
Abstract
In regular culture conditions with leukemia inhibitory factor (LIF), the majority of mouse embryonic stem cells (mESCs) are maintained in a self-renewal stage; very few mESCs have differentiated morphology. When LIF is withdrawn, mESCs tend to differentiate; this differentiation process can be enhanced by the introduction of exogenous fibroblast growth factor (FGF). Here, we show that even in the presence of exogenous FGF1, mESCs can maintain self-renewal and expression of pluripotency markers in the presence of LIF. To elucidate the mechanism in which LIF dominates over the FGF1, extracellular signal-regulated kinase 1/2 (Erk1/2) signaling of mESCs cultured in a medium containing FGF1 or LIF/FGF1 was examined. The results demonstrate that Erk1/2 was activated by FGF1 in the absence of LIF; however, the FGF1-induced Erk1/2 phosphorylation was suppressed when LIF was introduced. Moreover, FGF1-Erk1/2 downregulation was inhibited by a signal transducer and activator of the transcription 3 (Stat3) inhibitor WP1066, suggesting that LIF-induced Stat3 activation plays an important role in the FGF1-Erk1/2 inhibition in mESCs. We further demonstrate that the binding affinity of phospho-Erk1/2 and Sprouty2 was increased via Stat3 activation. Binding of phospho-Erk1/2 and Sprouty2 blocks the activation of Erk1/2 signaling, thus inhibiting the downstream differentiation process in mESCs. Our findings demonstrate, for the first time, that LIF-induced Stat3 phosphorylation plays an important role in promoting the binding of phospho-Erk1/2 and Sprouty2, and thus inhibiting FGF-induced differentiation.
Introduction
In mESCs, the activation of extracellular signal-regulated kinase ½ (Erk1/2) signaling will promote differentiation and suppress proliferation [2]. When the fibroblast growth factor (FGF) binds to its receptor, FGFR, the growth factor receptor binding protein 2 (Grb2) will bind to the autophosphorylated receptor. This binding will in turn lead to subsequent phosphorylation of Raf, Mek1/2, and Erk1/2 [3,4]. The phosphorylation/activation of Erk1/2 signaling leads to the expression of transcription factors, such as Ets, c-Jun, c-Fos, and Elk [3]. The activation of Erk1/2 can also downregulate the expression levels of Tbx3 and Nanog, leading mESCs toward differentiation [5,6].
Sprouty2 was first identified in Drosophila as an antagonist of the FGF signaling that patterns apical branching of Drosophila airways [7]. Further study showed that overexpression of Sprouty2 would lead to the inhibition of FGF-induced Erk1/2 signaling [8 –10]. When the Sprouty2 protein was activated, it could bind with protein phosphatase 2A. Binding of Sprouty2 will lead to protein dephosphorylation, so Sprouty2 can act as the Erk1/2 inhibitor [11].
It has been proven that leukemia inhibitory factor (LIF)-induced Jak/signal transducer and activator of transcription 3 (Stat3) signaling is important to the maintenance of mESCs self-renewal [2,12 –14]. The LIF receptor is a heterodimeric receptor, including LIFR and gp130 [15]. The binding of LIF to LIFR and gp130 will lead to the activation of Stat3 signaling. Briefly, the Stat3 will be phosphorylated by tyrosine kinase Jak, then the phosphorylated Stat3 will form homodimers, translocate to the cell nucleus, and subsequently upregulate the expression of c-Myc [12,16] and suppressor of cytokine signaling 3 (SOCS3) [17]. LIF can also upregulate the expression of Klf4, Sox2, and Oct4, thus maintaining self-renewal of mESCs [5]. On the other hand, Jak will also lead to the phosphorylation of SH2 domain-containing tyrosine phosphatase 2 (SHP2) and its downstream Erk1/2 signaling. However, SOCS3 upregulated by LIF-induced Stat3 activation can bind to the gp130 Y757 and block the binding of SHP2 to gp130, thus inhibiting the subsequent Erk1/2 phosphorylation stimulated by LIF-gp130 [17 –19].
It has been reported that activation of Erk1/2 signaling could promote differentiation and suppress proliferation in mESCs [2]. Ying et al. also indicate that inhibition of Erk1/2 signaling using both the FGFR inhibitor and Erk1/2 inhibitor can maintain mESCs in an undifferentiated stage without LIF [20]. Together, these reports suggest that Erk1/2 suppression is important for blocking mESCs differentiation. Under regular culture conditions with LIF, mESCs were maintained in a self-renewal and pluripotent stage. However, under this culture condition, there are 2 ways for Erk1/2 signaling activation to induce differentiation: the binding of LIF to gp130 and the binding of endogenous FGF to FGFR [6]. The inhibition of LIF-gp130-induced Erk1/2 signaling through SOCS3 has been reported [17 –19]. However, whether the FGF-induced Erk1/2 phosphorylation is blocked in mESCs regular culture condition with LIF has not been reported. To clarify how mESCs react to FGF1 stimulation in the presence of LIF, signaling pathways of mESCs cultured in a medium containing both LIF and FGF1 were examined. Here we showed that LIF suppresses FGF1-induced Erk1/2 phosphorylation through the binding of Sprouty2 to phospho-Erk1/2.
