
Abstract
Previous studies indicate that the release of proteases, including the gelatinase matrix metalloproteinase (MMP)-9, from mature granulocytes plays a crucial role in cytokine-induced hematopoietic stem and progenitor cell (HSPC) mobilization. However, studies with MMP-9-deficient mice revealed that HSPC mobilization was normal in these animals, suggesting that additional proteases must be active at clinically relevant cytokine concentrations. In the present study, we provide evidence that the collagenase MMP-8 is involved in stem cell mobilization. A rapid release of MMP-8 from isolated neutrophil granulocytes can be observed during an in vitro culture. During granulocyte colony-stimulating factor-induced HSPC mobilization, highly elevated serum concentrations of MMP-8 were observed on days 4 to 6 of the mobilization regimen, concomitantly with elevated MMP-9 serum levels and higher numbers of circulating CD34+ cells. Elevated serum concentrations of both proteases were also found in umbilical cord blood serum. In functional assays, adhesion of HSPC to osteoblasts as an essential component of the endosteal stem cell niche is negatively influenced by MMP-8. The chemokine CXCL12, which is critically involved in stem cell trafficking, can be proteolytically processed by MMP-8 treatment. This degradation has a strong inhibitory influence on HSPC migration. Taken together, our data strongly suggest that MMP-8 can be directly involved in hematopoietic stem cell mobilization and trafficking.
Introduction
D
Cytokine-induced HSPC mobilization can be regarded as a multistep process with a crosstalk between adhesive structures and cytokines [1,10]. During mobilization, adhesive interactions between HSPC and their microenvironmental niches have to be modulated, and proteolytic enzymes are ideal candidates for fulfilling this function. In fact, a highly proteolytic microenvironment characterized by the matrix metalloproteinase (MMP)-9, MT1-MMP, carboxypeptidase M, cathepsin G and K, and neutrophil elastase has been found in the bone marrow after treatment with mobilizing agents [11]. MMP-9 has been suggested to be directly involved in mobilization, since antibodies against MMP-9 were capable of preventing the interleukin-8 (IL-8)-induced mobilization of HSPC in rhesus monkeys [12]. In humans, elevated serum concentrations of MMP-9 in the peripheral blood have been observed after granulocyte colony-stimulating factor (G-CSF)-induced stem cell mobilization [13 –16]. On the other hand, experiments in mice yielded only low serum levels of MMP-9 after IL-8-induced mobilization [17], and MMP-9-deficient mice did not show any alterations in cytokine-induced mobilization [18]. It is therefore likely that MMP-9 is not the only secreted MMP playing a role in stem cell mobilization.
The MMPs comprise a large family of proteolytic enzymes that can degrade extracellular matrix (ECM) molecules, membrane-bound adhesion molecules, cytokines, and chemokines, thus altering the cell–cell or cell–matrix interactions [19 –21]. The members of the MMP gene family can be divided into several subfamilies, including collagenases, gelatinases, stromelysins, matrilysins, and membrane-bound MMPs, which display different substrate specificities and tissue distribution [22,23]. The two gelatinases, MMP-2 and MMP-9, are the most intensively studied MMPs, also in the bone marrow [24,25]. MMP-8 is a major collagenase synthesized by polymorphonuclear cells. MMP-8 and MMP-9 share some substrate specificities, as both enzymes degrade naïve collagen type I, VII, and X, although with different efficacies. Moreover, both enzymes digest denatured collagens or aggrecan [26]. Although it has been shown that neutrophils seem to be indispensable for hematopoietic stem cell mobilization [27], the release of MMP-8 during this process has yet to be studied in detail.
In the present study, we analyzed the expression of MMP-8, in addition to MMP-9, in the human bone marrow. A potential release of MMP-8 was analyzed in vitro via mobilizing agents, but also in vivo during G-CSF-induced mobilization. While only a very low number of circulating hematopoietic stem cells were found in normal peripheral blood, an enhanced number of stem cells can be observed in umbilical cord blood (UCB). To elucidate whether MMP-8 release is a general phenomenon that accompanies an elevated stem cell number in the peripheral blood, serum concentrations of MMP-8 were also determined in the sera of healthy donors and UCB. The functional implication of MMP-8 release was investigated in cell migration assays, cell adhesion studies, and proteolytic digestion of the ECM molecules and chemokines.
Design and Methods
Human primary cells, blood, and serum samples
Bone waste of endoprosthesis surgery was obtained from the Department of Orthopedic Surgery (University of Tübingen) after written consent by the patients. Human primary osteoblasts were isolated from the bones according to an established protocol published recently [28]. The cultured osteoblasts were routinely checked for the expression of osteogenic markers such as osteopontin, alkaline phosphatase, or Runx2.
Bone marrow aspirates and UCB were obtained from hematologically healthy donors after informed consent from the Department of Medicine II and from the Department of Gynecology and Obstetrics, University of Tübingen, respectively, according to the guidelines of the local ethics committee. Cord blood mononuclear cells (CBMNCs) were isolated by Percoll (1.077 g/mL) density-gradient centrifugation (Linaris) and washed with Dulbecco's phosphate-buffered saline (DPBS; Invitrogen). The CBMNC population was labeled with anti-CD34-conjugated microbeads according to the manufacturers' instructions (Miltenyi Biotec). CD34+ HSPC were enriched by magnetic cell separation using MACS columns (Miltenyi Biotec).
For the isolation of neutrophil granulocytes, peripheral blood was obtained from healthy donors. Neutrophils were isolated using polymorphprep (Axis-Shield) according to the manufacturer's instructions. G-CSF-mobilized peripheral blood samples were collected from 4 healthy donors (average age 22.7 years, range 17 to 31 years) who received rhG-CSF (Aventis) subcutaneously at 10 μg/kg/day for up to 7 consecutive days. All blood samples were obtained from donors after informed consent. Donor blood samples were collected at the beginning (day 0) and on days 4, 5, 6, and 7 of G-CSF treatment. After centrifugation, serum samples were stored at −70°C until used. Concomitantly, with the serum collection, white blood cell counts and the percentage of CD34+ cells were routinely determined. UCB samples (n=27) were obtained from full-term deliveries. The UCB sera were directly separated by centrifugation with 1,000 g for 10 min at 4°C and stored at −70°C until used. The peripheral blood sera were obtained from healthy volunteers.
Antibodies, proteases, chemokines, and ECM proteins
The monoclonal antibodies specific for MMP-8 (clone 115-1302) and MMP-9 (clone 6-6B) were obtained from Merck Millipore and Calbiochem, respectively. The affinity-purified polyclonal rabbit antibody against latent human MMP-8 was kindly provided by Prof. Tschesche (University of Bielefeld, Germany).
MMP-8 and MMP-9 (obtained from Calbiochem) were activated by incubating the samples with 1 mM 4-aminophenylmercuric acetate (APMA; Sigma-Aldrich) in an assay buffer (50 mM Tris–HCl, pH 7.5, 200 mM NaCl, 10 mM CaCl2, and 50 μM ZnCl2) for 3 h at 37°C. The chemokines IL-8 and MIP-1α (CCL3) were purchased from Biozol and CXCL12 from PeproTech. The basement membrane component laminin (LM)-511/521 derived from the human placenta, used for membrane coating, was obtained from Sigma-Aldrich. Recombinant human LM-511 was purchased from Biolamina.
