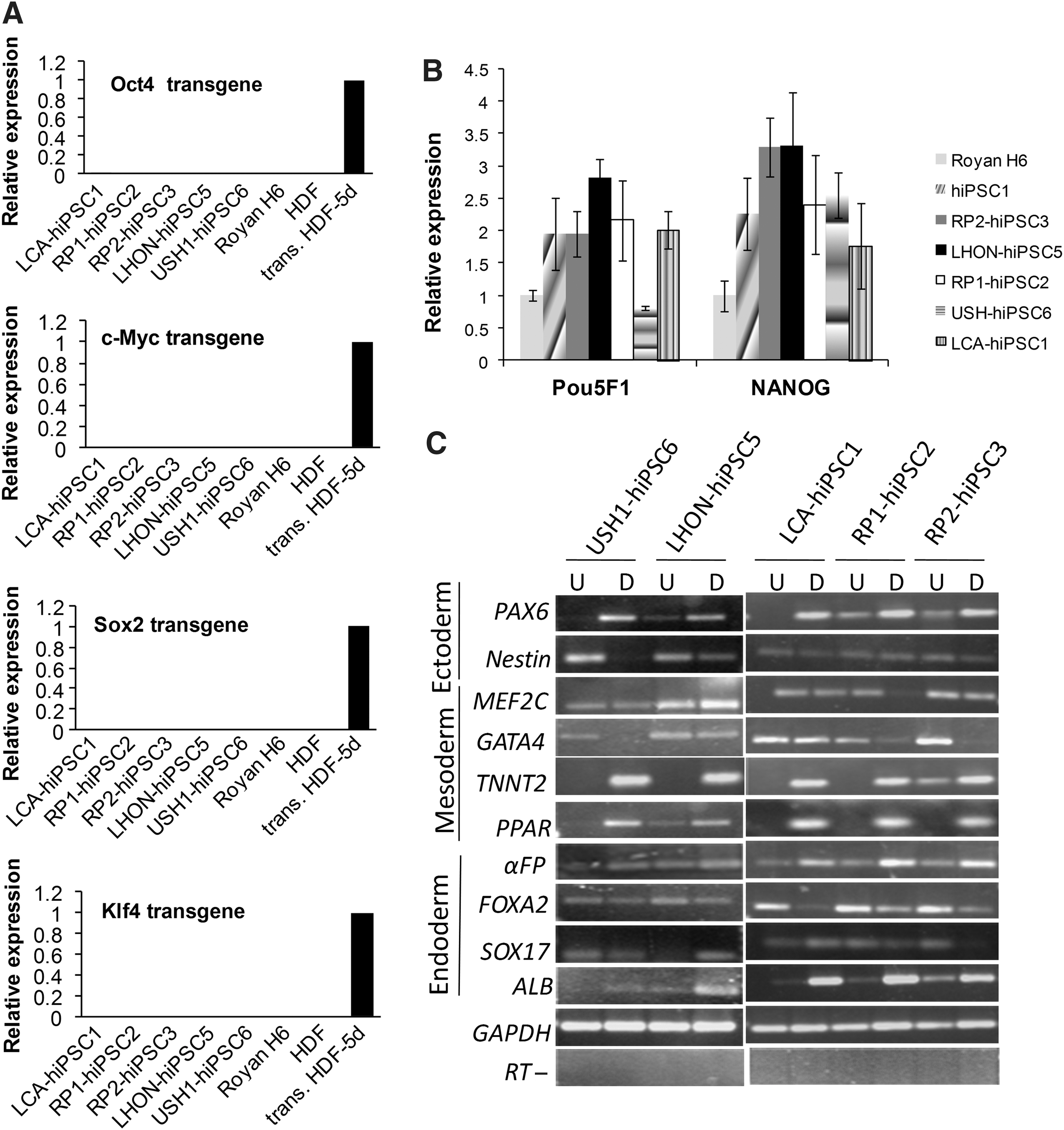
The expression of gene markers in retinal disease-specific hiPSCs.
Next, we evaluated RPE differentiation of these patient-specific hiPSC lines by the noggin+RA protocol and assessed the differentiated cells by morphology and qRT-PCR for expression of markers of RPE development during differentiation (Fig. 5). We found similar results in expression of RPE markers to those seen above.

Differentiation of retinal disease-specific hiPSCs into RPE.
Discussion
Here we describe a novel method for generating large quantities of RPE cells from hiPSCs and hESCs in adherent, serum- and feeder-free conditions, with the use of noggin+RA gradient, which renders the process clinically compliant. The hiPSC-derived pigmented cells demonstrate the morphology and marker profile of bona fide RPE cells.
The differentiation protocol described here differs from other published protocols by the serial application of noggin, bFGF, RA, and Shh. Similar to other published protocols, the restriction of hESCs and iPSCs to the neural lineage was initiated by the administration of noggin to inhibit BMP signaling [36,37]. However, with regards to media composition, this protocol differs from others in the application and a temporally changing concentration of RA (0.6 to 0.2 μM) exposure. Additionally, the absence of the selective inhibitor of TGF-β superfamily type I receptors, SB431542, resulted in more RPE. In keeping with this observation, it was reported that the presence of activin, a member of the TGF-β superfamily, augmented RPE differentiation [24]. Finally, the presence of exogenous Shh was another feature of our RPE differentiation protocol. This is in agreement with the presumed roles of noggin, bFGF, RA, and Shh in RPE development in vivo. In the embryo, RA and Shh are presumed to direct the differentiation of the optic vesicle into RPE. Several lines of evidence converge to suggest that both FGF and Shh are involved in the regulation of the initial patterning decisions in the optic vesicle that lead to the RPE and neural retinal fates [39 –41]. Shh misexpression, for example, leads to the appearance of pigmented cells in the neural retina [42]. Similarly, inhibition of the hedgehog pathway at the optic cup stage by treating Xenopus embryos with cyclopamine has been shown to lead to the lack of pigmentation and reduction in the expression levels of RPE markers [39]. Another factor shown to be an important developmental signal important for the patterning of the optic vesicle domains is RA [43]. RA is produced in the surface ectoderm over the mouse eye field at E8.75 and later in the dorsal RPE starting at E9.5 [44]. Overall, the combination of these factors significantly promotes differentiation into neural precursors with a forebrain phenotype and consequently into progenitors expressing eye field and subsequently RPE markers.
