
Abstract
Human adult mesenchymal stem cells (MSCs) support the engineering of functional tissue constructs by secreting angiogenic and cytoprotective factors, which act in a paracrine fashion to influence cell survival and vascularization. MSCs have been isolated from many different tissue sources, but little is known about how paracrine factor secretion varies between different MSC populations. We evaluated paracrine factor expression patterns in MSCs isolated from adipose tissue (ASCs), bone marrow (BMSCs), and dermal tissues [dermal sheath cells (DSCs) and dermal papilla cells (DPCs)]. Specifically, mRNA expression analysis identified insulin-like growth factor-1 (IGF-1), vascular endothelial growth factor-D (VEGF-D), and interleukin-8 (IL-8) to be expressed at higher levels in ASCs compared with other MSC populations whereas VEGF-A, angiogenin, basic fibroblast growth factor (bFGF), and nerve growth factor (NGF) were expressed at comparable levels among the MSC populations examined. Analysis of conditioned media (CM) protein confirmed the comparable level of angiogenin and VEGF-A secretion in all MSC populations and showed that DSCs and DPCs produced significantly higher concentrations of leptin. Functional assays examining in vitro angiogenic paracrine activity showed that incubation of endothelial cells in ASCCM resulted in increased tubulogenic efficiency compared with that observed in DPCCM. Using neutralizing antibodies we concluded that VEGF-A and VEGF-D were 2 of the major growth factors secreted by ASCs that supported endothelial tubulogenesis. The variation in paracrine factors of different MSC populations contributes to different levels of angiogenic activity and ASCs maybe preferred over other MSC populations for augmenting therapeutic approaches dependent upon angiogenesis.
Introduction
T
Among numerous progenitor cell types reported to have potential in the development of tissue engineering products, mesenchymal stem cells (MSCs) have been proposed as a prominent candidate. Human MSCs are self-renewing clonal precursors of cells derived from the mesoderm germ layer and exhibit numerous unique characteristics favorable for their use in tissue engineering, including (i) rapid proliferation enabling ex vivo expansion to support generation of large tissue constructs; (ii) wide differentiation capacity including differentiation toward adipocytes [2], osteocytes [3], chondrocytes [4], cardiomyocytes [5], smooth muscle cells [6], as well as a variety of connective tissues [7]; (iii) immunoprivileged nature that limits antigen presenting and costimulatory capacity, a characteristic likely to increase immune tolerance of the implanted constructs [8]; and (iv) secretion of a broad spectrum of growth factors and cytokines known to be angiogenic and cytoprotective [9 –11]. Apart from their clonogenicity and multipotency, MSCs also play supportive roles in tissue regeneration beyond their differentiation ability through promotion of angiogenesis and cell survival in a paracrine manner [12 –14].
MSCs have been successfully isolated from bone marrow [15,16], adipose tissue [2], cord blood [17], and dermis tissue [18]. However, due to lack of definitive cell surface antigens for specific classification, MSCs used in various studies inevitably represent a heterogeneous population. To determine whether MSC populations from these different tissues behave in a similar manner or reflect variations in the microniche from where they are derived, comparative analysis has been performed on basic cell characteristics and functional abilities in MSCs derived specifically from these tissues. Studies employing microarray-based comparisons of gene expression of human MSCs derived from adipose tissue (ASCs), bone marrow (BMSCs), and umbilical cord blood demonstrated that all 3 MSC populations have elevated expression of genes implicated in extracellular matrix production, morphogenesis, and development compared with fibroblasts [19]. A comparison of the immunological properties of BMSCs and ASCs further revealed a similar expression of immunologically relevant surface markers, including class I and II major histocompatibility complex and CD40/CD40L [20]. Both cell types exhibit comparable immunomodulatory effects that suppress mixed lymphocyte reaction and lymphocyte proliferative response to mitogens [21]. The differentiation capacity of BMSCs was more efficiently directed toward bone and cartilage, while ASCs preferentially differentiate into adipocytes, most likely due to a set of signature genes that are regulated differentially between these cells during maturation or lineage commitment and possibly influenced by the tissue-specific microenvironment regulating their biology [22,23]. In terms of angiogenic growth factors likely to increase angiogenic potential, murine ASCs are reported to secrete higher amounts of vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF) than murine BMSCs [24]. In contrast, human ASCs expressed comparable levels of VEGF mRNA when compared with human BMSCs [25]. Interestingly, despite similar VEGF-A mRNA levels, human ASCs exhibited greater proangiogenic activity than human BMSCs, an effect thought to be mediated by the matrix metalloproteinases (MMP)-3 and MMP-9 [25]. In addition, ASCs have been shown to exhibit greater therapeutic potential than BMSCs in attenuating murine brain ischemic injury [24] and improving limb perfusion recovery postischemia in a murine hindlimb ischemia model [25]. Collectively these studies suggest that variation in paracrine factor secretion profile of different MSC populations may provide an opportunity to select for a particular cell type that would benefit specific treatments, may that be tissue engineered constructs or patient-specific cell therapy.
To date, the basal paracrine activities of human ASCs in promoting angiogenesis have not been fully characterized or directly compared with angiogenic capacity of BMSCs, dermal sheath cells (DSCs), and dermal papilla cells (DPCs). Therefore, the aims of this study were to compare the transcriptional and translational profile of MSC populations derived from adipose tissue, bone marrow, and dermal tissue, and to compare their proangiogenic activity in vitro as well as assess the potential responsible paracrine factors.
Materials and Methods
Collection of human tissue for isolation of MSCs
Human tissues used for MSC isolation, including abdominal subcutaneous adipose tissue and scalp specimens, were excess normal tissues discarded during surgical protocols. Tissues were collected with informed consent according to the National Health and Medical Research Council guidelines and with approval from the St. Vincent's Health Human Research Ethics Committee. To minimize the potential variation resulting from experimental artefacts, such as serum concentration and cell passaging method, all MSC populations were cultured under identical conditions in vitro.
Primary culture of human adipose–derived stem cells
ASCs were isolated from freshly excised human abdominal subcutaneous adipose tissue (donor age between 35 and 58 years) according to the method described by Zuk et al. [2]. Briefly, adipose tissue (∼200 mL) was minced into 1 mm3 pieces, washed extensively with equal volumes of phosphate-buffered saline (PBS), and incubated at 37°C in the shaking water bath for 60 min in 0.075% type I collagenase (Worthington Biochemical, Lakewood, NJ). After tissue digestion, cells were collected by centrifugation at 300 g for 10 min to remove adipocytes and the cell pellets were resuspended in Dulbecco's modified Eagle's medium low-glucose medium (DMEM-lg; Invitrogen, Carlsbad, CA) containing 10% fetal calf serum (FCS) and 1% antibiotic-antimycotic solution containing penicillin, streptomycin, and amphotericin B (Invitrogen). Connective tissue debris was removed by filtering resuspended cells through a 100 μm nylon mesh and centrifuged at 700 g for 5 min, after which red blood cells were lysed by resuspending the cell pellet in 0.16 M NH4Cl and incubated at room temperature for 5 min. Finally the cell suspension in DMEM-lg was plated into T75 tissue culture flasks and incubated overnight at 37°C in a humidified atmosphere containing 5% CO2. Following overnight incubation, the flasks were washed extensively with PBS to remove nonadherent cells. ASCs between passages 3 to 6 were used in the experiments.
