
Abstract
A deficiency of maternal thyroid hormones (THs) during pregnancy may have severe impacts on fetal brain development. However, the cellular targets of THs and their underlying mechanisms are still unclear. In this study, we found that maternal hypothyroidism during pregnancy in mice inhibited neurogenesis in the embryonic telencephalon and caused learning and memory impairment in the offspring. To explore the underlying mechanisms, we treated cultured mouse embryonic neural stem cells (eNSCs) with a physiological level of 3, 5, 3′-triiodo-L-thyronine (T3). We found that T3 promoted the neuronal differentiation of eNSCs, while inhibiting astrocytic differentiation. In addition, the proliferation and maintenance of eNSCs were inhibited by T3. Furthermore, the TH receptor alpha 1 (TRα1) was detected in the eNSCs both in vivo and in vitro. Silencing TRα1 protein expression with specific siRNA eliminated the effects of T3 on eNSCs. We also found that T3 decreased STAT3 phosphorylation and STAT3-DNA binding activity through TRα1. The over expression of STAT3 attenuated the promotive effects of T3 on neuronal differentiation of eNSCs. Taken together, these results suggest that T3 promotes the neuronal differentiation of eNSCs by inhibiting STAT3 signaling activity through TRα1 and contributes to early neurogenesis in the embryonic telencephalon. Our studies reveal the physiological effects of TH in regulating eNSCs differentiation and suggest that eNSCs are one of the major cellular targets in the central nervous system by which TH influences early brain development. These findings also provide new insights into the mechanisms of neurological deficits caused by TH deficiency during embryogenesis.
Introduction
T
The 2 circulating THs are 3, 5, 3′-triiodo-L-thyronine (T3) and tetra-iodothyronine (T4); T3 is active, while T4 is thought to be the precursor of T3. It has been established that THs appear in the fetal brain very early in gestation [10,11]. They are present in embryonic fluids 4 weeks after conception in humans and as early as embryonic day (E) 10 in the rat embryo/trophoblast unit [12]. Free T4 (FT4) rapidly increases to adult levels, and its level is determined by the maternal availability of FT4. T3, which is generated from T4, reaches adult levels in the cerebral cortex by mid-gestation, and is partly bound to specific nuclear receptor isoforms [13].
T3 affects the development of both neurons and glia, which contribute to the harmonious development of the central nervous system (CNS) [14,15]. Specifically, T3 plays a critical role in myelination, because it acts on both the production and maturation of oligodendrocytes and on the expression of genes that encode for myelin-specific proteins [16,17]. However, the majority of the previous demonstrated effects of T3 occur during late gestation or after birth; its effects on the development of embryonic neural stem cells (eNSCs) during the early to middle gestation period are still unknown. In our previous work, we found that supraphysiological doses of T3 inhibit eNSCs proliferation and maintenance [18]. However, the physiological effects of T3 on eNSCs remain to be explored. Therefore, it is unclear why THs of maternal origin are present in the fetal brain before they are required for development [19].
Current studies on eNSCs have aided in the establishment of new models of brain development and human diseases. Embryonic NSCs originate from the neuroepithelial progenitors, which are derived from the neural tube at E8.5–E9.5 in mice [20]. NSCs are thought to give rise to the majority of the cells of the brain, including neurons, astrocytes, and oligodendrocytes; and this occurs in a strictly stage-dependent manner during development [21]. The balance among the proliferation, maintenance, and differentiation of eNSCs during embryogenesis is regulated by various growth factors, hormones, and corresponding signaling molecules [22,23]. Among the various signaling pathways, the JAK/STAT3 pathway, along with its antagonists SHP-2 and the SOCS3 [24 –28], the MAPK (Erk1/2) pathway [29,30], and the phosphatidylinositol 3-kinase (PI3-K)/Akt [31,32] pathway are responsible for the signaling transduction of the majority of growth factors and hormones. The activity or inactivity of these signaling pathways plays an important role in regulating NSC proliferation, maintenance, and differentiation.
The influence of THs on the development of the CNS is thought to be primarily mediated by nuclear thyroid hormone receptors (TRs), which belong to the nuclear hormone receptor superfamily [15]. In mammals, there are 2 main TR subtypes, TH receptor alpha (TRα) and TH receptor beta (TRβ), which are encoded by 2 different genes, THRA and THRB, respectively. Alternative splicing of TRα mRNA gives rise to TRα1 and TRα2. The TRβ locus encodes the TRβ1, TRβ2, and TRβ3 proteins [15,33,34]. Only TRα1, TRβ1, and TRβ2 proteins are considered to be true TRs. Previous studies have demonstrated that THs induce cerebellar neuronal migration through TRs and the MAPK pathway [35]. TH can also modulate intracellular protein trafficking through different isoforms of TRs. This involves the translocation of nuclear factor superfamily members, including STAT3 and STAT1a, from the cytoplasm to the nucleus [36,37]. However, the exact types and time of TRs that appear in the eNSCs have not been well explored, so the roles and underlying mechanisms of TRs in regulating eNSC fate are unclear. Thus, further studies are needed to clarify this issue and to better understand the role of TH in brain development.
Here, we characterized the effects of maternal hypothyroidism on neurogenesis in the embryonic telencephalon. In addition, we investigated the physiological effects of T3 on eNSCs and explored the underlying mechanisms that drive these effects. We also characterized the expression of TR isoforms in eNSCs both in vivo and in vitro and uncovered a role for TRα1 in regulating eNSC fate.
Materials and Methods
Animal treatments
Timed pregnant BALB/c mice were used in these experiments. All the experiments conformed to the guidelines on the handling and training of laboratory animals of both the National Institutes of Health and the Third Military Medical University. Hypothyroidism was induced by propyl-thio-uracil (PTU; Sigma-Aldrich) treatment that was administered orally, added daily to the drinking water (500 mg/L PTU, 0.075 g/L sodium bicarbonate, and 50 g/L sucrose) throughout the pregnancy. An intraperitoneal injection of PTU (10 mg/kg) was also given at E0 (the first day of pregnancy) [38,39]. The free T3 (FT3) and FT4 levels in the serum of the pregnant mice were measured at E13.5. The FT3 and FT4 levels in the offspring were measured 1 month after birth.