Materials and Methods
Reagents
Recombinant human FGF1 was prepared in our laboratory, following the published protocol [21,22]. A synthetic inhibitor of Stat3 signaling, WP1066, was purchased from Merck. The Mouse ESGRO® leukemia inhibitory factor (LIF) was purchased from Millipore.
Cell culture
Sox1-eGFP knock-in mESCs (46C) obtained from Dr. Austin Smith (University of Cambridge, UK) [1] were cultured on a gelatin-coated dish in the Dulbecco's modified Eagle's medium (DMEM) (Gibco) with 10% knockout serum replacement (Gibco), 1% fetal bovine serum (FBS) (Gibco), and 1000 U/mL Mouse ESGRO® LIF. The culture condition has been described in detail previously [1].
The regulation of LIF and FGF1
The 46C mESCs were cultured in 2 different medium groups, including the basal medium with FGF1 (100 ng/mL) or LIF (1000 U/mL)/FGF1 (100 ng/mL). The Basal medium contains the DMEM, 10% knockout serum replacement, and 1% FBS. Cells were passaged every 2 days. After 2 passages (4 days), alkaline phosphatase (AP) staining and western blotting were performed to examine the pluripotency.
AP staining
The 46C mESCs were fixed with 4% paraformaldehyde. Then AP staining was performed using an Alkaline Phosphatase Detection Kit (Millipore) following the manufacturer's suggested protocol.
Western blotting analysis
ES cells were lysed in the RIPA buffer [50 mM Tris pH 7.5, 150 mM NaCl, 10 mM EDTA, 1% NP-40, 0.1% sodium dodecyl sulfate (SDS)] (Millipore) plus a cocktail of protease inhibitors (Merck). Denatured proteins were separated by 10% SDS-polyacrylamide gel electrophoresis (PAGE), and then transferred to polyvinylidene difluoride (PVDF) membranes (Amersham). Samples were detected with specific antibodies, including Oct4 (1:2000), Sox2 (1:2000), Erk1/2 (1:4000), phospho-Erk1/2 (1:2000), Stat3 (1:1000), and phospho-Stat3 Y705 (1:1000). The Oct4 antibody was purchased from Novus; The Sox2 antibody was purchased from Millipore; and Erk1/2, phospho-Erk1/2, Stat3, and phospho-Stat3 Y705 antibodies were purchased from Cell Signaling. Chemiluminescence of immunoreactive bands was detected using secondary horseradish peroxidase-conjugated antibodies (Millipore) and enhanced chemiluminescence (ECL) reagents (Millipore).
Signaling of LIF and FGF1
To analyze the phosphorylation state of Erk1/2 and Stat3 in 46C mESCs cultured in a medium containing FGF1 or LIF/FGF1, total proteins from both groups were collected at different time points. Protein lysates were analyzed by western blotting.
Stat3 signaling inhibition
To block the Stat3 phosphorylation, a Jak/Stat3 signaling inhibitor, WP1066 was introduced. After 1 h of WP1066 (5 μM) treatment, the culture condition was changed into medium containing WP1066/FGF1 or WP1066/LIF/FGF1. Cells were harvested and cell lysates were extracted at different time points (1, 2.5, 4, 6, 8, 10, and 12 h) and analyzed by western blotting.
Immunoprecipitation
The 46C mESCs protein lysates (250 μg) were incubated with the phospho-Erk1/2-specific antibody (Cell Signaling) for 4 h; then the protein A/G agarose beads (Santa Cruz) were added and incubated for another 4 h. All procedures were performed at 4°C. Samples were then washed with the RIPA buffer, and 20 μL of the sample buffer was added. After incubation at 100°C for 10 min, samples were collected, separated by 10% SDS-PAGE, and then transferred to PVDF membranes (Amersham). PVDF membrane-transferred samples were detected with the antibody specific for Sprouty2 (Santa Cruz).
Cell transfection
The wild-type or mutant Stat3 genes—including Stat3 Y705F pRc/CMV (Y705F) [23], Stat3 Flag pRc/CMV (WT-Flag) [24], and Stat3 pcDNA3 (WT) [25]—were obtained from Addgene (
Results
mESCs did not differentiate in the presence of LIF and FGF1
To test whether mESCs can differentiate in the presence of LIF and FGF1, 2 groups were designed. The 46C mESCs were cultured in FGF1 (100 ng/mL) with/without the LIF (1000 U/mL) medium. Cell morphology, AP staining, gene expression, and protein expression levels were evaluated during the experiment. After 4 days of culture, cells in the LIF/FGF1 group showed normal morphology (Fig. 1C), just like those in the control group (medium containing LIF, Fig. 1A). Their AP stainings were positive, with a purple color (Fig. 1B, D). However, in the FGF1 group, mESCs exhibited differentiated morphology and AP staining was negative, with a brown color (Fig. 1E, F).