Primary culture of human DSCs and DPCs
Microdissection of hair follicles (HFs) and dermal papilla was performed as described previously [26,27]. Briefly, under a dissecting microscope, a small scalp specimen (1 cm3) was separated at the dermis-subcutaneous interface by scalpel, leaving the mid-to-lower portion of the HF exposed. Using blunt forceps, the fat tissue around the HF was compressed to partially extrude the upper portion of the HF from the subcutis, which enabled fine forceps to grip and extract intact HF for isolation of DPCs and DSCs. For isolation of DSCs, the whole HF was transferred to a 35 mm Petri dish containing DMEM-lg and cultured at 37°C in a humidified atmosphere containing 5% CO2. The explants were undisturbed for 7 days, during which the DSCs migrated from the mesenchymal layer of the HF to the base of the culture dish, where they proliferated. The HF was then removed on day 7 and the adherent cells continued to proliferate (passage 0) to establish DSC culture. To isolate DPCs, the HF was gripped with forceps at the supra-bulbar region and placed on the base of a Petri dish. A gentle force was applied to the forceps to compress the bulb. A small transect was made at the base of the stalk by scalpel blade to release the proximal sheath, allowing the papilla to emerge freely from the bulb when the forceps were compressed. This allowed easy aspiration of the HF papilla, and released it from the HF capsule or epithelium. Up to 12 HF papillae were transferred to a single 35 mm Petri dish, and by using a fine needle, a single scratch through the center of the HF papilla was made to anchor them to the bottom of culture dish, as well as breaking the basal lamina to facilitate emigration of cells during the subsequent cell explantation phase. The dermal papilla explant was cultured in DMEM-lg and incubated at 37°C in a humidified atmosphere containing 5% CO2 for 2–3 weeks, until the outgrowth cells were confluent. DSCs and DPCs between passages 4 to 7 were used in the experiments.
Primary culture of human bone marrow–derived stem cells
Human BMSCs purchased from a commercial supplier (Lonza, Berkshire, UK) were cultured in DMEM-lg, with fresh medium replaced every 2–3 days. A total of 2 founding cultures from different batches of cells were established and BMSCs between passages 4 to 8 were used in the experiments.
Collection and concentration of MSC-conditioned medium
ASCs, BMSCs, DSCs, and DPCs were cultured to 80%–90% confluence in T75 culture flask, then washed extensively with PBS, and replenished with 12 mL serum-free DMEM-lg (for cytokine array) or endothelial basal media-2 (EBM-2, Lonza; for angiogenesis bioassay) for 72 h prior to harvesting the media for further experimentation. Collected media samples were centrifuged at 3,000 rpm for 10 min to remove cell debris, and filtered through a 0.2 μm filter. For cytokine arrays, media were concentrated by a factor of 50× using Amicon® Ultra-15 centrifugal filter devices with 3 kDa molecular weight cut-off (Millipore, Billerica, MA).
Characterization of MSCs
Flow cytometric characterization
For flow cytometric characterization of CD markers on ASCs, DSCs, and DPCs, 5×105 cells were resuspended in 50 μL of binding buffer (PBS+2.5% FCS) and incubated with fluorescent-conjugated antibodies against CD34 (phycoerythrin, PE), CD45 (allophycocyanin, APC), CD73 (PE), CD90 (fluorescein isothiocyanate, FITC), and CD105 (APC) (Becton Dickinson Biosciences, North Ryde, NSW, Australia) on ice for 30 min protected from light. Control unstained cells were incubated with the appropriate isotype control antibody at the same concentration (BD Biosciences, San Jose, CA). Cells were analyzed using an FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). APC, FITC, and PE were detected in the FL-1, FL-2, and FL-4 channels, respectively. Cells were gated to exclude cell debris and 10,000 events were acquired using CellQuest Pro software (v5.2; BD Biosciences) and data were analyzed using FlowJo software (v8.8.6; FlowJo, Ashland, OR).
Adipogenic, osteogenic, and chondrogenic differentiation of ASCs
To demonstrate the differentiation ability of ASCs, cells were cultured under specific culture condition. For adipogenesis, confluent cells were cultured in adipogenic medium that was composed of 0.5 mM isobutylmethylxanthine, 10 μM insulin, 1 μM dexamethasone, and 0.2 mM indomethacine that were made up in high-glucose DMEM with 10% FCS and 1% antibiotic/antimycotic. Two weeks after adipogenic induction, Oil Red O staining was used to reveal the accumulation of lipid droplets in intracellular vacuoles, where cells were fixed with 4% paraformaldehyde (PFA) for 30 min at room temperature and stained with Oil Red O solution (0.5% Oil Red O solution in 2-propanol diluted with distilled water at a ratio of 3:2) for 1 h. Cell nuclei were counterstained with hematoxylin for 5 min. For osteogenic differentiation, confluent cells were cultured in osteogenic medium that was composed of 10 mM β-glycerophosphate, 100 nM dexamethasone, and 200 μM ascorbic acid-2-phosphate that were made up in high-glucose DMEM with 10% FCS and 1% antibiotic/antimycotic. Four weeks after osteogenic induction, von Kossa staining was used to assess the level of calcium precipitate. Cells were fixed in 4% PFA and rinsed with distilled water before incubation with 5% silver nitrate under ultraviolet light for 3 h. Cells were then treated with 2.5% sodium thiosulfate for 5 min before nuclei were counterstained with eosin for 5 min. A 3-dimensional cell culture method (cell spheroids) was used to induce chondrogenic differentiation, where 2×104 cells were resuspended in chondrogenic medium (StemPro® chondrogenesis differentiation medium; Gibco Invitrogen, Carlsbad, CA) containing 20% methylcellulose to form a single spheroid in nonadherent U-shaped bottom 96-well microplates (BD Biosciences). Two weeks after chondrogenic induction, spheroids embedded in OCT compound (Tissue-Tek® Sakura Finetek, Inc., Torrance, CA) were snap frozen by immersing in a dry ice–isopentane bath for 1 min. Frozen sections were cut at 10 μm thickness and stained for the presence of proteoglycan with 0.1% toluidine blue solution (pH 3.0) for 15 s.