Assay of FT3 and FT4 serum levels
FT3 and FT4 levels in the serum were determined with a commercially available radioimmunoassay kit (Srkbio), according to the manufacturer's instructions.
Water maze
The Morris water maze was performed as previously described with some modifications [40]. Briefly, the mice were trained for 6 days with 4 trials per day and an inter-trial interval of 5 min. The trial was considered complete once the mouse found the platform or if 60 s elapsed. The mice were returned to the pool 24 h after training for a probe trial. For this test, the platform was removed from the pool, and the mouse was allowed 60 s to search for it. A computer-assisted tracking system was used for data collection.
Embryonic NSCs culture, differentiation, and T3 treatments
The telencephalons of E13.5 mice were isolated for eNSC preparation as previously described [41,42]. The cells were cultured in a complete medium comprised of a 1:1 (v/v) mixture of Dulbecco's modified Eagle's medium (DMEM) and F12 medium (Gibco) supplemented with fibroblast growth factor-basic (bFGF) (20 ng/mL; Sigma-Aldrich), epidermal growth factor (EGF) (20 ng/mL; Sigma-Aldrich), and N2 (1×) supplements (Gibco) under floating conditions with a half-medium change every 3 days. To induce differentiation, bFGF and EGF were removed from the medium. T3 (Sigma-Aldrich) was added to the culture medium every 3 days at a final concentration of 0.3 nM.
Transfection of plasmids and siRNAs
Two plasmids (pEX_EF1_STAT3-YFP and an empty vector pDS_EF1-XB-YFP; ATCC) and 2 siRNA duplexes (5′-GGA CUU GGU UCU AGA UGA UTT-3′ and 5′-AUC AUC UAG AAC CAA GUC CTT-3′; GenePharma) were used. The plasmids and siRNA duplexes were transfected using Lipofectamine LTX or Lipofectamine 2000 (Invitrogen), respectively, according to the manufacturer's instructions. An oligonucleotide that is not homologous to any known genes was used as a negative control.
Cell proliferation, TUNEL, and neurosphere assay
NSC proliferation was assessed by a colorimetric cell-counting kit-8 (CCK-8) (Dojindo) as previously described [18]. The TUNEL assay was performed using an in situ cell death detection POD kit according to the manufacturer's instructions (Roche) [18]. The neurosphere assay was performed as previously described [43].
RNA isolation, reverse-transcription PCR, and real-time PCR analysis
Total RNA was extracted from both the E13.5 mouse telencephalon and cultured eNSCs using the Trizol reagent (Takara). TRα and TRβ cDNAs were amplified by reverse transcription-PCR (RT-PCR) using the following primers: TRα, 5′-TTC AGC GAG TTT ACC AAG ATC A-3′ and 5′-GTC ATC CAG GTT AAA GGC AGA G-3′ and TRβ, 5′-AAA AGC AAG GAC TCT GAC TTG G-3′, and 5′-GGA TGG AGA CTT TTC TGA ATG G-3′ (Invitrogen). Real-time PCR was performed for beta-3 tubulin (Tuj1), glial fibrillary acidic protein (GFAP), 2′, 3′-cyclic nucleotide 3′-phosphodiesterase (CNPase), and Sox2 with beta-actin as an internal control. The real-time PCR was conducted using a Bio-Rad IQ5 Detection System with the SYBR Green PCR Master mix (Takara). Gene-specific primers were used to amplify GFAP (5′-AGC CAA GGA GCC CAC CAA AC-3′ and 5′-TCT ATA CGC AGC CAG GTT GTT CTC-3′), Tuj1 (5′-CGC CAT GTT CAG ACG CAA G-3′ and 5′-CTC GGA CAC CAG GTC GTT CA-3′), CNPase (5′-TGC ACT GTA CAA CCA AAT TCT GTG A-3′ and 5′-TTG AAG GCC TTG CCA TAC GA-3′), Sox2 (5′-AAC CGA TGC ACC GCT ACG A-3′ and 5′-TGC TGC GAG TAG GAC ATG CTG-3′), and beta-actin (5′-CAT CCG TAA AGA CCT CTA TGC CAA C-3′ and 5′-ATG GAG CCA CCG ATC CAC A-3′) (Takara).
Immunohistochemical and immunocytochemical staining
To detect the Tuj1 and TRα1 protein expression in the telencephalon, the whole embryonic brain was fixed with paraformaldehyde (4% in PBS), and 10 μm-thick consecutive sections were taken throughout the brain. The sections were stained with antibodies against Tuj1 (1:50, R&D) or TRα1 (1:50) and corresponding secondary antibodies followed by diaminobenzidine (DAB) staining. The Tuj1-positive cells in the ventricular zone (VZ) and the subventricular zone (SVZ) were counted as previously described using a Leica microscope [44,45]. For each sample, every 10th section throughout the telencephalon was analyzed. In total, 8 sections were analyzed for each condition (the control and PTU-treated) from at least 3 embryos from 3 litters obtained in parallel experiments. For double staining with antibodies against Sox2 and TRα1, the sections were incubated with primary antibodies (mouse anti-mouse Sox2, 1:50; rabbit anti-mouse TRα1, 1:50) at 4°C for 24 h followed by fluorescently conjugated secondary antibodies (Alexa Fluor® 488 goat anti-mouse and Alexa Fluor 647 goat anti rabbit, 1:100, Invitrogen) at room temperature for 1 h.
For double staining with antibodies against Nestin and TRα1 in NSCs, neurospheres that were formed 6 days in vitro (DIV) were allowed to adhere to poly-L-lysine-coated glass coverslips (12 mm). For staining with antibodies against Tuj1, GFAP and CNPase in differentiated cells, neurosphere-forming cells were cultured for 3 DIV, dissociated and incubated at 37°C for 24 h, adhered to coverslips, and further cultured for 6 days (to induce the differentiation of neurons and astrocytes) or 9 days (to induce the differentiation of oligodendrocytes) in the differentiation medium. Immunolabeling was performed according to previously described methods. Primary antibodies against Nestin (1:100, Chemincon), TRα1 (1:100), Tuj1 (1:100), GFAP (1:100, Dako), and CNPase (1:100, Millipore) were used. Hoechst 33342 (5 μg/mL; Sigma-Aldrich) was used as a nuclear staining. The cells were counted in 6 different fields of each coverslip using a 20× objective. The number of positive cells for each antibody was first calculated as the total number of cells and then expressed as the percentage of Tuj1-, GFAP-, and CNPase-positive cells.