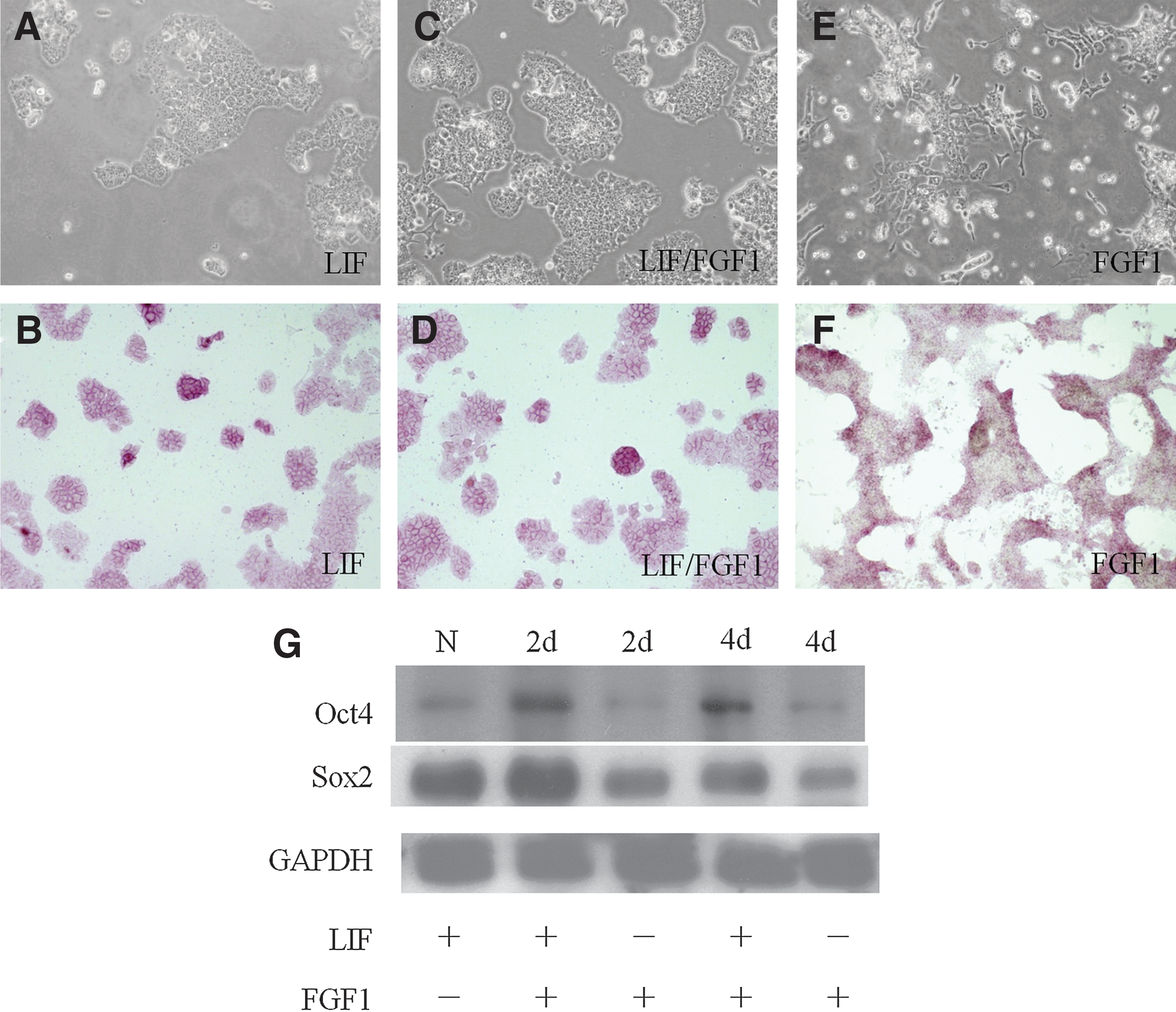
46C mouse embryonic stem cells (mESCs) did not differentiate in the presence of leukemia inhibitory factor (LIF) and fibroblast growth factor (FGF)1. The 46C mESCs were cultured in medium containing LIF
Gene expression levels of markers for pluripotency and 3 germ layers were evaluated by reverse transcription-polymerase chain reaction (RT-RCR) analysis. Our data showed that after 4 days of FGF1 treatment the expression level of Oct4 was downregulated, while the neuroectoderm marker Sox1 was upregulated in 46C mESCs. The cells cultured in the medium containing LIF and FGF1 still maintained the expression of Oct4, and the expression level was similar to that of the control group (data not shown).