Real-time reverse transcription–polymerase chain reaction
MSCs derived from adipose tissue, bone marrow, and dermis grown to 80%–90% confluence were incubated with serum-free DMEM-lg for 72 h before RNA was isolated. cDNA was synthesized using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). Real-time reverse transcription–polymerase chain reaction was then conducted with Taqman® technology using the following primers (Applied Biosystems; Assay-On-Demand primers): stromal-derived factor-1 (SDF-1, Hs00930455_m1), VEGF-A (Hs00900054_m1), VEGF-C (Hs00153458_m1), VEGF-D (Hs01128659_m1), basic fibroblast growth factor (bFGF, Hs00266645_m1), HGF (Hs00300159_m1), insulin-like growth factor-1 (IGF-1, Hs01547656_m1), nerve growth factor (NGF, Hs00171458_m1), angiogenin (ANG, Hs02379000_s1), and interleukin-8 (IL-8, Hs00174103_m1). Endogenous eukaryotic 18S ribosomal mRNA (18S, Hs99999901_s1) was amplified as the house keeping control. Reactions were carried out in 96-well plates with the Applied Biosystems 7900HT Fast Real-Time PCR system, with duplicates for each sample, and at least one of nonreverse transcribed control and no-template control per run. The machine ran at 95°C for 20 s to activate the AmpliTaq Gold polymerase and continued with 50 cycles of 1 s at 95°C and 20 s at 60°C for primer dissociation and annealing/elongation, respectively. Relative expression level of candidate factor in ASCs was normalized to 1 and all results were expressed as relative fold change in gene expression of each cell type compared with the ASCs.
Cytokine arrays
Cytokine array (RayBio® Human Cytokine Antibody Array G Series 3; RayBiotech, Inc., Norcross, GA) was carried out according to the manufacturer's instruction. In brief, the glass chip was air dried and blocked with blocking buffer for 30 min at room temperature to prevent nonspecific binding. The chip was then incubated with 100 μL of 50× concentrated conditioned medium (CM) containing equal amounts of protein (2 mg/mL) overnight at 4°C. After extensive washing with wash buffer to remove unbound materials, the chip was incubated with a cocktail of biotin-conjugated antibodies against different individual cytokines for 2 h at room temperature. The chip was then washed and incubated with 1,500-fold diluted fluorescent dye–conjugated streptavidin for 2 h at room temperature in the dark. Unbound reagent was washed out and excess water was removed by centrifuging the glass chip at 1,000 rpm for 3 min and air dried before the chip was scanned at an excitation frequency of 532 nm using GenePix™ 4000B (Axon Instruments, Sunnyvale, CA). Signal intensity was quantified by densitometry using ImageJ software (U.S. National Institute of Health, Bethesda, MD), where median signal values of the unknown were subtracted with median background values and expressed as fold change compared with the internal positive signal (biotinylated protein for normalization of streptavidin signal).
In vitro angiogenesis bioassays
Human microvascular endothelial cells (HMECs; Lonza) were used to examine the ability of MSC-derived CM to regulate various aspects of angiogenesis in vitro. HMECs were cultured and expanded in endothelial growth medium-2 MV (EGM-2 MV; Lonza) that consisted of EBM-2, 5% FCS, human bFGF, VEGF, IGF-1, human epidermal growth factor, hydrocortisone, ascorbic acid, and GA-1000.
HMEC proliferation assay
To examine the effects of CM on HMEC proliferation in vitro, HMECs were seeded on 12-well plates at a concentration of 1×104 cells/well. After overnight incubation to allow cell adhesion, the medium was changed to CM obtained from ASCs (ASCCM), BMSCs (BMSCCM), DSCs (DSCCM), or DPCs (DPCCM) supplemented with 5% FCS for 3 days. HMECs cultured in EBM-2 with 5% FCS and EGM-2 MV alone were considered as negative and positive controls, respectively. Viable cell number was enumerated using trypan blue exclusion and a hemocytometer.
Scratch/wound healing assay
Twenty-four-well microplates were assembled with culture-insert chambers (Ibidi; Biovalley, Marne La Vallee, France) containing 2.8×104 HMECs in EGM-2 MV media in each of the 2 compartments per insert. After overnight incubation, the culture-inserts were removed and the medium was replaced with CM supplemented with 5% FCS, EBM-2 with 5% FCS, or EGM-2 MV. Cell growth over the cleared area was photographed at 0 and at 24 h incubation time. The area covered by HMECs at the designated time points was calculated with ImageJ and expressed as a percentage compared with 0 h time point.
Tube formation assay
Forty-eight-well microplates were coated with 100 μL per well of Growth Factor Reduced Matrigel (GFR-Matrigel; BD Biosciences) and incubated for 30 min at 37°C to solidify. HMECs were resuspended in EGM-2 MV and plated at 3×104 cells/well for 1 h at 37°C. Medium was then aspirated without disrupting the adherent cells and washed with PBS once before replenished with CM supplemented with 5% FCS, EBM-2 with 5% FCS, or EGM-2 MV. After 24 h, the formation of tube-like structures was examined microscopically. Photographs at ×40 magnification were taken of each well, and the number of rings formed, branch points, and total length of formed tubes were measured using ImageJ software. All experiments were performed in duplicate. To assess the mechanisms of tube formation regulated by ASCCM in this assay, HMECs were assigned to 1 of 5 experimental groups: (i) positive control (EGM-2-MV); (ii) ASCCM+5% FCS; (iii) ASCCM+5% FCS+VEGF-A neutralizing antibody (4 μg/mL); (iv) ASCCM+5% FCS+VEGF-D neutralizing antibody (8 μg/mL); and (v) ASCCM+5% FCS+VEGF-A neutralizing antibody (4 μg/mL)+VEGF-D neutralizing antibody (8 μg/mL).
Statistical analysis
All experiments were performed at least 3 times and data were expressed as mean±SEM. Statistical significance was determined using either one-way or two-way ANOVA, followed by a Bonferroni multiple comparison post hoc test when appropriate using Prism5 software (GraphPad, La Jolla, CA). A P value of less than 0.05 was considered to be statistically significant.
Results
Isolation of MSCs from human tissues
ASCs were isolated from the stromal vascular fraction of human abdominal adipose tissue with 100% successful cell isolation rate and formed a monolayer after initial plating on tissue culture plastic. A consistent isolation procedure with routine yield of 2 to 4×106 cells per 100 mL of adipose tissue processed was achieved. Under microscopic examination, the adherent cells displayed a fibroblast-like spindle-shaped morphology that was consistent with previous descriptions of ASCs [2] (Fig. 1). To confirm the purity of ASCs from the stromal vascular fraction, cells between passages 4 to 6 were examined by flow cytometry for expression of CD markers and most were found to be positive for CD73, CD90, and CD105 and negative for CD34 and CD45 [5]. Furthermore, ASCs demonstrated in vitro ability to differentiate into adipogenic, osteogenic, and chondrogenic lineages (see Supplementary Fig. S1; Supplementary Data are available online at

Phase-contrast photomicrographs showing morphological characteristics of human mesenchymal stem cells from adipose, bone marrow, dermal sheath, and dermal papilla (ASCs, BMSCs, DSCs, and DPCs) (magnification ×40 and ×100). All MSC populations were cultured under identical conditions to avoid variations incurred by methodology differences.