Immunofluorescence staining
Six-micrometer bone marrow cryostat sections were fixed with methanol at −20°C for 10 min and washed with DPBS. The tissue samples were incubated for 1 h with the primary antibodies diluted in DPBS containing 0.1% bovine serum albumin (BSA). After washing with DPBS, bound antibodies could be detected by Cy3™-conjugated goat anti-mouse or Cy3™-conjugated goat anti-rabbit antibodies (Dianova) and FITC-conjugated anti-mouse-IgM antibody (BD-Pharmingen). The cell nuclei were identified by counterstaining with 4′,6-diamino-2-phenylindol-dihydrochloride (DAPI; 1 μg/mL). The first antibody was omitted for control staining. Photographs were taken on a Zeiss Axiophot microscope (Carl Zeiss).
FACS analysis
Expression of MMPs of human bone marrow cells was studied by a dual-color FACS analysis. Unfractionated bone marrow aspirates were treated with an ammonium chloride-containing buffer to remove all erythrocytes. Then, the bone marrow cells were briefly washed with PBS.
For the detection of intracellularly localized MMPs, 2×105 cells were incubated for 20 min with Cytofix/Cytoperm (BD Biosciences). The Perm/Wash solution (BD Biosciences) was used for all washing procedures. After incubation for 15 min with mAbs against MMP-8 or MMP-9, the cells were washed, and a phycoerythrin-conjugated anti-mouse-IgG1 (Jackson ImmunoResearch) as a secondary antibody was applied for 20 min. Subsequently, the cells were washed and stained with a CD-15-FITC-conjugated mAb (clone HI98; Bio Legend) for 15 min. The labeled cells were washed and analyzed for cell surface or intracellular antigen expression using an FACScan flow cytometer and FACScan Research software (Becton Dickinson).
Zymography
UCB and normal blood serum samples were diluted in a 6×loading buffer [300 mM Tris–HCl, pH 6.8, containing 12% sodium dodecyl sulfate (SDS), 60% glycerol, and 0.6% bromophenol blue] in the absence of a reducing agent and heated at 60°C for 10 min. The samples were separated in 10% polyacrylamide gels containing 0.1% of denatured gelatin (Biochrom). After electrophoresis, gels were washed 3 times in a renaturating buffer (2.5% Triton X-100) for 20 min to remove SDS. After rinsing twice in a developing buffer (50 mM Tris–HCl, pH 7.6, containing 200 mM NaCl, 5 mM CaCl2, 1 μM ZnCl2, 0.05% Brij35, and 0.05% NaN3), the gels were incubated at 37°C for 72 h in the same buffer under gentle agitation. Gels were stained for 1 h at room temperature in 40% methanol and 10% glacial acetic acid containing 0.1% (w/v) Coomassie Brilliant Blue R250 (CBB; Merck), and destained in the same solution without CBB until clear bands appeared against the blue background. Pro-MMP-9 (M r 92000) and MMP-9 (M r 84000) were identified by comparing them to a known gelatinolytic activity from the control samples containing the commercially available purified MMP-9 protein.
MMP-8 and MMP-9 ELISA
The amounts of MMP-8 and MMP-9 in the cell culture supernatants or serum probes were quantitatively determined using the Biotrak™ MMP-8 or MMP-9 ELISA system commercially available from GE Healthcare. The assay procedure followed the recommended instructions of the manufacturer. All samples were analyzed in duplicate and compared with standard internal curves that were newly performed for each experiment. MMP-8 and MMP-9 can be measured in the range 0.25–4 and 1.0–64 ng/mL, respectively.
Cell adhesion assays
For the quantitative determination of adhesive cell–cell interactions, primary osteoblasts were seeded in 48-well plates and grown for 72 h to confluency. About 1×106 MACS-isolated CD34+-HSPC were incubated with 2 μg of the fluorescent dye BCECF-AM [2′, 7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester; Sigma-Aldrich] for 15 min at 37°C. The labeled HSPC were washed twice with serum-free RPMI containing 1 mM CaCl2, 1 mM MgCl2, and 50 μM MnCl2. The confluent monolayer of primary osteoblasts was washed twice with PBS, and 1×105 CD34+ cells were allowed to attach to the osteoblasts for 1 h. Subsequently, 100 ng/mL of activated and nonactivated recombinant MMP-8 or MMP-9 (Calbiochem) was added to the cells for 1 h. HSPC without treatment were used for control experiments. After washing with PBS (supplemented with Mn2+, Ca2+, and Mg2+), the fluorescence of attached HSPC was measured with Fluoroskan Ascent (Thermo Scientific, Dreieich, Germany). The percentage of adherent CD34+ cells was calculated as follows:
The cell–cell adhesion assays were carried out in triplicate. After quantification, CD34+ cells attached to the primary osteoblasts were fixed and stained with 0.1% crystal violet to visualize cell adhesion of the CD34+ cells under a Zeiss Axiovert light microscope. Photographs of representative fields were taken.
Cell adhesion to the ECM protein LM-511 was carried out as described previously [29]. Briefly, 1 μL of LM-511 or LM-511 digested by MMP-8 was spotted onto plastic dishes and immobilized by air-drying at room temperature. Nonspecific binding of CD34+ cells to the plastic dishes was prevented by preincubation with 1% BSA/PBS. Then, the CD34+ cells were allowed to attach for 1 h in a serum-free medium (supplemented with Mn2+, Ca2+, and Mg2+). Nonadherent cells were removed by gently rinsing the dishes with prewarmed PBS. Specific cell adhesion was evaluated under a Zeiss Axiovert microscope.
Analysis of LM-511 digestion by activated MMP-8
Three hundred nanograms of LM-511 were incubated with 100 ng of MMP-8 in an assay buffer containing 1 mM APMA for 0–24 h at 37°C. At different time points, degradation was stopped by adding a 6×loading buffer containing 0.2 M dithiotreitol (DTT) and 12% β-mercaptoethanol. For comparison, 100 ng MMP-9 or PBS was added and incubated for 24 h. Samples were loaded and run on 3%–8%-gradient Tris–acetate gels (Invitrogen). Then, the gels were fixed for 10 min in 50% MeOH and 5% glacial acetic acid, washed twice for 5 min with H2O, incubated in a 0.02% sodium thiosulfate solution for 1 min, and washed again with H2O for 1 min. Gels were incubated in a silver-staining solution containing 0.1% AgNO3 for 10 min and washed shortly with H2O. The staining was developed with 3% sodium carbonate, 0.05% formaldehyde, and 0.000016% sodium thiosulfate until the desired staining intensity was reached. The reaction was stopped using 50 mM EDTA, pH 8, for 10 min. For further analysis by peptide mass fingerprint, gels were stained with CBB R250.