The pluripotency-associated transcription factors, Oct4 and Sox2, were examined by western blotting. The LIF/FGF1 group maintained their protein expression of pluripotent markers, including Oct4 and Sox2, while the FGF1-treated group showed downregulated expression of Oct4 and Sox2 after 4 days of culture (Fig. 1G). The flow cytometry analysis also indicates that LIF/FGF1-treated 46C mESCs maintained a similar percentage of SSEA1 expression as a normal group cultured in the medium containing LIF, while the FGF1-treated group showed a 10% decrease in SSEA1 expression. The 46C mESCs cultured without LIF and FGF1 for 2 and 4 days also showed a slight decrease in SSEA1 expression (Supplementary Fig. 1; Supplementary Data are available online at
LIF-induced Stat3 signaling in 46C mESCs is not affected by FGF1
We further examined whether the signaling pathways of 46C mESCs are different when the cells were cultured in the medium containing either FGF1 or LIF/FGF1. It has been proven that LIF can stimulate the Jak/Stat3 signaling pathway and its downstream transcription factors, including c-Myc and SOCS3, in mESCs [17,26]. In this study, we cultured the 46C mESCs in the medium containing FGF1 or LIF/FGF1, and the protein extracts were analyzed by western blotting. It was found that Stat3 phosphorylation can be stimulated by LIF (Fig. 2A). The Stat3 phosphorylation stimulated by LIF was not downregulated when cells were cultured in the medium containing both LIF and FGF1 (Fig. 2A). On the other hand, the Stat3 phosphorylation was downregulated in the absence of LIF (Fig. 2B), suggesting that the Stat3 phosphorylation was affected by LIF, but not by FGF1.
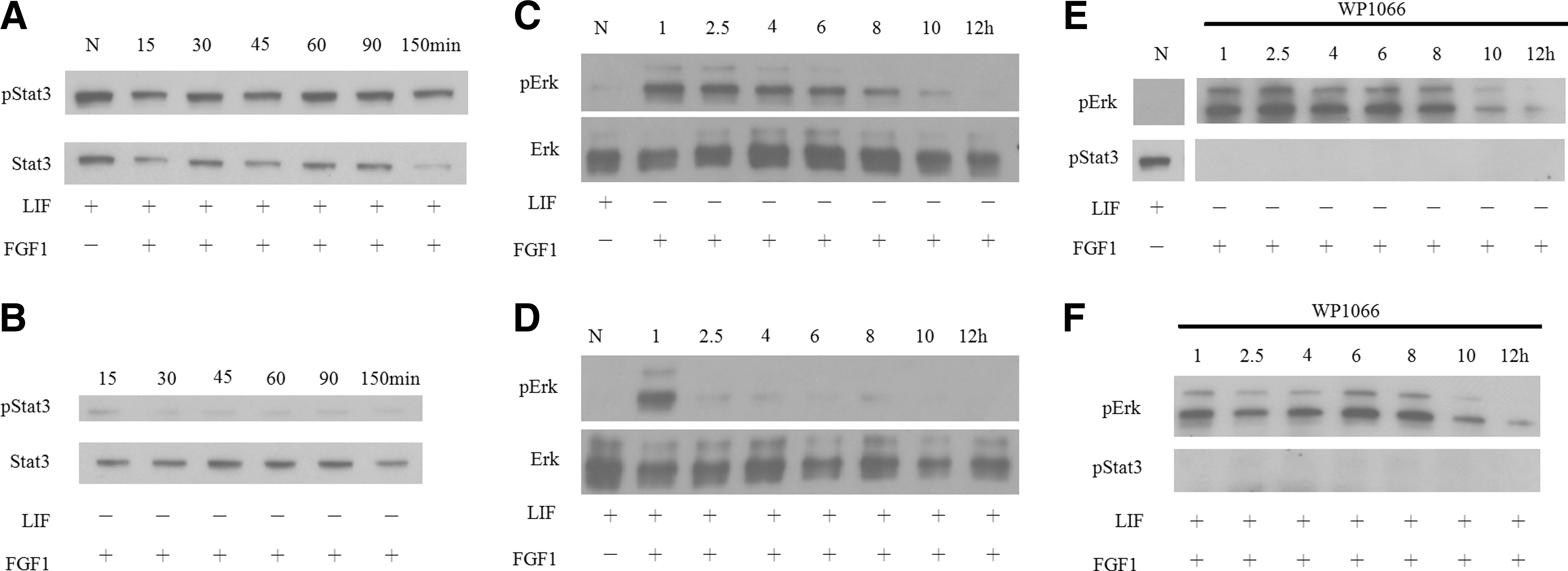
The LIF-induced signal transducer and activator of transcription 3 (Stat3) phosphorylation was not affected by FGF1, while FGF1-induced extracellular signal-regulated kinase 1/2 (Erk1/2) phosphorylation was downregulated by LIF-induced Stat3 signaling in 46C mESCs. The 46C mESCs were cultured in medium containing LIF/FGF1
FGF1 upregulated the Erk1/2 phosphorylation in mESCs
Previous reports have shown that FGF1 stimulates the Erk1/2 signaling and its downstream transcription factors to promote differentiation in mESCs [26]. Western blotting was performed to examine the activation of Erk1/2 in 46C mESCs. We showed that the Erk1/2 phosphorylation was upregulated by FGF1 (Fig. 2C). In the presence of FGF1, the activated Erk1/2 signaling was sustained up to 10–12 h (Fig. 2C). Western blotting data revealed that the phosphorylation state of Erk1/2 and Stat3 in 46C mESCs was very similar to those of reports in other mESCs [26]. The phosphorylation levels of Erk1/2 in the 46 mESCs without the stimulation of LIF and FGF1 were also upregulated, but displaying a slight different kinetic time course (Supplementary Fig. 2).