Characterization of paracrine factor secretion by ASCs, BMSCs, DSCs, and DPCs
To determine which proangiogenic factors were secreted by the MSCs, cells were grown to 80%–90% confluence and incubated with serum-free medium for 72 h prior to collection of CM. Media were then analyzed using a broad-spectrum cytokine antibody array that tested for the presence of 42 different cytokines and growth factors (see Supplementary Table S1 for detail). The level of each protein detected was expressed relative to the internal control of each sample, allowing direct semiquantitative comparisons between each sample. Most of the angiogenic factors tested, including ANG, VEGF, TGF-β1, and PDGF-BB (see Table 1 for complete list of factors), were detected in the CM of all MSC populations and the level of protein was found to be comparable between populations. Furthermore, BMSCs produced significantly lower levels of the chemotactic factor growth-related oncogene (GRO) as compared with ASCs, DSCs, and DPCs, while GRO-α was not detected at all. The secretion pattern of chemotactic factors, such as monocyte chemotactic protein (MCP), RANTES, stem cell factor (SCF), and SDF-1, exhibited variations between batches of CM within each MSC population. Most of the IL family proteins were not detected in any cell population; however, all cell populations secreted relatively high levels of IL-6 and produced detectable amounts of IL-8 that were comparable between cell types. Interestingly, leptin, a factor that has been associated with adipocytes, was not detected in ASCCM, while both DSCs and DPCs secreted significant levels of leptin protein.
Conditioned medium collected at the end of a 72-h incubation period was concentrated 50 times. Human cytokine antibody array G series 3 was used to measure and compare the amount of growth factors and cytokines released by ASCs, MSCs, DSCs, and DPCs. Median intensity of each growth factor or cytokine was expressed as the relative ratio of median internal positive control. Results from 6 (ASCs and BMSCs) and 3 (DSCs and DPCs) independent cultures are shown for each cell population.
P<0.05 one-way ANOVA between cell populations.
−, mean values<0.05;+, 0.05≤mean value<0.5;++, 0.5≤mean value<1.0;+++, mean value>1.0; MSCs, mesenchymal stem cells; ASCs, MSCs derived from adipose tissue; DSCs, dermal sheath cells; IGF-1, insulin-like growth factor-1; VEGF, vascular endothelial growth factor; IL, interleukin; SDF-1, stromal-derived factor-1; GRO, growth-related oncogene; MCP-1, monocyte chemotactic protein; SCF, stem cell factor; TNF, tumor necrosis factor; DPCs, dermal papilla cells; PDGF-BB, platelet-derived growth factor-BB; TGF-β1, transforming growth factor-β1.
Gene expression of selected paracrine factors in ASCs, BMSCs, DSCs, and DPCs
A multitude of growth factors play vital roles in regulation of different aspects of the angiogenesis process and a panel of candidates had been selected based on their ability to promote endothelial cell proliferation and migration, including VEGF-A, bFGF, NGF, ANG, VEGF-C, VEGF-D, SDF-1, IGF-1, HGF, and IL-8 [29 –31]. Consistent with the cytokine array data, basal expression of the angiogenic factors VEGF-A, bFGF, NGF, and ANG was detected in the different populations of MSCs at comparable levels (Fig. 2A). When the level of expression for each angiogenic factor was examined using the average ΔCt value across all 4 MSC populations, ANG expression level was less than bFGF, VEGF-A, and NGF (see Supplementary Table S2). This result contradicted our cytokine array analysis, where accumulation of ANG in CM was more prominent than VEGF-A in general.

Gene expression of selected paracrine factors in MSC populations. Expression of angiogenic factor mRNAs was comparable between cell populations
All cell populations expressed detectable levels of VEGF-C and VEGF-D, factors that are critical in lymphangiogenesis through interaction with the VEGF receptor-3 (Fig. 2B). While expression of VEGF-C was comparable between all MSC populations, VEGF-D was expressed abundantly only in ASCs. Expression of the chemotactic factor SDF-1 measured at basal conditions was found to be inconsistent between batches of a cell type, supporting the results obtained in the cytokine arrays (Fig. 2C). ASCs expressed the highest levels of IL-8 whereas expression was almost undetectable in other cell populations (Fig. 2C). HGF and IGF-1 are known to be both angiogenic and cytoprotective. Interestingly, expression of IGF-1 was significantly higher in ASCs than the other MSC populations, while expression of HGF tended to be higher in both ASCs and BMSCs compared with DSCs and DPCs (Fig. 2D).
MSC-CM promotes angiogenesis of HMECs in vitro
To investigate the bioactivity of MSC-derived factors on HMECs in vitro, CM was generated under serum-free conditions in EBM-2 from each MSC population. In a proliferation assay, HMECs cultured in ASCCM showed a trend to increase proliferation rate that was almost comparable with the positive control group (ASCCM 1.63±0.30 vs. positive control 2.12±0.45; n=4, P>0.05) while HMECs cultured in BMSCCM, DSCCM, and DPCCM did not proliferate any more than the cells in negative control (BMSCCM 1.04±0.45, DSCCM 0.77±0.48, DPCCM 0.86±0.42 vs. negative control 0.90±0.02; n=4, P<0.05) (Fig. 3).