Peptide mass-fingerprint analysis
After SDS–polyacrylamide gel electrophoresis, the relevant protein bands stained with CBB R250 were excised. After destaining and drying, gel slices were reduced with 2 mM DTT and alkylated with 20 mM iodacetamide (Sigma-Aldrich), according to the standard protocols [30]. Trypsin digestions were carried out at 37°C overnight by adding 0.5 μg of trypsin (Promega) per gel slice in 50 mM NH4HCO3. Peptide fragments were extracted twice using 1% (v/v) formic acid in 50% (v/v) acetonitrile. Supernatants were pooled and desalted on a C18 column (ZipTip; Millipore). Eluted peptides were analyzed on a linear-trap quadrupole Orbitrap XL mass spectrometer (Fisher Thermo Scientific) coupled to Nano-HPLC NanoLC 2D (Eksigent) with a nanoflow electrospray ionization source. For database searches (Swiss-Prot protein sequence database, Homo sapiens,
Matrix-assisted laser desorption/ionization–time-of-flight analysis of CXCL12 degradation by activated MMPs
For matrix-assisted laser desorption/ionization–time-of-flight (MALDI-TOF) analysis, 100 ng of CXCL12 were incubated in an assay buffer for 2, 8, and 24 h at 37°C with or without 64 ng of activated MMP-8 or MMP-9. Degradation was stopped by immediate freezing at −80°C. Five microliters of sample was acidified with 5 μL of 0.2% trifluoroacetic acid (Sigma), and 10 μL was loaded on a C18 column (ZipTip; Millipore). After on-column washing, proteins were eluted with 5 μL 50% acetonitrile/0.1% trifluoroacetic acid, and 1 μL was spotted onto a Bruker AnchorChip 800 μm with prespotted 2,5-dihydrobenzoic acid (Bruker; 20 mg/mL in 30% acetonitrile/0.1% TFA). A Bruker Ultraflex III MALDI-TOF mass spectrometer (Bremen) was used for mass spectrometry analysis in a reflector mode by collecting 2,000 shots per spot. Protein Calibration Standard I (Bruker) was used for calibration.
Cell migration assay
A ChemoTx 96-well plate with a 3-μm pore size (NeuroProbe) was coated with 20 μL of LM-511/521 (100 ng/mL) for 30 min. The supernatants were removed, and the membrane was allowed to dry for 30 min. The cells (5×104 cells/30 μL/well) were resuspended in a cell migration medium (SFEM; Stem Cell Technologies) and added to the upper side of the membrane. Three hundred microliters of a cell migration medium that contains 100 ng/mL of the undigested or digested chemokine CXCL12 were added to the lower chambers. About 5×104 cells were added to one lower chamber as a control for the maximum number of applied cells. After incubation for 16–20 h at 37°C in a humidified environment containing 5% CO2, the cells from the lower chamber were collected, and the pellets were directly transferred to −80°C. The cell migration was quantified by an analysis of DNA content in each sample using the CyQUANT® Kit from Invitrogen. The fluorescence of the maximum number of applied cells was set to 100%.
Stimulation of neutrophils with IL-8 and MIP-1α
Isolated neutrophils were resuspended in cold serum-free RPMI in a concentration of 1×106 cells/mL and cultured at 37°C/5% CO2 for 0, 10, or 30 min with 100 ng/mL IL-8 or MIP-1α or without stimulus. Supernatants were centrifuged at 2,100 rpm for 10 min to remove cell debris and then concentrated 20×by using 10-kDa size-exclusion ultrafiltration devices (Vivaspin 500; Sartorius Stedim). Until usage, the concentrated supernatants were stored at −20°C.
Immunoblotting
The concentrated cell culture supernatants were diluted in a DTT-containing sample buffer and run on 8% SDS polyacrylamide gels. Polyvinylidene difluoride (PVDF) membranes were used for blotting. After transfer, the membranes were blocked with Tris-buffered saline containing 0.1% Tween-20 and 5% skimmed milk powder. The membranes were incubated overnight at 4°C with the primary antibody against human MMP-8. Bound antibodies were detected with HRP-conjugated anti-mouse antibodies (Dako) and the chemiluminescence reagent Immobilion Western (Merck Millipore).
Statistical analysis
All values are expressed as mean±standard deviation. The statistical significance was determined by a two-tailed parametric t-test or one-way ANOVA using GraphPad Prism 4 software (Version 4.02). Differences were considered to be significant for P<0.05 (*).
Results
Expression of MMP-8 by human bone marrow-derived cells and peripheral blood neutrophils
The expression of the gelatinase MMP-9 and the collagenase MMP-8 was analyzed in situ by immunofluorescence staining of the bone marrow-cryostat sections as well as by intracellular FACS labeling of isolated bone marrow mononuclear cells (BMMNC) (Fig. 1). Prominent staining for MMP-8 and MMP-9 was also detected on individual cells or cell clusters within the bone marrow-cryostat sections (Fig. 1A, E). By merging the signals for CD15 (Fig. 1B, F) with the signals obtained for both MMPs, an almost complete overlap could be observed in these sections (Fig. 1C, G). A strong expression of MMP-8 or MMP-9 was found on the subpopulations of the CD15+ granulocytic cells by dual-color flow cytometry analyses (Fig. 1D, H).
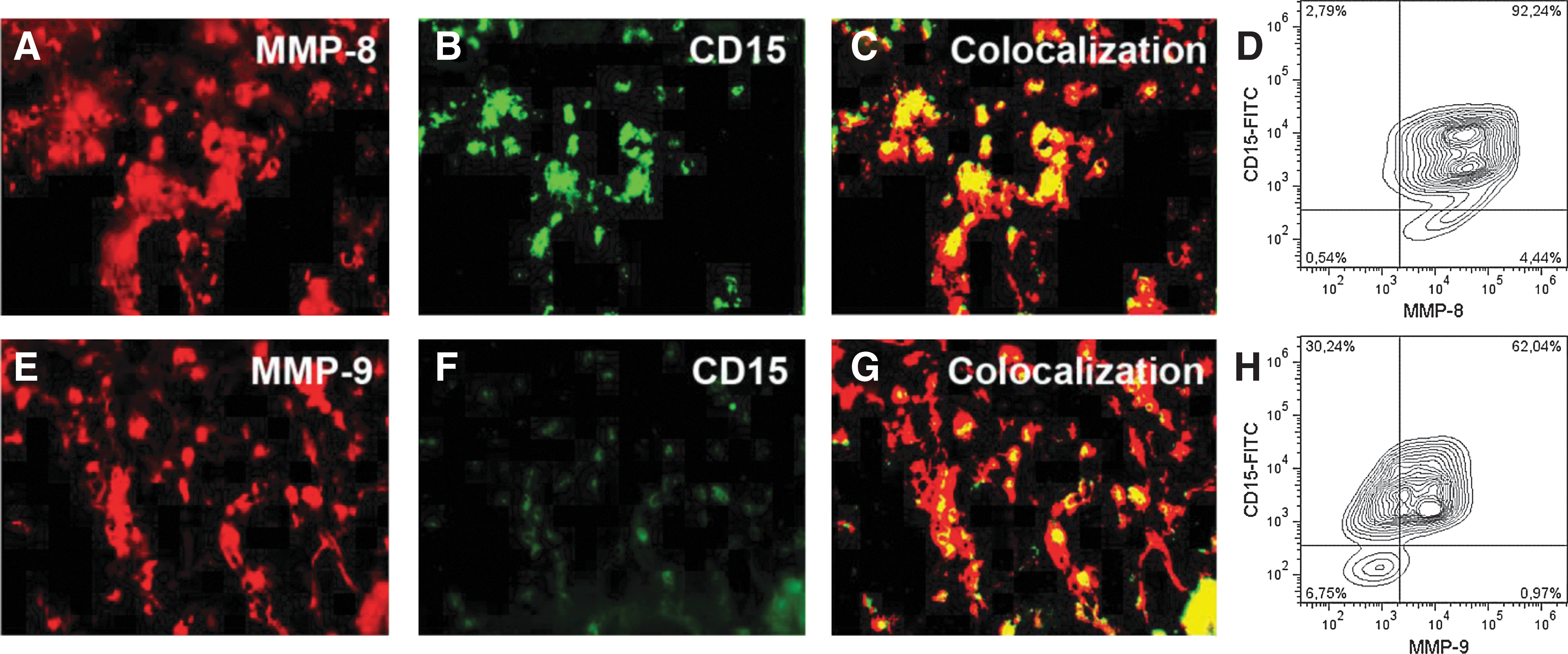
Expression of matrix metalloproteinase (MMP)-8 and MMP-9 in the human bone marrow. Double immunofluorescence staining of bone marrow-cryostat sections with antibodies against human MMP-8 and CD15
To study the influence of mobilizing chemokines in vitro, freshly isolated peripheral blood neutrophils were treated with IL-8 or MIP-1α. At defined time intervals, the cell culture supernatants were collected, concentrated, and subjected to immunoblot analyses. As shown in Supplementary Fig. S1 (Supplementary Data are available online at
Release of MMP-8 during G-CSF-induced mobilization and in UCB
To detect a potential release of MMPs in vivo, serum samples were analyzed during cytokine-induced hematopoietic stem cell mobilization (Fig. 2). The MMP-8 serum levels of 4 healthy donors before and during administration of G-CSF were quantified by an MMP-8-specific ELISA. Compared to baseline levels (day 0), a significant increase of MMP-8 was detected in all serum samples that were taken during cytokine treatment (Fig. 2A). The dramatic increase of MMP-8 serum concentrations during G-CSF treatment was accompanied by an increase in the number of CD34+ cells in the peripheral blood.