Western blot and EMSA
The Western blot and EMSA were performed as previously described and quantified with an Odyssey infrared imaging system (LI-COR; Lincoln) [18].The primary antibodies used for Western blot were p-Erk1/2 (1:2,000; Cell Signaling Technology, CST), t-Erk1/2 (1:2,000; CST), p-Akt (1:2,000, CST), t-Akt (1:2,000; CST), p-STAT3 (1:2,000; CST), t-STAT3 (1:2,000; CST), and β-actin (1:5,000; Sigma-Aldrich), and the Odyssey-specific IRDye® 680 or 800 donkey anti-rabbit and IRDye 680 donkey anti-mouse (1:5,000; LI-COR) secondary antibodies were used. The following oligonucleotides were used for the EMSA (5′ to 3′, sense strand): consensus STAT3, GAT CCT TCT GGG AAT TCC TAG ATC and mutant STAT3, GAT CCT TCT GGG CCG TCC TAG ATC (LI-COR).
Statistics
All data were expressed as the mean±standard error of mean (SEM). Data were analyzed using the Student's t-test or a 1-way analysis of variance in combination with Fisher's post hoc test. P<0.05 was considered significant. Unless otherwise stated, all data were collected from at least 3 independent duplicate experiments.
Results
A deficiency of maternal THs inhibits the neurogenesis of the embryonic telencephalon and leads to a learning and memory impairment in the offspring
The effects of TH on brain development during embryogenesis in vivo were studied in pregnant females with hypothyroidism; this mouse model was generated using PTU [38,39]. The effects of PTU treatment were monitored by measuring FT3 and FT4 levels in the serum of pregnant mice. After treatment with PTU for 13 days, a significant reduction in the levels of both hormones was observed (Fig. 1A). However, the FT3 and FT4 levels in the serum of the offspring were not affected by PTU as detected 1 month after birth (Fig. 1B). Next, the degree of neurogenesis in the mouse telencephalon was evaluated at E16.5, which is the peak of embryonic neurogenesis in mice. Neurogenesis was assessed using the neuron-specific maker Tuj1. The number of Tuj1-positive cells in the VZ/SVZ of the telencephalon was much lower in PTU-treated mice when compared with control mice (Fig. 1C and Supplementary Fig. S1; Supplementary Data are available online at
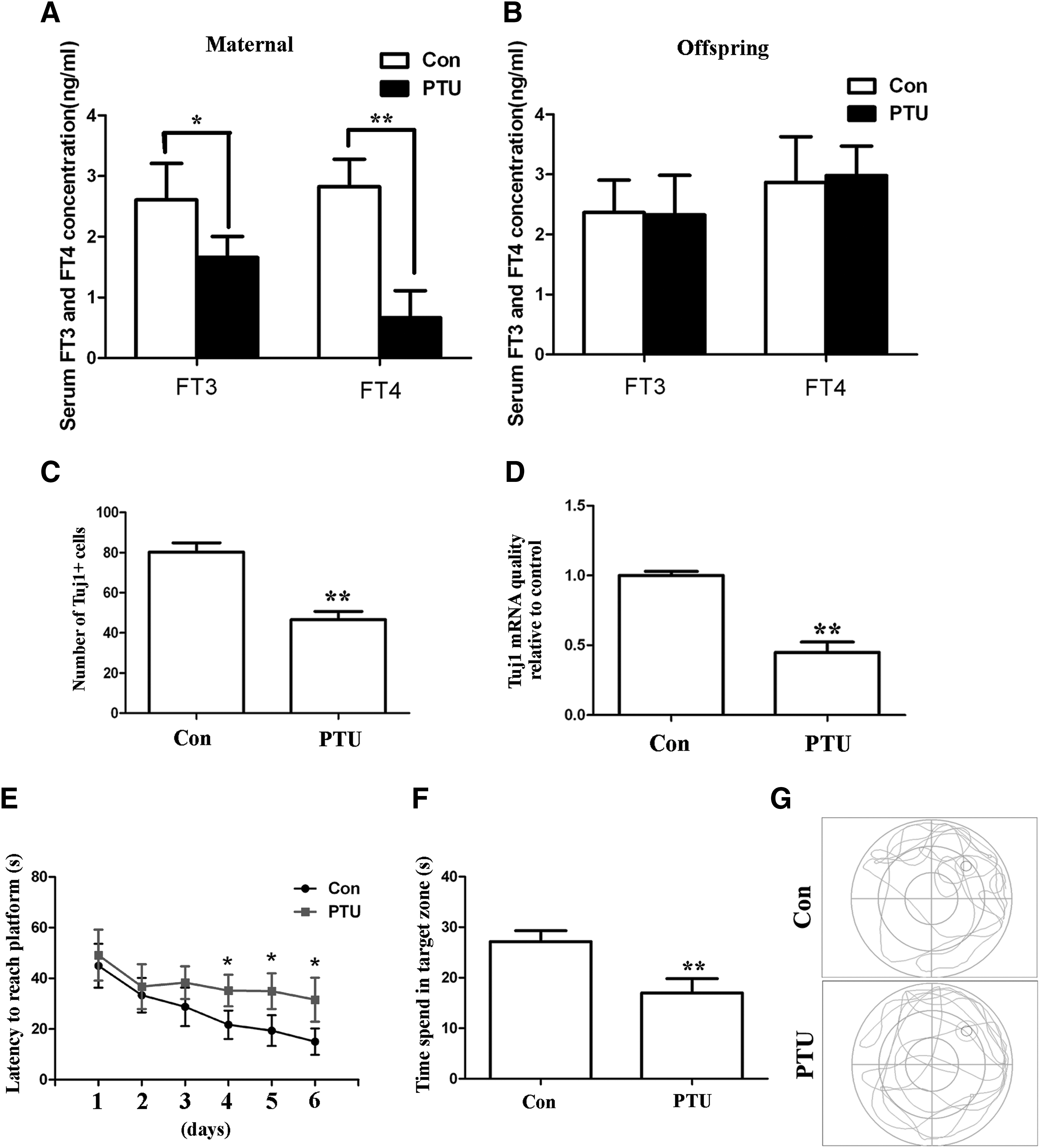
Hypothyroidism during pregnancy inhibits neurogenesis in the embryonic telencephalon and impairs learning and memory in the offspring.