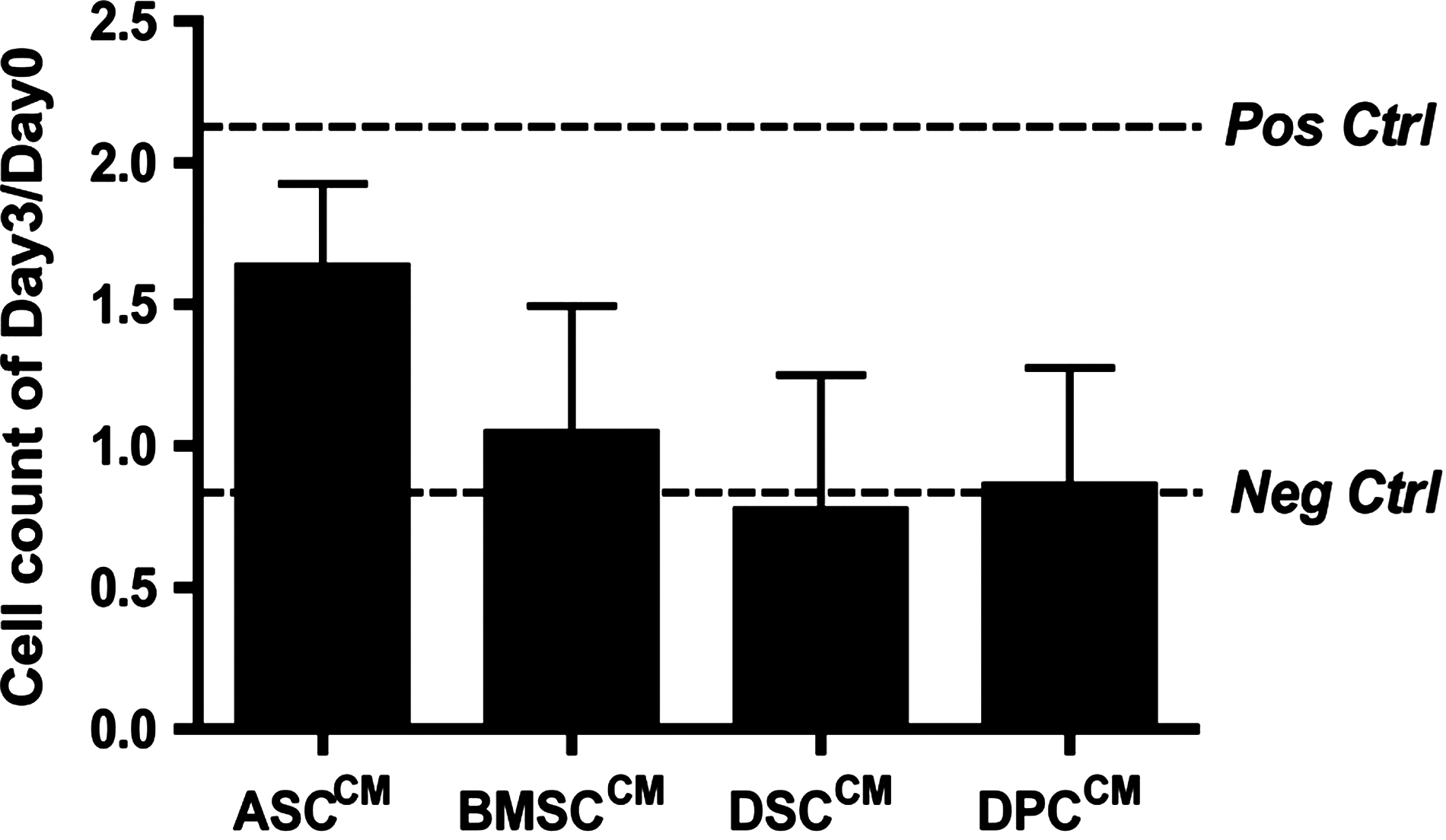
Effect of MSC-conditioned medium (CM) on human microvascular endothelial cell proliferation. CM derived from MSC populations showed comparable effects on proliferation of human microvascular endothelial cells (HMECs) in vitro (P=0.49). Pos ctrl, positive control; Neg ctrl, negative control.
To examine the ability of MSC-CM to promote migration of HMECs in a 2-dimensional culture, a wound-healing assay was used. HMECs cultured in EGM-2 MV medium (positive control) significantly healed to 63.2%±8.4% while negative control only healed to 12.8%±6.0% of the area (Fig. 4). The CM from each MSC population induced a moderate effect on repopulating the gap compared with the controls, and they were comparable between 4 MSC populations (ASCCM=36.9%±6.7%, DSCCM=35.9%±7.0%, BMSCCM=31.1%±6.0%, and DPCCM 24.3%±6.2%; n=6, P=0.51).
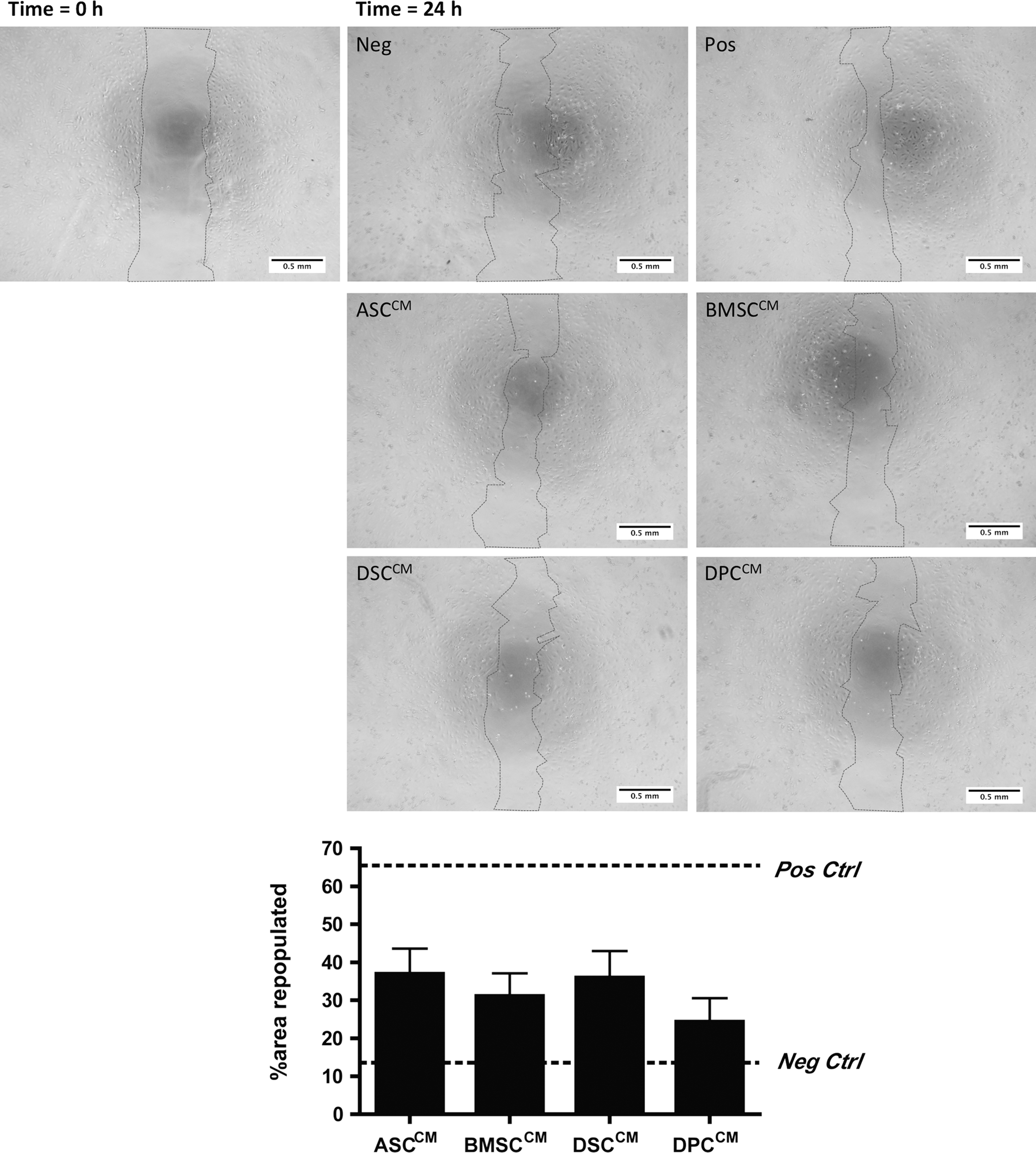
Paracrine activity of various MSC populations showed similar efficacy at promoting migration and proliferation of HMECs to recover the unpopulated area in the wound-healing assay (P=0.51). HMECs cultured in endothelial growth medium-2 MV (Pos ctrl) recovered the area at a significantly higher percentage as compared with cells cultured in endothelial basal media-2+5% fetal calf serum (Neg ctrl). CM derived from all MSC populations induced an intermediate re-coverage of the “wound,” ranging from 24% to 36%. Magnification ×40. Pos ctrl, positive control; Neg ctrl, negative control.