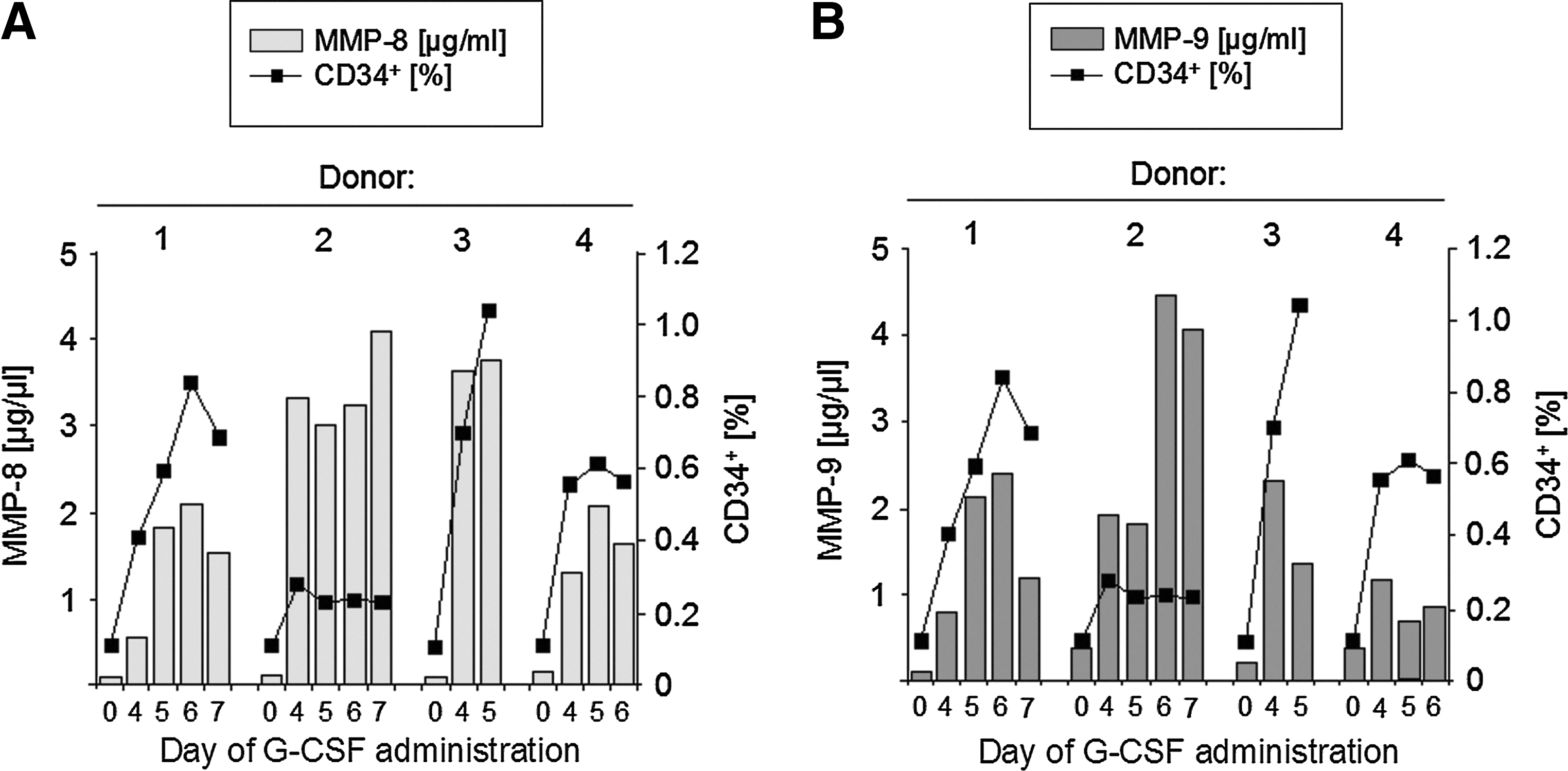
Quantification of MMP serum levels during granulocyte colony-stimulating factor (G-CSF) treatment.
We also quantified the MMP-9 serum concentrations of the 4 donors during G-CSF mobilization with an MMP-9-specific ELISA. Without cytokine treatment, MMP-9 serum levels were found to be very low (<0.4 μg/mL). A drastic increase up to 4 μg/mL could be observed during administration of G-CSF (Fig. 2B). Again, in accordance with increased MMP-9 serum levels, an elevated number of mobilized CD34+ cells could be observed (Fig. 2B), indicating an involvement of the MMPs, MMP-8 and MMP-9, in HSPC mobilization.
Since UCB contains an elevated number of circulating CD34+ cells too, the serum concentrations of MMP-8 and MMP-9 were analyzed by ELISA in the UCB sera and compared to the normal peripheral blood sera. Although the observed increase in MMP concentration was not as high as during cytokine-induced stem cell mobilization, a more than 10-fold increase for MMP-8 could still be detected in the UCB sera (Fig. 3A). A zymographic analysis confirmed the data obtained by ELISA for MMP-9 (Fig. 3B). Here the amount of MMP-9 in normal blood seemed to be under the detection limit, but strong signals for MMP-9 can be detected in the UCB sera.