To determine whether these changes in the telencephalon would cause any neurobehavioral dysfunctions later in life, the Morris water maze was used to assess the learning and memory of 2-month-old offspring. Over the course of the training period, the mice required progressively less time to escape to the platform in both the PTU-treated group and the control group. However, the escape latencies in the PTU-treated group remained much longer than in the control group (Fig. 1E). During the probe trail, the mice in the control group spent a longer time in the target zone than the PTU-treated mice (Fig. 1F) and showed a stronger spatial bias for the area of the pool where the platform was located during training (Fig. 1G). These results suggest a learning and memory impairment in the offspring of the PTU-treated mice.
T3 promotes the neuronal differentiation of eNSCs in vitro
New neurons in the telencephalon are generated from eNSCs [46]. To further examine the effects of THs on embryonic neurogenesis, we cultured eNSCs from E13.5 mouse telencephalon. The characteristics of eNSCs were identified as previously described [47] (Supplementary Fig. S2). Then, we treated eNSCs with T3 at a final concentration of 0.3 nM, which is close to the physiological level indicated by previous reports [48 –50]. The effects of T3 on eNSC differentiation were investigated by immunostaining with the neuron-specific marker Tuj1, the astrocyte-specific marker GFAP, and the oligodendrocyte-specific marker CNPase. In accordance with the results in vivo, we found that T3 treatment increased the percentage of Tuj1-positive cells (Fig. 2A, B). We also found that the number of CNPase-positive cells was increased, while the GFAP-positive cells were markedly decreased after T3 treatment (Fig. 2A, B). In addition, the same set of lineage markers were examined by real-time PCR. The results showed that the Tuj1 and CNPase mRNA levels were robustly up-regulated, while the GFAP mRNA levels were significantly down-regulated after T3 treatment (Fig. 2C). Furthermore, T3 increased the number of Tuj1-positive neurites as detected by inducing single eNSCs differentiation for 3 days, and the T3-induced differentiated neurons appeared with more branch points and longer neurites (Fig. 2D and Supplementary Table S1). To detect the effects of T3 on the differentiation of neurospheres, subcultured eNSC spheres were immunochemically stained with Tuj1 and GFAP 7 days after differentiation. After T3 treatment, the Tuj1-positive cells looked robust with high density of neurites (Fig. 2E).
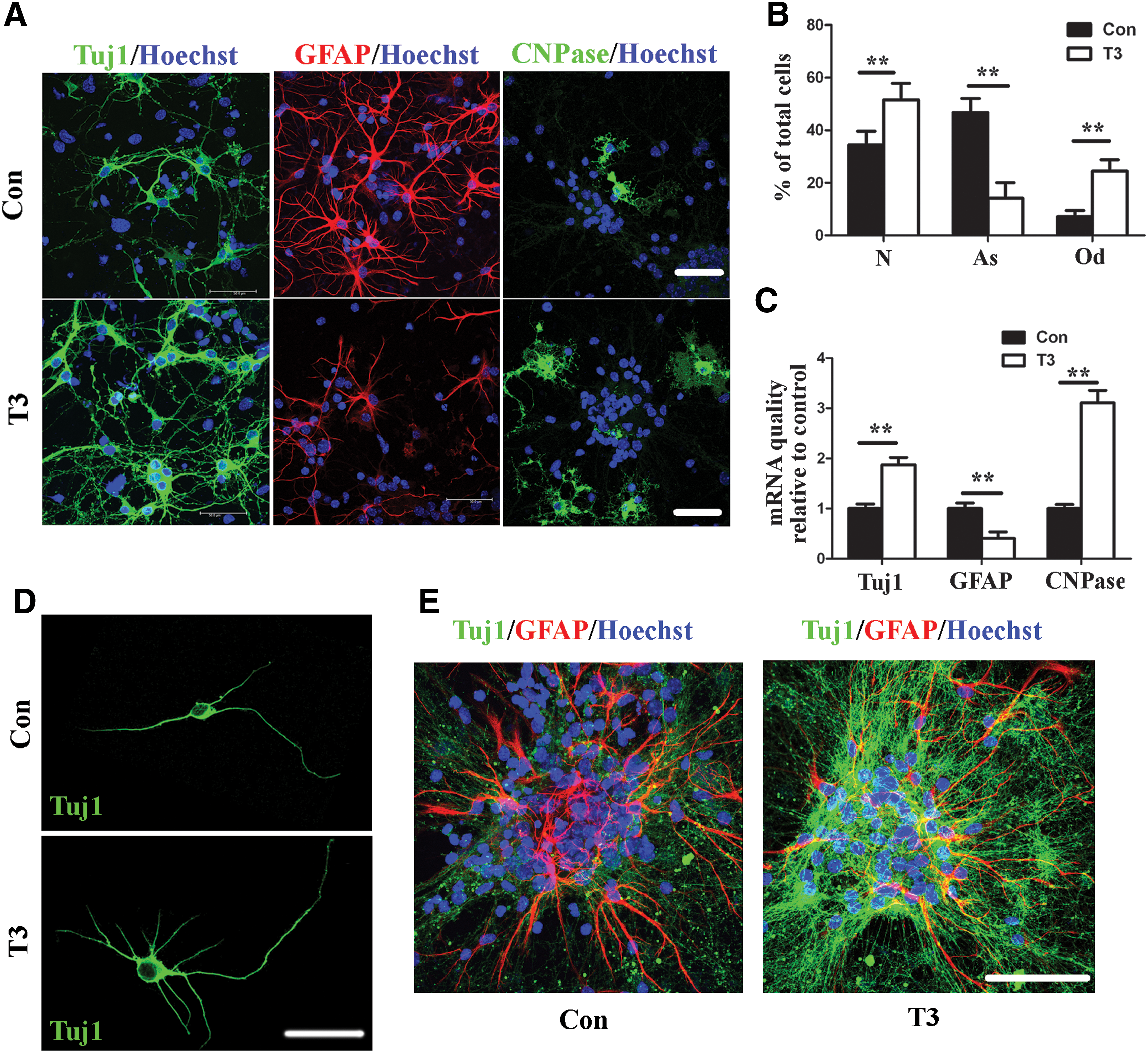
The effects of the physiological level of T3 on eNSCs differentiation.