LIF downregulated the FGF1-Erk1/2 phosphorylation through Stat3
The 46C mESCs were cultured in the LIF/FGF1-containing medium for 1, 2.5, 4, 6, 8, 10, and 12 h; then, total protein lysates were extracted. In the presence of LIF and FGF1, the FGF1-induced Erk1/2 phosphorylation was downregulated (Fig. 2D), while the Stat3 phosphorylation was maintained (Fig. 2A). The Erk1/2 phosphorylation lasted more than 4 h and started to reduce gradually after 6 h in FGF1-treated cells. In contrast, the phospho-Erk1/2 levels lasted for only 1 h in the LIF/FGF1-treated group (Fig. 2C, D). The phosphorylation of Erk1/2 in D3 mESCs was also evaluated. It was found that in the LIF/FGF1 treatment group, the phosphorylation of Erk1/2 was suppressed at a basal level, similar to the normal condition, while in the FGF1 group, the phosphorylation of Erk1/2 lasted for at least 4 h (Supplementary Fig. 3). This observation is similar to what we observed for 46C mESCs. These data suggested that in the presence of both LIF and FGF1, FGF1-induced Erk1/2 signaling was downregulated by LIF in at least 2 different types of mESCs.
We further explored why FGF1-Erk1/2 signaling was downregulated in the presence of LIF. To examine whether activation of Stat3 downregulated the FGF1-Erk1/2 signaling, the JAK/Stat3 inhibitor WP1066 was introduced [27,28]. Cells were cultured with WP1066 for 1 h, then shifted to the medium containing WP1066/FGF1 (Fig. 2E) or WP1066/LIF/FGF1 (Fig. 2F) and subsequently cultured for 1, 2.5, 4, 6, 8, 10, and 12 h. The phosphorylated Stat3 was inhibited after 1 h of WP1066 exposure even under the stimulation of LIF (Fig. 2F). This result also showed that in the presence of LIF and FGF1, inactivating Stat3 phosphorylation will relieve the downregulation of Erk1/2 (Fig. 2E). Erk1/2 phosphorylation exhibited similar patterns in the FGF1, WP1066/FGF1, and WP1066/LIF/FGF1 groups (Fig. 2C, E, and F). The downregulation of Erk1/2 by LIF seemed to be rescued by blocking Stat3 activation through WP1066. These data revealed that the FGF1-induced Erk1/2 activation could be downregulated by LIF-induced Stat3 phosphorylation.
To further determine whether this mechanism is specific to FGF1, the phosphorylation levels of Erk1/2 in 46C mESCs treated with the epidermal growth factor (EGF) or LIF/EGF were also analyzed. Our data showed that, similar to the LIF/FGF1 treatment, the EGF-induced Erk1/2 phosphorylation was downregulated when mESCs were treated with both LIF and EGF. The Erk1/2 phosphorylation lasted more than 10 h in EGF-treated cells (Supplementary Fig. 4A). In contrast, the phospho-Erk1/2 levels lasted for only 2.5 h in the LIF/EGF-treated group (Supplementary Fig. 4B). We further showed that, in the presence of LIF and EGF, inactivating Stat3 phosphorylation using WP1066 will also relieve the downregulation of Erk1/2 (Supplementary Fig. 4C, D).
LIF increased the binding affinity of phospho-Erk1/2 to Sprouty2
Previous studies have reported that overexpression of Sprouty2 will lead to the downregulation of FGF-induced Erk1/2 signaling [8 –10]. Here we further determined whether the LIF-Stat3-mediated Erk1/2 inactivation was affected by the increased Sprouty2 expression. The Sprouty2 expression did not vary until 8 h of culturing in the medium containing either FGF1 or LIF/FGF1 (Fig. 3A). These results suggest that the Sprouty2 expression level is not a major factor for LIF-Stat3-mediated Erk1/2 inactivation in mESCs. We further examined whether LIF downregulated Erk1/2 through the binding of Sprouty2 by immunoprecipitation. The 46C mESCs were cultured in the medium containing FGF1 or LIF/FGF1 for 1 h, and their total cell lysates were immunoprecipitated with the phospho-Erk1/2-specific antibody. Combining pull-down and western blotting analyses, we detected the binding of phospho-Erk1/2 and Sprouty2 in the LIF/FGF1-treated cells, but not in the FGF1-treated group (Fig. 3B). This study was repeated in D3 mESCs. Our data showed that, in D3, the phosphorylation of Erk1/2 activated via FGF1 can be suppressed by LIF, as described above (Supplementary Fig. 3). The result also indicates that the binding affinity of phospho-Erk1/2 and Sprouty2 was increased by LIF in D3 mESCs (Supplementary Fig. 5). These data suggest that LIF increased the binding affinity of phospho-Erk1/2 and Sprouty2 in 2 different mESCs.