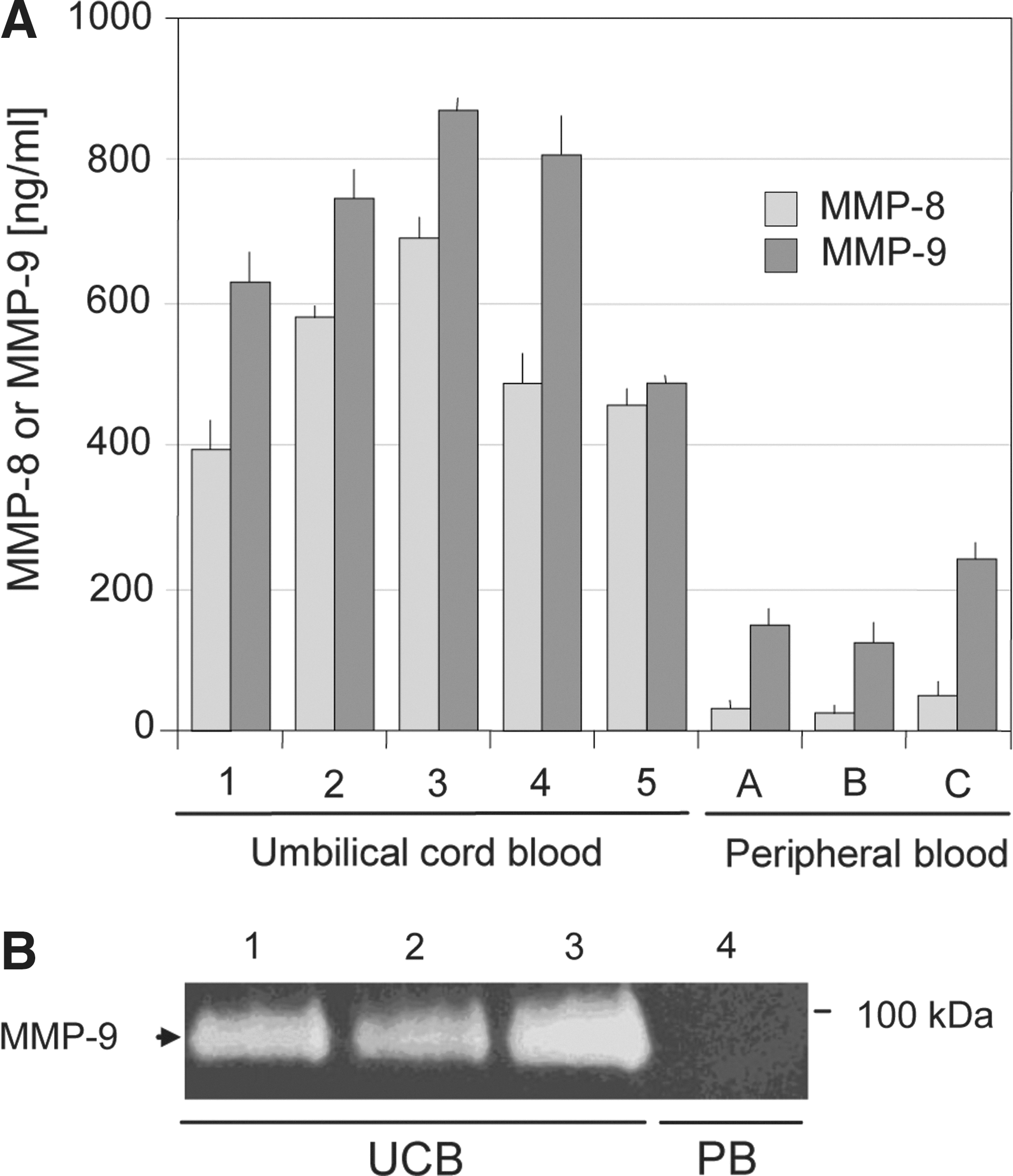
ELISA and zymographic analysis of gelatinolytic activities in umbilical cord blood (UCB) and normal blood samples.
Activated MMP-8 strongly influences HSPC cell adhesion
Cell binding of HSPC to isolated osteoblasts was analyzed in the absence or presence of activated MMPs. Cord blood-derived CD34+ HSPC strongly attached to adherent primary osteoblasts (Fig. 4A, control). Addition of activated MMP-8 or MMP-9 to the bound cells drastically reduced cell adhesion of HSPC to osteoblasts (Fig. 4A). The observed reduction of HSPC binding was quantified by using BCECF dye-labeled HSPC (Fig. 4B). Addition of nonactivated MMP-8 and MMP-9 to the pre-existing adhesive interactions did not interfere with cell binding, but for activated MMP-8, a reduction of cell adhesion of up to 76% was observed. MMP-9 unveils a less-intense effect of 34% decreased cell binding compared to control. A preincubation of HSPC with activated MMP-8 or MMP-9 also led to a drastic inhibition of HSPC binding to primary osteoblasts (Supplementary Fig. S2). Again, this effect could not be observed with nonactivated proteases.
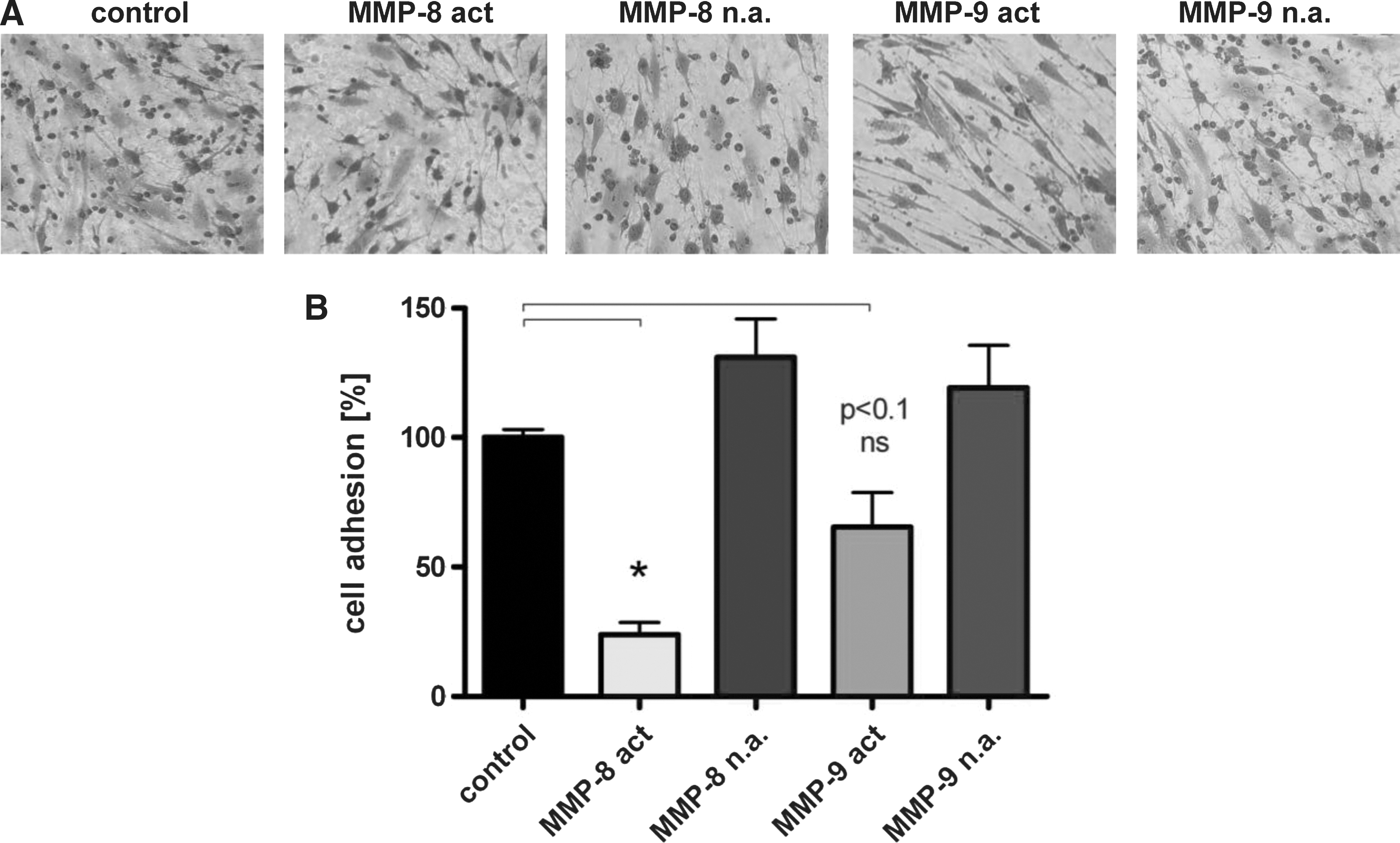
Cell–cell interactions between hematopoietic stem and progenitor cells (HSPC) and osteoblasts.