To investigate the paracrine effect of MSCs to induce formation of vascular networks in a 2-dimensional HMEC culture, cells seeded on GFR Matrigel®-coated wells were cultured in MSC-CM. Formation of HMEC tubes was then assessed by counting the complete rings formed, frequency of branching points with HMECs adjoining from at least 3 directions (branch points), and total length of networks formed (Fig. 5). HMECs grown in EBM-2 only (negative control) did not form tubular structures, whereas HMECs grown in EGM-2 MV (positive control) formed tubules, branches, and rings. ASCCM, BMSCCM, and DSCCM were able to stimulate the formation of HMEC tubular networks and sustained the structure for at least 24 h at a comparable level to that observed in a positive control. However, DPCCM was not as efficient as the CM harvested from other MSC populations and was assessed to be significantly less efficient than ASCCM in supporting the tube-like structures of HMECs as determined by the number of branch points (31.9±1.4 vs. 12.0±3.5; n=4, P=0.03), rings formed (16.8±2.6 vs. 4.5±2.4; n=4, P=0.03), and total tube length (in pixels; 16460±938 vs. 7151±1827; n=4, P=0.01).
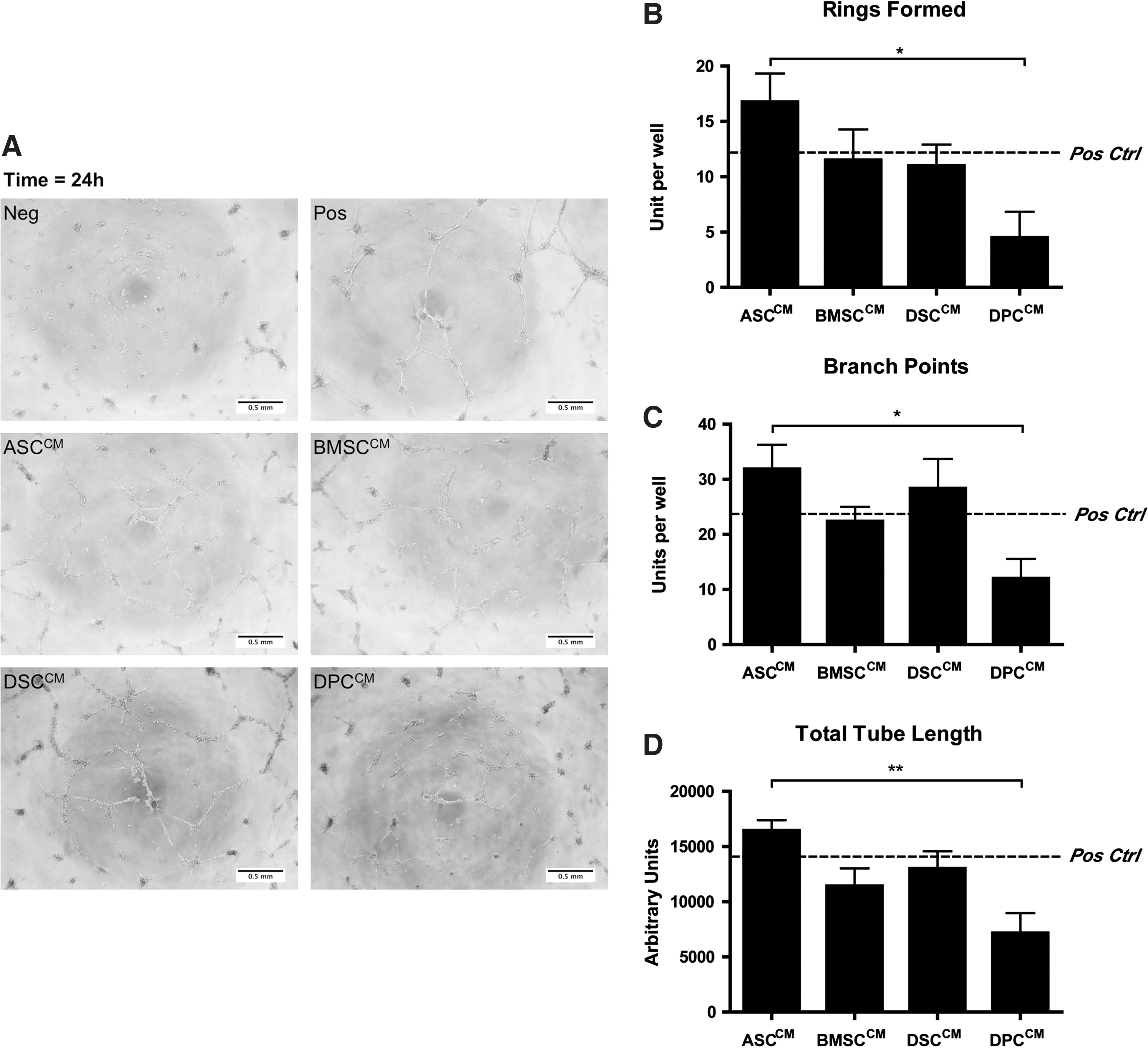
CM derived from MSC populations was able to support the formation of tubular structures in HMECs, which was not evident in the negative control
The proangiogenic effect of ASCCM was abolished in the presence of VEGF-A and VEGF-D neutralizing antibodies
To determine whether VEGF-A and VEGF-D were the responsible paracrine factors contributing to the proangiogenic effect of ASCCM, HMECs were cultured in ASCCM with or without neutralizing antibodies against VEGF-A and VEGF-D. The tube formation assay was employed as ASCCM elicited the strongest response on HMEC tubulogenesis rather than HMEC proliferation and migration assays. In the presence of VEGF-A, VEGF-D, or both neutralizing antibodies, the effect of ASCCM HMEC tube formation was significantly reduced (Fig. 6) as evidenced by fewer rings formed (67.1%±2.1%, 76.9%±1.0%, and 58.9%±3.6%, respectively; P<0.05 vs. ASCCM, n=3) and reduced total tube length (87.3%±2.0%, 89.4%±0.9%, and 82.2%±1.9%, respectively; P<0.05 vs. ASCCM, n=3). A significant reduction in number of branch points was only observed when both neutralizing antibodies were included as compared with ASCCM group (71.1%±6.8%; P<0.05 vs. ASCCM, n=3).