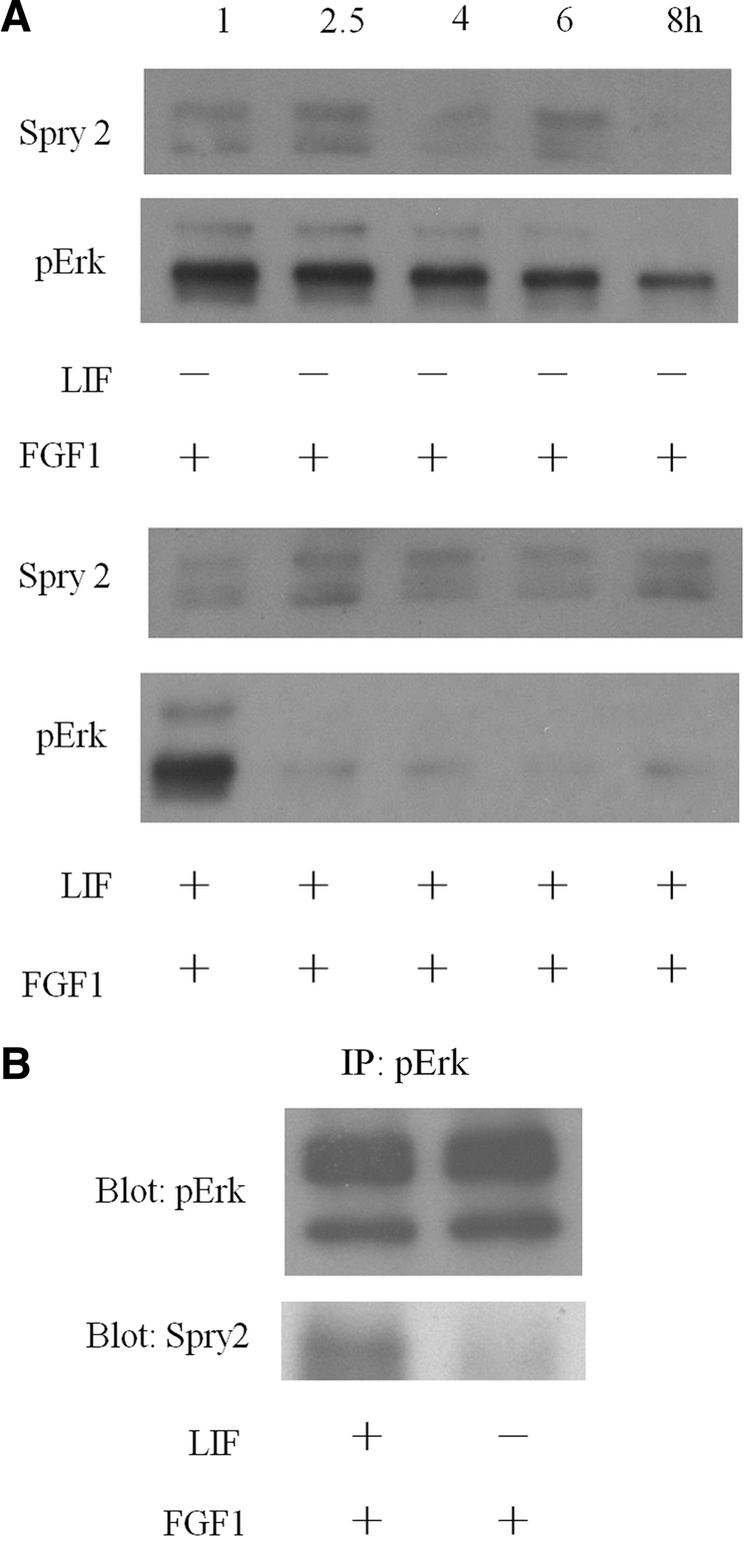
LIF increases the binding affinity of phospho-Erk1/2 and Sprouty2 (Spry2) in 46C mESCs. Protein extracts from mESCs cultured in medium containing FGF1 or LIF/FGF1 on different time points (1, 2.5, 4, 6, and 8 h) were analyzed by western blotting using Spry2-specific antibody
Stat3 phosphorylation contributed to the binding of phospho-Erk1/2 to Sprouty2
To determine whether LIF-induced Stat3 activation can affect the binding of phospho-Erk1/2 and Sprouty2, we examined the binding of phospho-Erk1/2 and Sprouty2 in the presence of a Stat3 inhibitor, WP1066. The 46C mESCs were treated with WP1066 for 1 h to inhibit the Stat3 phosphorylation. After 1 h of inhibition, the conditions were changed to the medium containing WP1066/FGF1 or WP1066/LIF/FGF1 for another hour. Total protein lysates were extracted after 1 h of incubation, after which immunoprecipitation was performed. The binding of phospho-Erk1/2 and Sprouty2 was not found in either the WP1066/FGF1 or the WP1066/LIF/FGF1 medium groups (Fig. 4A). In Fig. 3B, we showed that in the presence of LIF, binding of phospho-Erk1/2 and Sprouty2 could be observed; however, this binding was blocked by WP1066 (Fig. 4A). These data together suggest that LIF enhanced the binding affinity of phospho-Erk1/2 and Sprouty2 through the activation of Stat3 signaling.