In addition, we also found that the physiological level of T3 inhibited eNSC proliferation and maintenance, as demonstrated by a decrease in absorbance at 450 nm in the CCK-8 assay (Fig. 3A), the mRNA expression of the NSC-specific marker Sox2 (Fig. 3B), and the number of primary and secondary neurospheres that formed in culture (Fig. 3C, D). However, no changes were found in the percentage of TUNEL-positive cells in eNSCs that were cultured for 4 or 6 days with T3 as compared with control eNSCs (Fig. 3E, F), suggesting that the inhibitory effects of T3 on eNSC proliferation and maintenance were not due to inducing cell apoptosis. Taken together, these results strongly suggest that T3 is a promotive factor for the neuronal differentiation of eNSCs.
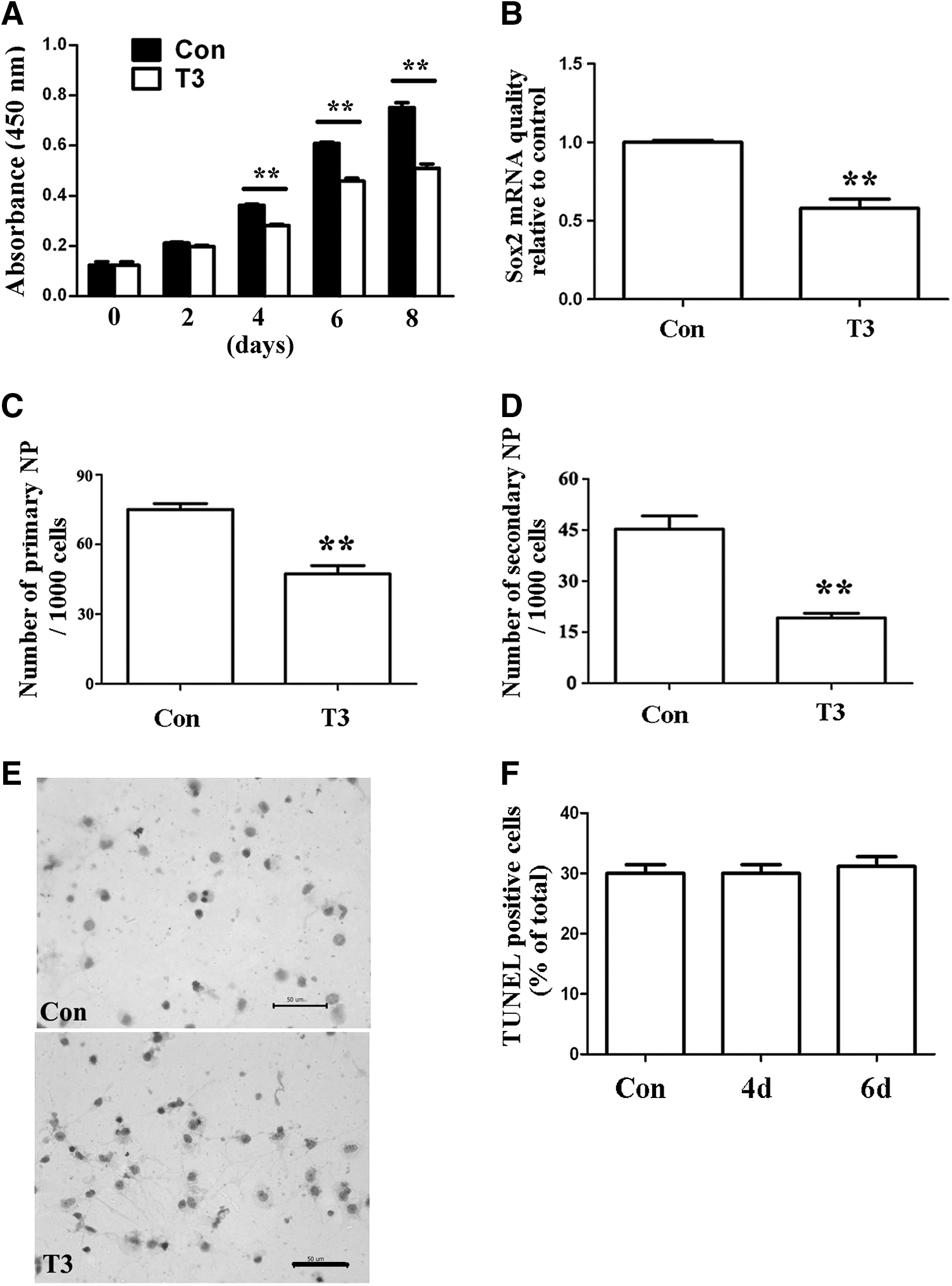
The effects of physiological level of T3 on the proliferation and maintenance of eNSCs.
TRα1 is required for the effects of T3 on eNSCs
To detect the role of TRs in regulating eNSC fate, the mRNA and protein expressions of the TRs were first characterized in both the E13.5 mouse telencephalon and cultured eNSCs. The TRα transcript was found both in cultured eNSCs and in the E13.5 telencephalon. In contrast, TRβ was not detected in either cultured eNSCs or the telencephalon (Fig. 4A). Since TRα2 does not bind T3, the TRα1 protein expression was examined in the telencephalon, and strong reactivity to TRα1 was found in this region (Fig. 4B). To determine whether these TRα1-positive cells in the telencephalon were eNSCs, double staining was conducted both in vitro and in vivo. An immunocytochemical analysis of TRα1 revealed nuclear staining in nestin-positive eNSCs in vitro (Fig. 4C). Additionally, double staining in the telencephalon revealed that TRα1 was present in cells that were positive for the NSC-specific marker Nestin in vivo (Fig. 4D, E). These results indicate that TRα1 is present in E13.5 NSCs both in vivo and in vitro.