Processing through MMP-8 of the ECM component LM-511
Interaction of HSPC in the endosteal stem cell niche is based on the cell–cell and cell–matrix interactions. Therefore, a reduced binding of HSPC to the primary osteoblasts under the influence of activated MMPs can be elicited by the degradation of adhesive components known to be secreted by osteoblasts in the niche. Hematopoietic progenitor cells can interact with ECM molecules such as the LM isoform LM-511, which is an essential part of the ECM of the human bone marrow [32 –34]). CD34+ HSPC adhered to LM-511 immobilized on a cell culture dish (Fig. 5A). Unexpectedly, proteolytic digestion of LM-511 with MMP-8 had no influence on this interaction. Degradation kinetic studies for 0–24 h revealed that MMP-8 actively and efficiently degraded LM-511, whereas MMP-9 did not (Fig. 5B). The original LM-511 substrate already showed a double band for the LM-α5 chain. Within the first 4 h of MMP-8 digestion, the upper band of the LM-α5 chain disappeared with a simultaneously enhanced staining signal for the second band (LM-α5 processed; Fig. 5B). However, a new band of about 250 kDa became visible already after 0.5 h of the degradation process. This 250-kDa product is clearly larger than the LM β1- and γ1-chains, and therefore is supposed to be a degradation product of the LM-α5 chain. It was stable over 24 h, and no further processing could be perceived. For the LM β1- and γ1-chains, we detected degradation products with molecular masses of around 180 kDa. Because a clear degradation process leading to stable products was observed for all 3 LM chains, we further analyzed the composition of the single-degraded chains in more detail by peptide mass-fingerprint analysis. Four peptides that represent distinct domains of the LM-α5 chain (Table 1) were selected. In contrast to the full length of the LM-α5 chain, peptides of the C-terminal globular domains LG4 and LG5 were missing in the lower band of the LM-α5 chain (peptide 4), suggesting that MMP-8 is involved in the processing of this part of the LM-α5 chain (Fig. 5C, step I). The analysis of the peptides of the 250-kDa fragment revealed that the peptides of a larger part of the N-terminus (peptide 1) were missing, disclosing this domain as the second processing area for MMP-8 (Fig. 5C, step II). However, the analysis of the 250-kDa fragment also showed that there was no further processing of the C-terminal end. Peptides of the globular domains LG1–3 (peptide 3) that are important for cell adhesion [35] can still be found, explaining the nonimpaired cell adhesion of CD34+ HSPC to MMP-8-digested LM-511. The reduced cell adhesion of HSPC to pOBs under the influence of MMP-8 and MMP-9 (Fig. 4) has to be caused by the degradation of other ECM components or adhesion molecules.
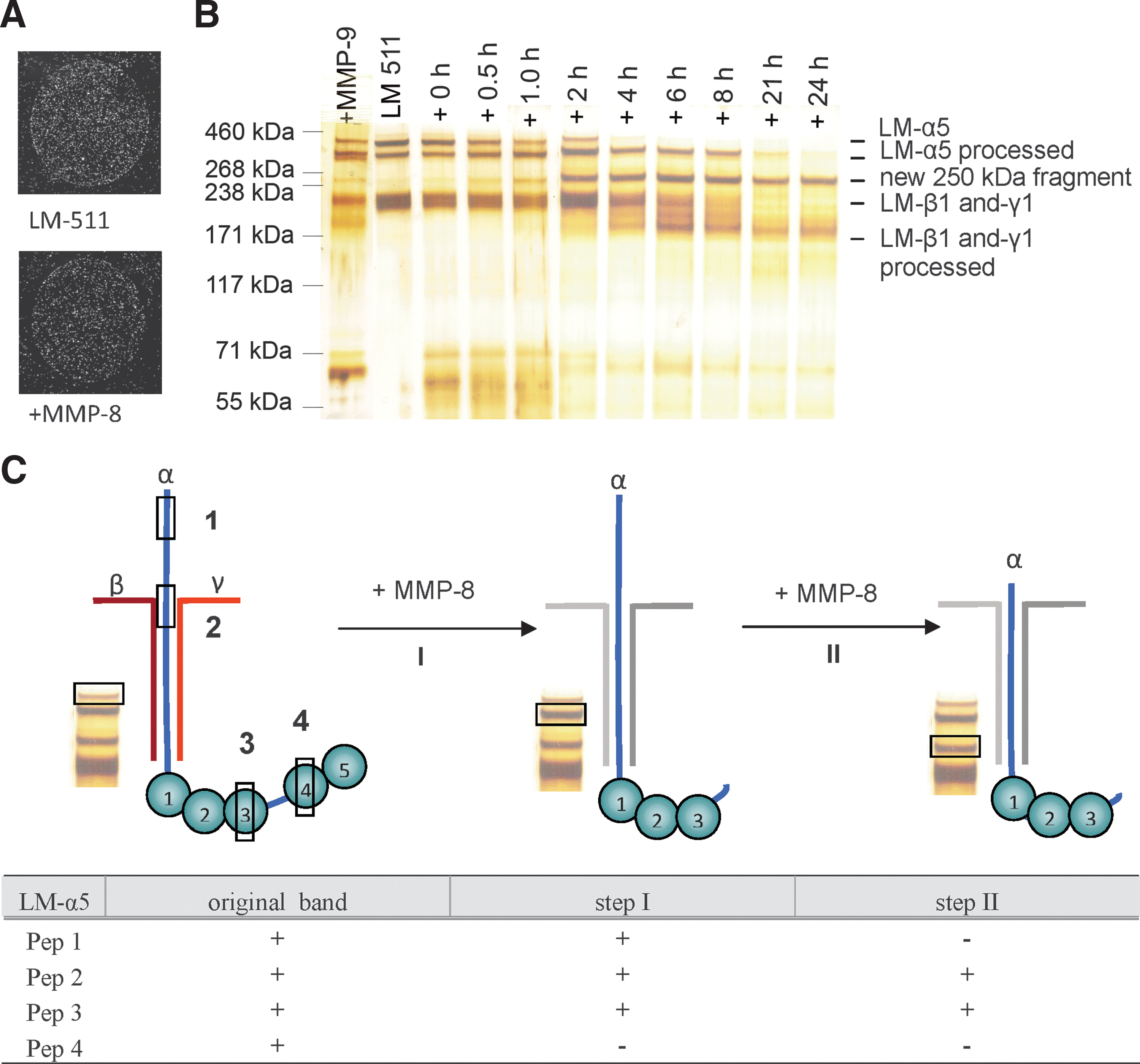
Adhesion of the HSPC to MMP-digested ECM components.