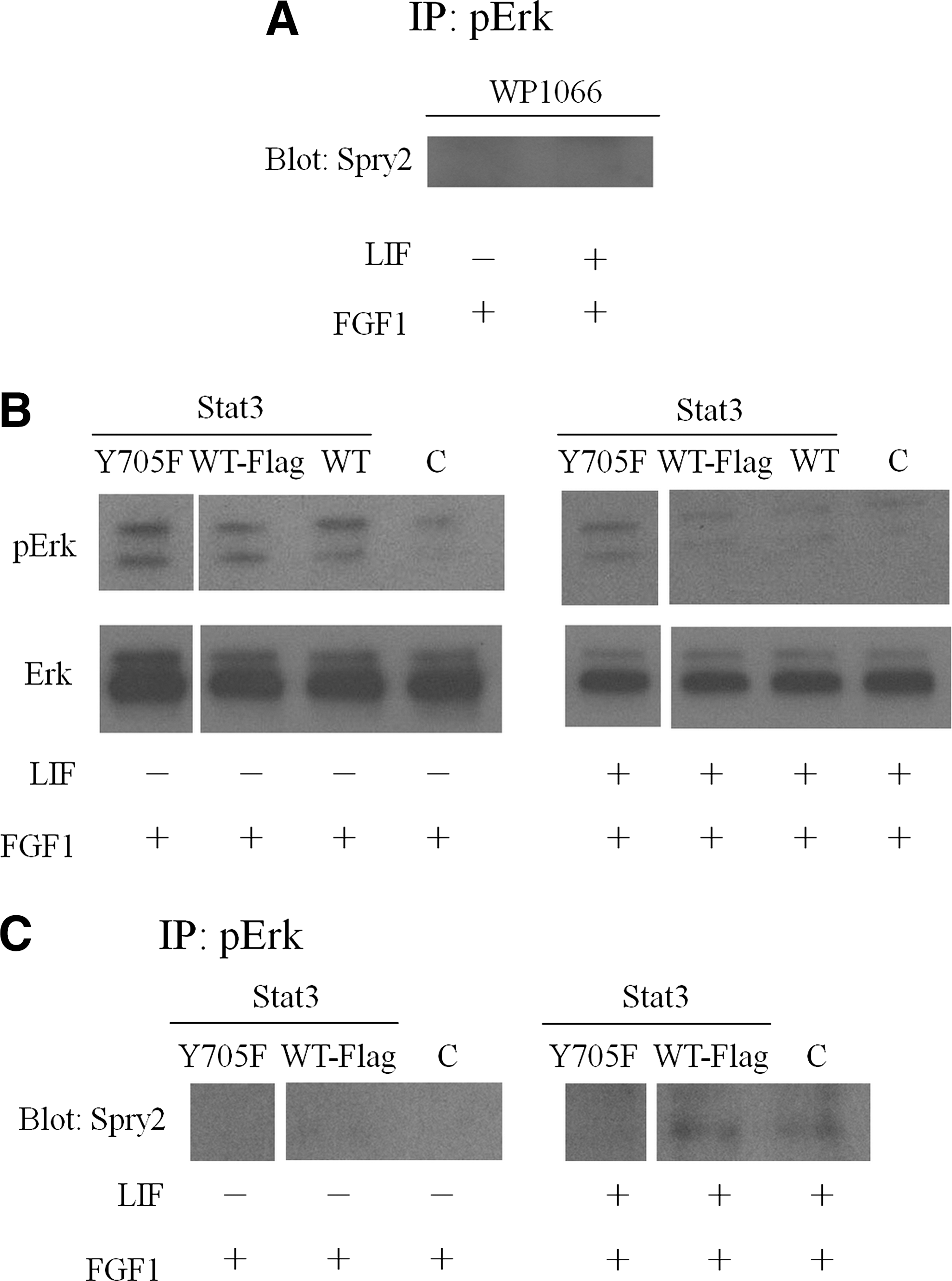
Stat3 phosphorylation contributed to the binding of phospho-Erk and Spry2. Stat3 inhibitor, WP1066, was used to block Stat3 phosphorylation in 46C mESCs. The cell lysates were immunoprecipitated with phospho-Erk1/2-specific antibody and the immunoprecipitates were analyzed by western blotting using Spry2-specific antibody
To further confirm these results, 3 wild-type or mutant Stat3 plasmids—Stat3 Y705F pRc/CMV (Y705F) [23], Stat3 Flag pRc/CMV (WT-Flag) [24], and Stat3 pcDNA3 (WT) [25]—were transfected into 46C mESCs, respectively. At 24 h after transfection, the media were changed into the FGF1 or LIF/FGF1 medium and then incubated for another 1 h. Total protein lysates were extracted for western blotting and immunoprecipitation analysis. The western blotting results revealed that the Y705F-transfected group showed no phospho-Erk1/2 downregulation in the LIF/FGF1 medium group, while in the control (no transfection) or wild-type Stat3 (WT and WT-Flag)-transfected group, LIF can still downregulate the FGF1-induced Erk1/2 phosphorylation, showing that nonphosphorylated Stat3 lost the function of Erk1/2 downregulation (Fig. 4B). It was also found that in the immunoprecipitation study, the binding of phospho-Erk1/2 and Sprout2 was not observed in the Y705F-transfected group. However, in the control and WT-Flag plasmid-transfected groups, the binding of phospho-Erk1/2 and Sprouty2 could still be observed (Fig. 4C). These data reveal that LIF-induced Stat3 phosphorylation at tyrosine 705 could promote the binding of phospho-Erk1/2 and Sprouty2, and thus blocking the downstream differentiation in 46C mESCs.