The reported yield of RPE cells after 4–8 weeks of spontaneous differentiation is relatively low; fewer than 1% of EBs contain pigmented cells [25]. Recently, it was demonstrated that the application of Wnt and Nodal antagonists casein kinase I inhibitor (CKI-7) and SB-431542 in serum-free and feeder-free floating aggregate cultures of hESCs and hiPSCs induced retinal progenitor MITF-positive colonies (32% of total colonies) and cells (22% of total cells) [29]. The highest yield of RPE-like pigmented cells at 8 weeks was 33%. Idelson et al. reported that 70% of the clusters contained pigmented areas [24] in the presence of activin A and nicotinamide, by floating aggregate culture and then replating. In our suspension system, after 60 days of differentiation, cells were MITF+(57%), RPE65+ (40%), CRALBP+ (30%) and BEST+ (39%). Interestingly, protein expression of RPE65 was only demonstrated in other studies after long-term culture (several months) [16], although mRNA has been detected under shorter culture conditions. RPE65 is an essential gene in visual function, mutations of which cause LCA [45,46].
In this study, we showed the generation of hiPSCs from adult HDFs of patients with retinal diseases by retroviral transduction of OCT4, SOX2, c-MYC, and KLF4 with the use of serum- and feeder-free culture conditions. Analysis of selected colonies demonstrated hESC-like morphology, expression of ALP, OCT4, SSEA3, TRA-1-60, and TRA-1-81, and Nanog, and the epigenetic silencing of OCT4, SOX2, c-MYC, and KLF4 transgenes in hiPSCs [47]. The established hiPSCs showed expression of 3 embryonic germ layer markers after spontaneous differentiation, supporting their pluripotent nature.
Moreover, after directed differentiation of these disease-specific hiPSCs toward RPE, the cells expressed RPE-related markers. The number of hiPSCs that contained RPE, however, was lower in comparison with normal hiPSCs. Similar results were observed by others when RP disease-specific hiPSCs differentiated into rod photoreceptor cells: the number of the patient-derived rod cells with distinct mutations decreased in vitro [30]. It is unclear if these results relate to the underlying disease associated with differentiated RPE [30] or a different propensity of the hiPSC lines for RPE differentiation [27].
In addition to the assays used in the current study (gene/protein expression), other assays such as quantitative ROS, phagocytosis, measurements of transepithelial resistance [48], enzyme-linked immunosorbent assays to detect polarized secretion of trophic factors (PEDF, vascular endothelial growth factor) [49] and a retinoid metabolism assay [50] could be evaluated for further qualification of differentiation. Since retinal disease-specific hiPSC-RPEs can be generated with relative ease from any individual, the immediate potential benefit of these hiPSCs not only will be in the exploration of disease etiology (for review, see reference 51), but also for drug development and screening a cell source for the study of ethnic/polymorphic variation. Recently, Takahashi et al. have established hiPSCs from 5 RP patients with distinct mutations in the RP1, RP9, PRPH2, or RHO genes [30]. Consequently, they differentiated hiPSCs into rod photoreceptor cells which had been lost in these patients. Interestingly, these patient-derived cells expressed markers for oxidation or endoplasmic reticulum stress, and showed different responses to vitamin E compared with what had been observed in clinical trials. Recently, it was also demonstrated that gyrate atrophy disease hiPSCs-derived RPE exhibited a disease-specific functional defect that could be corrected either by pharmacological means or following targeted gene repair [19].
These data suggest that hiPSCs may be a powerful resource for the production of RPE with which to study basic developmental biology [52]. Another possible application of hiPSCs involves cell-replacement therapy in eyes with retinal degenerative diseases due to primary RPE dysfunction [26]. Diseases involving the RPE are attractive targets for hiPSC-based therapies, due to easy differentiation of RPE cells from hiPSCs, noninvasive monitoring of the site and simple evaluation of retinal function. The proof-of-principle for the therapeutic potential of hiPSC-RPE has been established by treating diseases in animal models [13 –15,24,25]. Recently, Advanced Cell Technology received United States Food and Drug Administration approval for a phase I/II clinical trial to treat patients with AMD using hESC-RPE after showing its safety in preclinical models [14].
However, several issues such as epigenetic variations and the mutational load in hiPSCs [7,53 –56] remain to be solved before hiPSC-derived cells can be safely applied in clinical settings (for reviews, see references 4 and 5). Improvements in the production of safe and non-immunogenic hiPSCs will increase the number of applications of hiPSC derivatives.
In conclusion, our findings indicate a new short, simple, and efficient protocol for differentiation of hiPSCs to RPE cells. Additionally, our results provide a proof-of-concept that multiple retinal-disease specific hiPSC lines can be generated and differentiated into RPEs in a relatively simple and straightforward manner to evaluate defects ex vivo.
Footnotes
Acknowledgments
This study was funded by grants provided from Royan Institute and the Iranian Council of Stem Cell Research and Technology.
Author Disclosure Statement
The authors declare they have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.