TRα1, but not TRβ, is expressed in cultured eNSCs and E13.5 mouse telencephalon.
The potential involvement of TRα1 in mediating the effects of T3 on eNSCs was assessed by eliminating TRα1 protein expression using a TRα1-specific siRNA (Fig. 5A). Notably, the TRα1-specific siRNA abolished the T3-induced alterations in the expressions of Tuj1, GFAP, and CNPase mRNA (Fig. 5B). TRα1 silencing also eliminated the effects of T3 on the neurite outgrowth (Fig. 5C and Supplementary Table S2). We also found a reverse effect of the TRα1-specific siRNA on T3-induced neuronal differentiation and neurite outgrowth of the eNSC spheres (Fig. 5D). In addition, the siRNA against TRα1 blocked the T3-induced decrease in eNSC proliferation (Fig. 5E). The T3-induced reduction in secondary neurosphere formation and Sox2 mRNA expression was also attenuated by the TRα1-specific siRNA (Fig. 5F, G). These results implicate TRα1 in the effects of T3 on eNSCs and further demonstrate that both T3 and its receptor, TRα1, are required for the neuronal differentiation of eNSCs.

TRα1 is involved in the effects of T3 on eNSCs.
T3 inhibits the STAT3 signaling activity through TRα1
To address the possible signal transduction mechanisms underlying the effects of T3, the involvement of the Erk1/2, Akt, and STAT3 signaling pathways were examined, because these pathways had been previously reported to be crucial in regulating eNSC fate [30,51,52]. Neither the protein expression nor the phosphorylation of Erk1/2 and Akt were altered after T3 treatment (Fig. 6A, B, C). However, the treatment of eNSCs with T3 resulted in a marked decrease in the tyrosine phosphorylation of STAT3, although total STAT3 protein expression was not affected (Fig. 6A, D). Next, a gel shift assay was performed to assess the STAT3-DNA binding activity using nuclear extracts from eNSCs. T3 treatment resulted in a significant decrease in the intensity of the band representing the STAT3-DNA binding complex (Fig. 6E, F), which suggested a role for T3 in inhibiting the STAT3-DNA binding activity. Taken together, these results suggest that T3 inhibits the activity of the STAT3 signaling pathway.
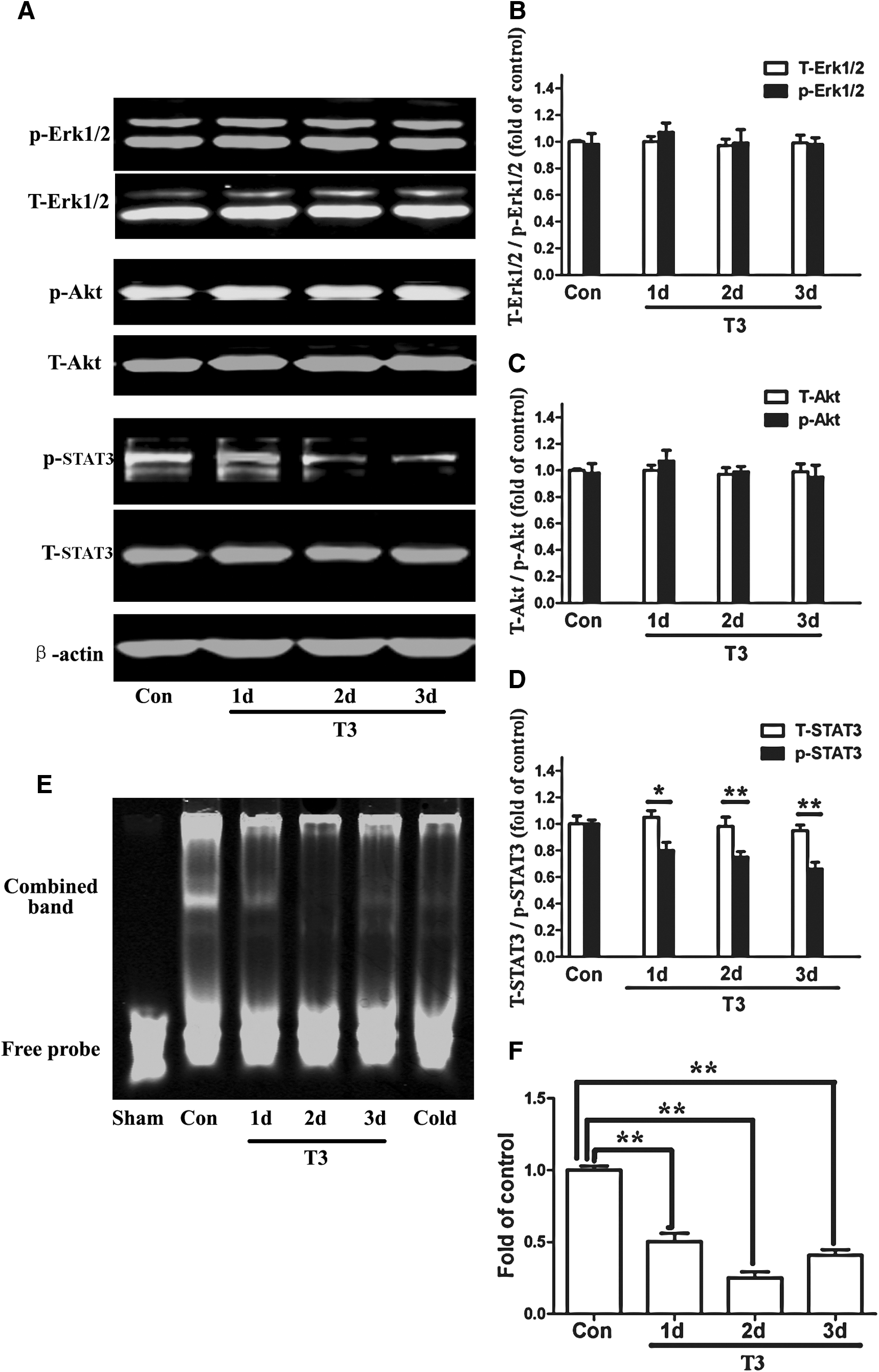
The effects of the physiological level of T3 on the Erk1/2, Akt, and STAT3 signaling pathway activity.