ASCCM supported the formation of tubular structures in HMECs as determined by the number of rings formed
Discussion
We performed a comparative analysis of paracrine factor profiles derived from MSCs of various tissue origins. To our knowledge, this study is the first to directly compare the basal paracrine factor profile of 4 different human MSC populations isolated from different sites: adipose tissue, bone marrow, and different parts of the HF (dermal sheath and dermal papilla). To confirm their paracrine activity, MSC-derived factors were tested and compared for their activities in angiogenesis bioassays in vitro. We also showed that VEGF-A and VEGF-D contribute to the proangiogenic paracrine effect of ASCs. From this data we find that ASCs may be among the most suitable MSC populations for regenerative applications that require angiogenesis, such as tissue engineering.
Cytokine antibody array was used to screen for potential factors critical for angiogenesis that were secreted into the CM. Although not offering absolute quantification of each growth factor (as compared with other methods such as enzyme-linked immunosorbent assays), the assay permitted relative comparison of protein levels between samples using internal positive control as reference and provides simultaneous information on multiple factors. When examining one of the major players in angiogenesis, VEGF-A, we observed a protein level in the CM that was similar between all MSC populations, a result confirmed by mRNA expression. Our observation is supported by data reported by Kim et al. [25], but is opposed to the studies from Peng et al. [32] and Ikegame et al. [24], where higher expression of VEGF-A was detected in BMSCs than ASCs in the prior study and conversely in the latter. Thus individual ASC populations may need to be assessed before their suitability is ensured. The discrepancy interpreted from various studies may be due to different isolation procedures and culturing methods employed as well as the absence of standardized detection methods utilized to identify different MSC populations. ASCs continue to be described as a heterogeneous population, varying from other MSCs [28] in that they do express CD34 [33], and with some cell surface markers also varying at different passage numbers [33].
Expression of several other growth factors was analyzed in this study because of their profound effect on angiogenesis, including ANG, bFGF, and NGF, which were found to be comparable between all MSC populations. Taken together with the VEGF data it was surprising that there was not comparable angiogenic activity between the MSC populations, as evidenced by the differences in tube formation assay between ASCs and DPCs. One explanation for the observed result may be the presence of other angiogenic factors whose expression we did not examine, such as angiopoietins, since the cytokine array employed in this study for the analysis of the CM could not identify all paracrine factors. Other plausible explanation for the differences observed may be the relative concentrations of paracrine factors as well as their multifaceted roles in biological processes apart from angiogenesis, such as deposition of extracellular matrix, cell adhesion, proliferation, and apoptosis. For example, presence of VEGF-A is essential for maintenance of cardiac function [34] whereas bFGF is a critical component of human embryonic stem cell culture medium as it maintains the cells in an undifferentiated state through mechanisms yet to be defined [35]. Similarly in addition to its angiogenic role, NGF is capable of activating the phosphatidylinositol-3-kinase [36] pathway that regulates both angiogenesis and cell survival [37]. Whether these factors contribute to the beneficial effects observed through mechanisms other than direct action on HMECs for promotion of angiogenesis remains to be determined. Lastly, the paracrine activity of MSCs to support the vasculature has been described in various studies yet none could be certain as which were the responsible factors. For example, overexpression of VEGF-A and HGF in MSCs demonstrated great potential in repair of the heart postinfarction [31], but gene delivery of VEGF-A to myocardium resulted in adverse effects [38]. This may indicate a unique role of supportive cell therapy using MSCs, where the regulated production of various paracrine factors works synergistically to produce the outcome and its effect cannot be replaced by single growth factor treatment. Therefore, the superiority of ASCCM we observed in the tube formation assay, despite similar expression profile of angiogenic factors examined, is likely to result from a combination of factors produced at specific concentrations that act directly or indirectly on vessel formation in this assay. A complete quantitative proteomic analysis of CM produced by MSC populations may offer more comprehensive information on the paracrine factors present, including factors acting on vessel stability, maturation, and regression such as angiopoietin-1/-2 (Agpt-1/-2) and plasminogen activator inhibitor-1 (PAI-1).
In vitro bioassays revealed similar effects of MSC paracrine activity from adipose-, bone marrow–, and dermis-derived MSCs at promoting the proliferation and migration of HMECs, which maybe explained by similarity in expression of angiogenic factors VEGF-A, bFGF, ANG, and NGF between these different MSC populations. However, ASCCM seemed to be more effective than DPCCM at tubulogenesis in the tube formation assay. This observation suggests differences in factors that are responsible for vessel stabilization, such as angiopoietin-1/-2 (Agpt-1/-2) [39] and PAI-1 [40], whose expression levels were not examined in the current study. Indeed, it was shown in the work by Ikegame et al. that ASCs and BMSCs produce comparable levels of Agpt-1 in vitro [24]. Additionally, researchers have demonstrated that MSCs overexpressing Agpt-1 were able to reduce pulmonary vascular endothelial permeability in a murine lipopolysaccharide-induced lung injury [41], as well as improving arteriogenesis in a porcine model of chronic myocardial ischemia [42]. For the current study, expression of Agpt-1 and PAI-1 among MSC populations and their role in formation of tube-like structure of HMECs in vitro remains to be verified and the possibility of other factors beyond these two also needs to be investigated.
In addition to VEGF, bFGF, ANG, and NGF, the expression of lymphangiogenic factors VEGF-C and VEGF-D was also detected in all MSC populations. The role of MSCs in lymphangiogenesis has been previously examined [43] and has attracted interest as a source of cells for treatment of lymphangiogenic disorders as well as attenuation of existing lymphangiogenic networks. Local administration of BMSCs on a weekly basis restores the lymphatic drainage in the mouse tail [43]. Implantation of ASCs in a gelatin hydrogel containing VEGF-C to the hindlimb footpad also reduced dermal edema depth in 4 weeks [44]. Furthermore, ASCs injected in hindlimb footpad differentiated into lymphatic endothelial cells [44]. In this study we examined the basal expression level of VEGF-C and VEGF-D, factors known to play a role in lymphangiogenesis [45]. VEGF-C was expressed comparably between different MSC populations, but the expression of VEGF-D was significantly higher in ASCs. It is thought that VEGF-C represents the major lymphangiogenic factor and VEGF-D is dispensable during embryonic development of lymphangiogenesis [46]. However, VEGF-D has also been shown to induce the strongest angiogenesis and lymphangiogenesis among all VEGFs when delivered into skeletal muscle via adenovirus [47]. While the implication of higher expression level of VEGF-D in ASCs compared with other MSC populations, as reported here, is unknown, it has previously been demonstrated that the expression of VEGF-D can be upregulated by hypoxia in BMSCs [48]. It remains to be determined whether the same response can be induced in ASCs.