MMP-8 affects the CXCL12-dependent migration of HSPC
Lodgment of HSPC in their niches is strongly influenced by the chemokine CXCL12, which can be proteolytically processed by activated MMP-9 to thereby affect the adhesive and migratory properties of HSPC [36,37]. The collagenase MMP-8 was tested for its ability to degrade CXCL12 by MALDI-TOF mass spectrometry (Fig. 6A). CXCL12 (amino acid sequence 1–68) incubated only with an assay buffer for 24 h exhibits a peak at m/z 7968 (Fig. 6A; i). Throughout the incubation with activated MMP-8 for 2, 8, and 24 h, a second peak at m/z 7644 arose in addition to the original peak in a time-dependent manner (Fig. 6A; ii–iv). The difference of 324 kDa corresponds to the amino acid sequence KPV removed from the N-terminus of CXCL12. As a positive control for CXCL12 degradation, activated MMP-9 was used, resulting in a peak at m/z 7969.2 corresponding to the unmodified substrate and a second product at m/z 7557.9 (Fig. 6A; v). The amino acid sequence KPVS, which is known to be processed by activated MMP-9 at the N-terminus, matched to the observed mass difference of 411.3 kDa measured by the MALDI-TOF analysis. The functional relevance of these findings was tested by performing a ChemoTx-migration assay with CD34+ HSPC. The cells were allowed to actively migrate toward a CXCL12-containing medium through a 3-μm pore-size membrane. Sixty percent of the cells migrated to the lower chamber within 20 h when nonprocessed CXCL12 was used as a stimulus. Processing of CXCL12 by MMP-8 or MMP-9 led to a significantly reduced cell migration of HSPC (Fig. 6B). MMP-8 and MMP-9 themselves did not demonstrate any chemotactic activity. These findings suggest that MMP-8 in cooperation with MMP-9 can influence the chemokine gradient during HSPC mobilization supporting migration out of the endosteal niche.
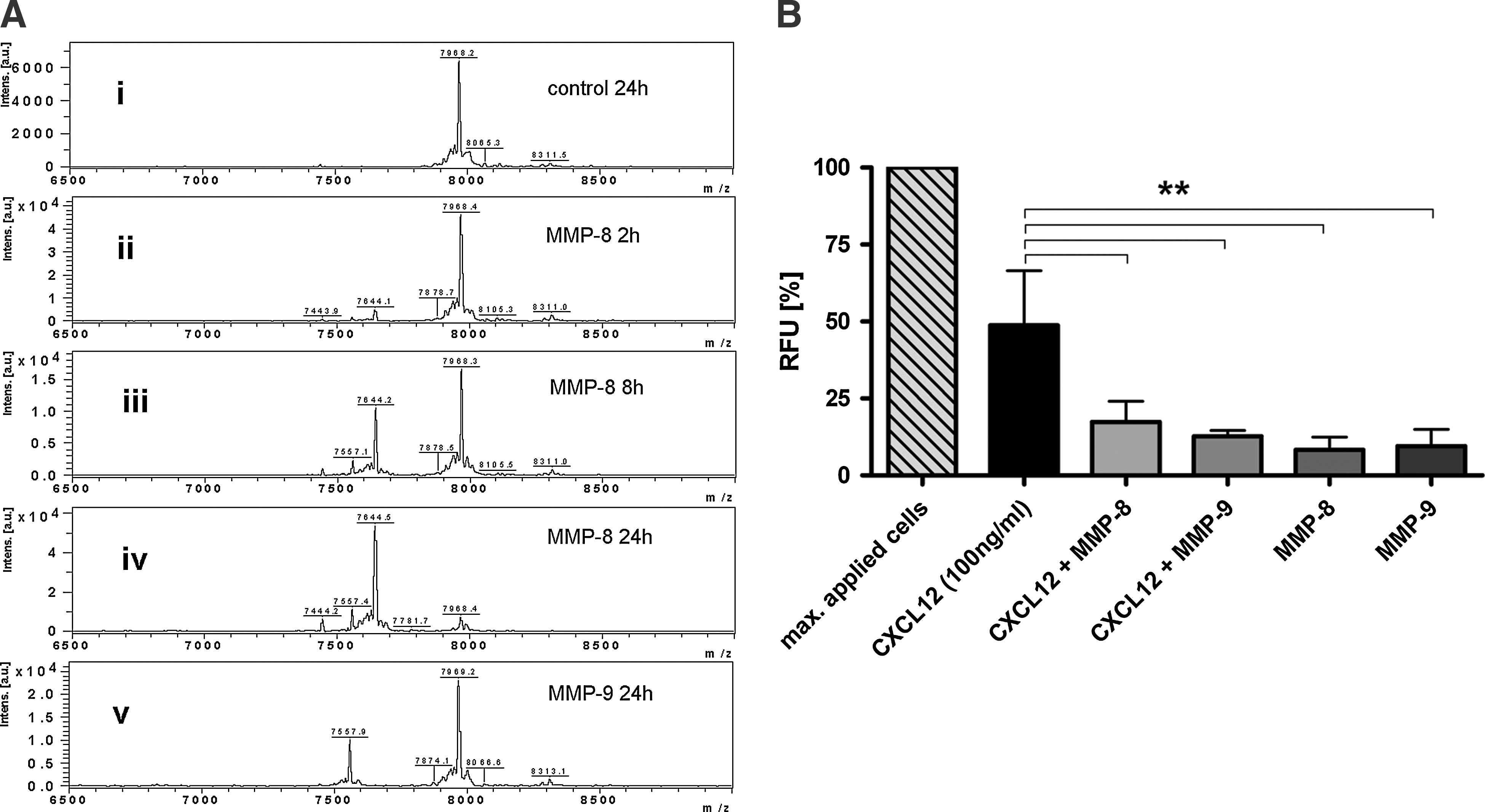
MALDI-TOF analysis of MMP-8-mediated CXCL12 degradation.
Discussion
In the present study, we introduce the collagenase MMP-8 as a new player in the proteolytic microenvironment of the human bone marrow during hematopoietic stem cell mobilization. MMP-8 is released during cytokine-induced mobilization as well as during physiological trafficking seen around birth. We elucidate the functional importance of MMP-8, since this ECM- and chemokine-degrading protease is able to digest niche-specific components such as LM-511 and CXCL12. Additionally, we disclose a role of MMP-8 in interfering with chemokine gradients influencing HSPC migration.