To assess the involvement of TRα1 in regulating the STAT3 signaling activity, a TRα1-specific siRNA was first used to silence TRα1 expression. Western blot analysis revealed that the TRα1-specific siRNA blocked the effects of T3 on STAT3 phosphorylation (Fig. 7A), suggesting that T3 down-regulated STAT3 phosphorylation through TRα1. Furthermore, the EMSA experiments revealed that the inhibitory effects of T3 on the STAT3-DNA binding activity were attenuated by TRα1-specific siRNA (Fig. 7B). Taken together, these results strongly indicate that TRα1 mediates the inhibitory effects of T3 on STAT3 phosphorylation and STAT3-DNA binding activity.

T3 regulates the STAT3 signaling activity through TRα1.
T3 promotes the neuronal differentiation of eNSCs by inhibiting the STAT3 signaling activity
Previous studies have demonstrated that STAT3 is crucial for the proliferation and maintenance of NSCs [43]. STAT3 has also been shown to induce NSC differentiation into astrocytes [24,51]. To further assess the role of STAT3 in regulating the neuronal differentiation of eNSCs, the STAT3-specific inhibitor Stattic was used to block the STAT3 signaling pathway. Immunostaining showed that the percentage of Tuj1-positive cells was markedly increased, while the percentage of GFAP-positive cells was significantly decreased after Stattic treatment (Fig. 8A, B). Only a slight increase in the percentage of CNPase-positive cells was observed (Fig. 8A, B). Real-time PCR analysis of these lineage markers verified the results from the immunostaining (Fig. 8C), which further confirmed that STAT3 inhibition differentiated eNSCs into neurons. In addition, we also found that Stattic treatment inhibited eNSC proliferation and maintenance (Supplementary Fig. S3). These results together confirm the critical role of STAT3 in regulating eNSC fate.

The role of STAT3 in regulating the effects of T3 on eNSCs.
Based on these results, we naturally asked a question whether STAT3 signaling participates in the neuronal promotive effects of T3 on eNSCs. Since T3 acts in an inhibitory manner on the STAT3 signaling pathway, we overexpressed STAT3 in eNSCs by transfecting the pEX_EF1_STAT3-YFP plasmid before T3 treatment. The STAT3 protein expression, phosphorylation, and nuclear translocation were increased after the transfection of this plasmid (Fig. 8D, E, F), suggesting an up-regulation of STAT3 signaling activity. Further investigations revealed that the alterations in the expression of Tuj1 and GFAP mRNA induced by T3 were eliminated by STAT3 overexpression (Fig. 8G). However, the CNPase mRNA levels were still significantly increased after T3 treatment despite the high activity of STAT3 signaling (Fig. 8G), which indicated that STAT3 may not be a major regulator of oligodendrocytic differentiation. The results from the CCK-8 and neurosphere assays indicated that STAT3 overexpression attenuated the inhibitory effects of T3 on eNSC proliferation and maintenance (Fig. 8H, I). Taken together, these results suggest that the low activity level of STAT3 signaling induced by T3 is responsible for the mechanism of T3-induced neuronal differentiation of eNSCs.
Discussion
Maternal hypothyroidism during pregnancy impairs brain function in the offspring of humans and other mammals, but little is known about the cellular targets and the underlying mechanisms of maternal hypothyroidism. To our knowledge, this is the first report revealing the promotive effect of TH and TRα1 on the neuronal differentiation of eNSCs. These findings significantly advance our understanding of TH activity during early brain development and provide exiting new insights into the mechanisms of neurological deficits caused by THs deficiency during pregnancy.
Hypothyroidism in humans is characterized by severe neurological consequences that are often irreversible, highlighting the critical role of THs in the brain [53]. Even subtle insufficiencies in maternal THs during the first and second trimesters of pregnancy may adversely affect neurodevelopment in the offspring, which can result in irreversible cognitive deficits [2,54,55]. This was supported by our experimental results that revealed a reduction in embryonic telencephalonic neurogenesis and a learning and memory impairment in the offspring. The maternal thyroid is the only source of THs for the fetal brain before the onset of fetal thyroid production, which occurs at E17.5–E18 in rats and mice. Even after the fetus begins to produce its own THs, the maternal thyroid continues to supply a significant proportion of fetal THs. Maternal FT4 can cross the efficient uterine-placental “barrier” and give rise to T3 [2,10]. We found that PTU, a potent inhibitor of TH synthesis [56], significantly decreased the levels of maternal FT4 and FT3 but had no effect on the THs levels of the offspring. Thus, the animal model generated by PTU accurately mimicked maternal hypothyroidism during pregnancy. PTU is widely used in the clinical treatment of hyperthyroidism, and it has been previously used to establish animal models of hyperthyroidism [38,39,57]. PTU treatment during pregnancy has not been reported to have any effects on the thyroid function of the offspring in humans [57]. In this study, although the TH levels in the offspring were normal, neurological development, as demonstrated by a learning and memory dysfunction, was abnormal. These results indicate that the cognitive deficit observed in our experiments results from a deficiency in fetal brain development during pregnancy that is caused by insufficient THs levels.