The proangiogenic effect of ASCCM on in vitro HMEC tube formation was significantly attenuated in the presence of VEGF-A and VEGF-D neutralizing antibodies, indicating a paracrine effect involving both factors. However, no synergistic effect was observed when both neutralizing antibodies were incorporated in the study. While both factors are known to be potent angiogenic factors activating the VEGF receptor-2 (VEGFR-2) [49,50], they induce differential activation upon binding to the receptor [50]. It was shown that VEGF-A induces stronger and more effective activation of phospholipase C-γ tyrosine phosphorylation compared with VEGF-D but the response induced by VEGF-D is more sustained and becomes as effective as VEGF-A after 60 min. Indeed, addition of VEGF-A or VEGF-D neutralizing antibody to ASCCM resulted in comparable inhibition of tube-like structure formation that was measured at 24 h. Despite the similar effects observed in vitro, VEGF-D–stimulated angiogenesis was less effective than VEGF-A in an in vivo mouse model of angiogenesis with sponge implantation, emphasizing the influence of multiple biological factors within the microenvironment [51]. Further investigation on the VEGF receptor signaling pathway pattern, especially activation of VEGFR-2, would provide more information on the signaling cascade induced by the paracrine factors.
When screening for the factors that most significantly differed in expression between MSC populations, we were surprised to find leptin to be produced significantly more by DPCs and DSCs, while BMSCs and ASCs secreted minimal or undetectable level of leptin, respectively. This finding supports the hypothesis that MSCs of different origin may exhibit similar expression of CD markers or differentiation ability, but they may still possess characteristics that are unique to their tissue of origin [23]. Leptin is a major differentiation marker of preadipocytes that plays a crucial role in regulation of metabolism and is strongly associated with adipose tissues [52 –54]. Despite its obvious role in adipose biology, production of leptin was not detectable in ASCCM, and the observation was consistent with the original characterization of ASCs by Zuk et al. [2], where leptin expression was detected specifically in processed lipoaspirate postadipogenic-induction. The absence of leptin in ASCCM suggests that that paracrine activity of ASCs changes once they become committed in a specific differentiation lineage as they acquire different patterns of paracrine factor to accommodate the change in cellular biology (unpublished data). On the contrary, leptin was found in high levels in both dermal follicular cell populations, which was also observed by Iguchi and colleagues [55]. This result correlated well with the role of leptin in various aspects of skin biology, where it has been demonstrated to participate in HF morphogenesis [56,57] and regulation of the hair cycle [58,59]. Furthermore, it plays a fundamental role in normal wound healing, potentially through its ability to induce neovascularization during tissue repair [60,61]. The observation of varied leptin expression between different MSC populations highlighted the potential impact of different paracrine factors when choosing a specific MSC population for a particular desired outcome.
Analysis of factors secreted in the CM from these MSC populations revealed very low or almost undetectable levels of inflammatory factors, apart from IL-6 and IL-8 that were both detected at comparably higher levels. IL-6 acts as an antiinflammatory cytokine through mediating an inhibitory effect on tumor necrosis factor (TNF)-α and IL-1 [62]. On the other hand, IL-8 has been identified as a key parameter in localized inflammation through regulating neutrophil infiltration and activation [63]. During the course of tissue engineering in vivo, which has been suggested as one of many applications for MSCs [64 –68], escalation of inflammatory responses caused by foreign materials can be detrimental to the outcome. Inflammatory factors, namely the ILs and TNF, may initiate the inflammation cascade through autocrine or/and paracrine mechanisms and generate a hostile environment within the niche. Therefore an appropriate choice of cell type for tissue engineering should prevent an excessive inflammatory response, although recent data have suggested a link between inflammation and angiogenesis [69]. In correlation with the hypothesized association of angiogenesis with inflammatory response, IL-8 has demonstrated an angiogenic property in the rat cornea model where human recombinant IL-8–containing hydroxyethyl-methacrylate induced vessel formation [70]. Furthermore, it was found that myocardial production of IL-8 and GRO was increased in acute ischemia of rat heart, causing homing of bone marrow CD34+ angioblasts that lead to myocardial neovascularization and functional cardiac recovery [71]. It is difficult to conclude whether the level of IL-6 and IL-8 detected in the cytokine array would be pathophysiologically important in vivo, but if their expression can be managed spatially and timely in a precise fashion, the presence of these ILs could contribute greatly to the process of angiogenesis.
Conclusion
While the potential of embryonic or induced pluripotent stem cells for tissue repair and tissue engineering has gained much interest in the last decade, the use of adult stem cells such as MSCs continues to be explored in this context. As the use of embryonic stem cells creates ethical controversies and induced pluripotent cells invoke the challenge of balancing between safety and efficacy because of gene insertion. On the other hand, MSC populations have been well characterized with great potential for paracrine-mediated benefits in various stem cell therapies, including angiogenesis and cytoprotection. In this study we compared the paracrine factor profile of 4 different MSC populations in order to determine the most suitable population for tissue engineering. We have demonstrated that MSCs arising from different tissue niches have unique paracrine factor profiles and such variation may contribute to different angiogenic activity of their CM. Of all MSC populations examined, ASCs represent an attractive cell type for promoting angiogenesis in tissue engineering–like applications. First, the source of adipose tissues is easily accessible and the cell population can be isolated successfully in relatively large quantity and expanded rapidly in vitro as compared with BMSCs and the dermal MSCs. Second, ASCs express a broad range of paracrine factors that are known to be angiogenic and exhibited an ability to promote angiogenesis in vitro through the secretion of VEGF-A and VEGF-D. In conclusion, ASCs maybe preferred over other MSC populations for specific benefits in terms of vascular support in various applications.
Footnotes
Acknowledgments
The authors would like to acknowledge support from the National Health and Medical Research Council for project grant (No. 509271), scholarship to S.H. (No. 567152) and A.A. (No. 567168), and fellowship to G.D. (No. 1003113). R.S. would like to acknowledge the support of Skin & Cancer Foundation of Victoria. The O'Brien Institute acknowledges the support of JO and JR Wicking Trust, Cass Foundation and Victorian State Government's Department of Innovation, Industry and Regional Development's Operational Infrastructure Support Program. We thank Mr. Keith Mutimer and Dr. Russel Knudsen for providing specimens. The authors would also like to acknowledge Dr. Keren Abberton for her critical review of the manuscript.
Author Disclosure Statement
No competing financial interest exists.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.