Several proteases such as neutrophil elastase, cathepsin G [11], and MMP-9 [38] have already been described to take part in the process of induced stem cell mobilization. However, studies with protease-deficient knockout mice also revealed that individual proteases alone do not account for the observed mobilization of HSPC [18], indicating that additional proteases such as MMP-8 might participate in this process. MMP-8-deficient mice do not show apparent abnormalities during the development or in adult life, but they are more susceptible to skin cancer [39]. Whether MMP-8-null mice suffer from any problems in stem cell mobilization is so far unknown. Elevated protein concentrations of MMP-8 were found in blood serum of the G-CSF-mobilized donors. We also confirmed elevated blood serum concentrations of the gelatinase MMP-9 upon G-CSF mobilization in healthy donors, which was reported earlier by Levesque et al. [11] and Carstanjen and coworkers [13].
During the last trimester of pregnancy, an increased number of CD34+ HSPC are mobilized from the fetal liver and can be found in the circulating blood, including UCB. ELISA analyses showed that, compared to normal blood serum, MMP-8 concentration was strongly enhanced in the cord blood serum. A similar elevated concentration of MMP-9 could be observed in cord blood serum samples.
MMP-8, which is mainly produced by neutrophils, is known to be secreted as an inactive zymogen that needs to be activated before it gains its proteolytic function [40]. Several proteases, including MT1-MMP [41], are used for this activation. Immunofluorescence and FACS staining show that MMP-8 expressed by CD15+ BMMNC is a newly identified protease of the proteolytic microenvironment of the human bone marrow. During inflammation, the levels of various cytokines are elevated [42]. It has been reported that inflammatory cytokines such as MIP-1α and IL-8 have a stimulatory influence on immune cells, especially neutrophils, leading to the secretion of MMP-9 by these cells [6,27]. During inflammation, an elevated number of cycling CD34+ cells can be observed, indicating stem cell mobilization as a stress response. Our studies showed that neutrophils, either nonstimulated or stimulated with MIP-1α or IL-8, strongly secret MMP-8 in an in vitro culture. Isolated primary osteoblasts, which are an important component of the endosteal stem cell niche, also express activated MMP-8 and MMP-9 (C. Steinl, M. Essl, and G. Klein, unpublished observation). Recent publications highlighted a role of the complement cascade in hematopoietic stem cell mobilization [43]. Upon stimulation of PMN with the complement cascade protein C5, the secreted gelatinases MMP-2 and MMP-9 and the membrane-bound MT1-MMP have been reported to be activated [44], and hereafter influence HSPC mobilization. Whether MMP-8 expression and activation are likewise provoked by this mechanism will be an important issue of future studies.
MMP-8 is best known for its ECM-degrading properties [40]. Since HSPC are located close to osteoblasts and stromal cells with their secreted ECM in the endosteal stem cell niche, a major issue of the present study was to gather an insight on whether MMP-8 plays a role in these adhesive interactions. In vitro cell adhesion assays using primary osteoblasts and HSPC unveiled that MMP-8 and MMP-9 have a strong influence on HSPC already attached to osteoblasts. This effect is most probably due to the degradation of the cell–cell and/or cell–matrix contacts by the activated proteases. Preincubation of HSPC with activated MMP-8 and MMP-9 presents an even stronger reduction of cell binding to osteoblasts. This effect could be explained by a degradation of important, but currently unidentified, cell adhesion molecules on the surface of HSPC and should be the subject of future studies. Mobilized HSPC that get in contact with elevated concentrations of these proteases are less adhesive and hence could move to the vascular niche and the blood circulation.
To our knowledge, MMP-8 has no clearly defined spectrum of substrate specificity. LM processing has been previously reported for the isoform LM-332, which is not found in the human bone marrow [45 –47]. Here we show for the first time that MMP-8 can also degrade the isoform LM-511, which is strongly expressed by human osteoblasts (C. Steinl, M. Essl, and G. Klein, unpublished observation). During this degradation, there is an incomplete breakdown of the single LM chains, since stable products were visible after 24 h of digestion. Peptide mass-fingerprint analysis suggests that larger parts of the N-terminus as well as the C-terminal LG4 and LG5 domains are removed from the LM-α5 chain, but the remaining parts, which are important for the triple-helical tertiary structure and the integrin-binding domains LG-1–3 [48,49], are still intact. The N-terminus of the LM α-chains is important for the 3-dimensional organization of LMs in the basement membranes [50]. However, since bone-lining osteoblasts do not deposit a structured basement membrane, the exact organization of LM-511 in the bone marrow is so far unknown, and therefore the functional consequences of the degradation of this matrix component at the N-terminus is also unknown. However, we could show that the processed LM-511 is still adhesive for HSPC after the enzymatic treatment, since the cell-binding site of LM-511 is still intact.
During mobilization of HSPC, the CXCL12/CXCR4 axis is highly relevant for the guidance of HSPC among the bone marrow endosteal niche, the vascular niche, and the blood circulation. Proteases play a central role in the maintenance of the dynamic chemokine gradient. Neutrophil elastase, carboxypeptidase M [51], cathepsin G, cathepsin X [28], MT1-MMP [52,53], MMP-2, and MMP-9 can be subsumed as a group of proteases that have been shown to take part in digesting CXCL12 with the loss of the migratory activity as a consequence. The N-terminus of CXCL12 is especially important for the guiding nature of this chemokine. Cho et al. recently evaluated the diverse characteristics of missing amino acids (aa) at the N-terminus [38]. MMP-2 and MMP-9 are known to remove 4 aa, cathepsin G 5 aa, and the neutrophil elastase 3 aa. In contrast to the data obtained from mice [39] and humans [54], we observed by the MALDI-TOF analysis that MMP-8 specifically removed 3 aa of the N-terminus of CXCL12. The first 2 aa of the N-terminus are important for the migratory activity, whereas the following 6 aa support receptor binding to CXCR4 [55]. As a consequence, MMP-8 removal of 3 aa influences receptor binding and migratory activity, exhibiting an important role of MMP-8 upon the stated CXCL12 gradient in the bone marrow [56]. Our migration assays on HSPC with either unprocessed CXCL12 or MMP-8-processed CXCL12 confirmed these findings.
In summary, we provide strong evidence that the collagenase MMP-8, which can be released from neutrophils, can drastically reduce the cell–cell interactions of HSPC with osteoblasts. Elevated MMP-8 concentrations in blood serum either from G-CSF-mobilized donors or from cord blood samples strongly support a role of MMP-8 in stem cell trafficking. The proteolytic degradation of the important chemokine CXCL12 by MMP-8 indicates that MMP-8 is a part of a dynamic network of chemoattractant-degrading proteases involved in stem cell trafficking.
Footnotes
Acknowledgment
The authors would like to thank Linda Yan (Pennsylvania State University, State College) for critically reading the manuscript and Dr. Sandra Behrend for her role in the initial design and conduct of this study. This work was supported by a contract research Adulte Stammzellen II of the Baden-Württemberg Stiftung, Germany (grant No. P-LS-AS/HSPA8-13 and No. P-LS-ASII/23), and from the DFG graduate school 794.
Author Disclosure Statement
The authors reported no potential conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.