Previous research on the effects of TH and embryonic neurogenesis are a major focus on the effects of TH and neurons, including the migration and maturation of neurons. The physiological effects of T3 on eNSCs are still unclear. The study of NSCs has provided fundamental insights into brain development and function and has suggested several potential therapies that address devastating neurological disorders. Thus, eNSCs provide a powerful model for exploring the possible cellular targets and the underlying mechanisms of THs during early brain development. During the early stages of embryogenesis, eNSCs primarily increase in number in the undifferentiated state. The proliferation of eNSCs will slow down during mid-to-late gestation, because in this stage, eNSCs are supposed to undergo differentiation and give rise to neurons. Our results suggest that TH is a critical regulator of this normal developing process of eNSCs. It is difficult to precisely measure the physiological concentration of T3 in the fetal brain during pregnancy. Previous studies have indicated that the concentration of T3 used in our in vitro experiments is close to physiological levels [48 –50]. We observed that in vitro treatment with T3 promotes the neuronal differentiation of eNSCs. We also observed that a lack of THs decreases neurogenesis in the VZ/SVZ zone of the telencephalon in vivo. Cortical projection neurons are generated from eNSCs in 2 areas, the VZ and the SVZ, of the telencephalon [46,58,59]. Our results reveal that one of the physiological functions of T3 is to promote a proliferation to differentiation switch in eNSCs and enhance the neuronal differentiation. We believe that this is the first report which establishes that T3 is a potent factor in inducing embryonic neurogenesis. Previous studies have mainly focused on the effects of T3 on remyelination and oligodendrocyte differentiation and maturation [38,60]. Some studies have revealed that T3 promotes neurogenesis in the adult hippocampus, olfactory bulb, and SVZ; these effects are likely due to the promotion of adult NSC proliferation and the inhibition of NSC apoptosis by T3 [39,50,61,62]. In contrast, other studies have demonstrated that T3 terminates neural progenitor cell proliferation and induces oligodendrocytic differentiation in the first few postnatal weeks [63,64]. The differences among these studies might result from the stage-dependent functions of TH in brain development. Here, we report a novel finding that eNSCs are one of the major cellular targets by which TH influences early brain development. Our findings proved that THs are critical for terminating eNSC proliferation and maintenance, while promoting the neuronal differentiation, which contributes to the neurogenesis in the telencephalon during early brain development.
TRs are thought to be critical in TH functions in the CNS. The question remains whether the TR is involved in the effects of T3 during early brain development. In the mammalian fetus, the TR appears in the brain well before the thyroid develops [65]; however, the exact timing and type of TR mRNA and protein expression have not been well established in eNSCs. We found that TRα1, but not TRβ, is expressed in eNSCs both in vivo and in vitro at E13.5. In rats, the TRα1 transcript has been detected at E11.5 in the neural tube and at E12.5 in certain areas of the diencephalon and the ventral rhombencephalon [15,34]. TRα1 is considered as playing a prominent role in TH activation in the CNS based on its high expression throughout the brain, where it accounts for ∼70%–80% of the total T3-receptor binding [1,2]. However, the non-TR-mediated effects of TH, also known as “nongenomic” or “nonnuclear” effects, have also been previously described in neural cells [36]. Our in vitro experiments suggest that the effects of T3 on eNSCs are TR mediated. However, it remains to be determined whether these effects will be recapitulated in the TR knockout mice.
In assessing the signal transduction mechanisms of TH, we found that T3 had no effect on the phosphorylation of Erk1/2 and Akt, but it significantly decreased the STAT3 signaling activity. The interactions among THs and the MAPK (Erk1/2), PI3-K/Akt, and STAT3 signaling pathways have been previously demonstrated in the liver, lung, and skin [36]. However, our results suggest that the situation is different in the developing brain. In the previous studies, we have observed that T3 inhibits STAT3 signaling activity in eNSCs in a hyperthyroidism model [18]. In this study, we detected that a physiological level of T3 also significantly decreased STAT3 signaling activity. In addition, we found that T3 regulated STAT3 signaling activity through TRα1. These findings indicate that T3, TRα1, and STAT3 interactions play an important role in the physiological functions of TH and normal brain development. STAT3 is highly expressed specifically in eNSCs at early stages of brain development (ie, the expansion phase of eNSCs), and this is critical for the proliferation and maintenance of eNSCs [43,66,67]. During the subsequent neurogenesis phase between E11.5–E16.5, the STAT3 activity is decreased [68]. It has also been observed that the down-regulation of the STAT3 gene promotes the neuronal differentiation of neighboring cells in a noncell-autonomous manner [43]. A decrease in the activity of STAT3 or its upstream factors, Jak2 and Jak3, inhibits NSC proliferation and maintenance and instead induces the differentiation of NSCs into neurons and oligodendrocytes [28,69]. In our experiments, we provided direct evidence that the inhibition of STAT3 activity promoted the neuronal differentiation of eNSCs. We also found that the overexpression of STAT3 in NSCs attenuated the effects of T3. These results strongly confirm that T3 exerts its neuronal promotive effects on eNSCs through inhibiting STAT3 signaling activity. However, the oligodendrocytic differentiation from eNSCs was only slightly regulated by STAT3 signaling, indicating that other mechanisms may be involved in this process. Previous studies have revealed that STAT3 is activated at a low level in eNSCs during the neurogenesis due to the presence of neuron-promoting ligands such as cardiotrophin 1 and neuropoietin [68]. Here, we found that T3-TRα1 binding resulted in a low level of STAT3 activity during the embryonic neurogenesis. STAT3 signaling has been shown to interact with Notch signaling pathways in eNSCs through the Notch ligand Dll1 [43]; therefore, the decline in STAT3 activity most likely leads to the promotion of neuronal gene expression through the Dll1/Notch pathway [42,43]. However, STAT3 plays diverse roles in embryonic neurogenesis, and the mechanism by which STAT3/TRα1 interactions regulate the differentiation of eNSCs into neurons and glia needs further investigation.
In conclusion, our investigations reveal that T3 promotes the neuronal differentiation of eNSCs through TRα1 by inhibiting the STAT3 signaling activity and contributes to the early neurogenesis in the embryonic telencephalon. Maternal hypothyroidism during pregnancy, which leads to THs deficiency in the fetal brain, decreases neurogenesis in the embryonic telencephalon by influencing the fate of eNSCs and leads to a dysfunction in the learning and memory of the offspring. This study provides exciting new insights into the role of THs in regulating the fate of eNSCs during early brain development. In addition, these findings underscore the importance of a further investigation into the roles of TH and TRs in early embryonic neurogenesis to gain new insights into the etiology and clinical treatment of the neurological deficits of the offspring that are caused by hypothyroidism during pregnancy.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 30901179) and the National Program on Key Basic Research Project (2011CB50370). The authors thank Miss Min Li, Wei Sun and Liting Wang for their technical assistance.
Author Disclosure Statement
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.