Discussion
The feeder-free culture system and the GFP-tagged neural progenitor cells make the 46C mESCs an ideal model for exploring signals of developmental factors on the pluripotency and cell differentiation. In this study, we found that exogenous FGF1 can efficiently drive the 46C cells differentiation only in the absence of LIF. The LIF/Stat3 signaling not only sustains the pluripotent gene expression, but also dominantly suppresses the differentiation signal elicited by FGF (Figs. 1, 2) or other growth factors, such as EGF (Supplementary Fig. 4).
To clarify the cross talk of LIF and FGF1 signaling, we first showed that the FGF1-induced Erk1/2 signaling was downregulated, while the LIF-induced Stat3 signaling was still maintained in both LIF- and FGF1-treated mESCs (Fig. 2). Our results here showed that specifically interrupting Stat3 phosphorylation by WP1066 rescued the Erk1/2 phosphorylation in the LIF/FGF1 culturing condition (Fig. 2), indicating that Stat3 activation is critical in LIF-mediated inhibition of FGF1-Erk1/2 signaling. Similar studies were also performed using EGF and LIF. Our data showed that LIF-induced Stat3 signaling can also override the EGF-induced Erk1/2 phosphorylation, suggesting that the inhibition of FGF1- or EGF-induced phospho-Erk1/2 by LIF-induced Stat3 phosphorylation/activation may be through similar mechanisms. It has been reported that when the FGF1 and other growth factors bind to their respective receptors, Erk1/2 signaling would be activated through Grb2, Ras, Raf, and Mek1/2 signaling cascade [26]. Since Erk1/2 activation via EGF is through a similar signaling cascade, we speculated that they would also be suppressed by LIF/Stat3. Our observations of the mESCs treated in the presence of LIF/EGF with the Stat3 inhibitor WP1066 (Supplementary Fig. 4) confirmed such a speculation.
Using immunoprecipitation western blot assay, we further revealed that Sprouty2 can bind to phospho-Erk1/2 only in the presence of LIF, indicating that LIF increased the binding affinity of Sprouty2 to phospho-Erk1/2 (Fig. 3 and Supplementary Fig. 5). This behavior was observed in both 46C and D3 mESCs, suggesting that it is likely a common phenomenon for mESCs, but not an anomaly for a particular type of mESC. In addition, using genetic and pharmacological inhibitors of Stat3 further confirmed that LIF-induced Stat3 phosphorylation was essential for the interaction of phospho-Erk1/2 and Sprouty2.
SOCS3 is important to self-renewal of mESCs and inhibition of FGF-Erk1/2 signaling [18]. Especially, SOCS3 was shown to bind to the LIF receptor and, thus, mediate the LIF-gp130-induced Erk1/2 phosphorylation [17,19]. However, the LIF-mediated inhibition of mESC differentiation is dependent on more than just SOCS3, because there were half SOCS3−/− mESCs still maintained in a self-renewal state in the presence of LIF [18]. Here, we speculate that Sprouty2 is an alternative route for the Erk1/2 inhibition in the LIF-treated mESCs. Downregulating both Spouty2 and SOCS3 may completely abrogate the self-renewal effect of LIF and additionally promote mESC differentiation.
Although the interaction between Stat3 and Sprouty2 requires further investigation, this Stat3-dependent Erk inhibition is notably not ubiquitous in mammalian cells. In several cancer cell lines—such as multiple myeloma, chronic lymphocytic leukemia, gastric cancer, lung cancer, and laryngeal carcinoma—both activated Stat3 and Erk1/2 coexisted for sustaining the cell proliferating [29 –34]. In addition, activation of Stat3 and Erk1/2 are required during neuronal differentiation of mESCs [35 –40]. However, we demonstrated that activated Stat3 signaling could suppress the Erk1/2 activity by recruiting the Sprouty2 to inhibit the differentiation process, indicating that activation of Stat3 and Erk1/2 is antagonistic in undifferentiated mESCs.
Our study provides an explanation for how LIF downregulates Erk1/2 phosphorylation. Binding of phospho-Erk1/2 and Sprouty2 inhibits Erk1/2 activation, thus blocking the downstream differentiation process. In other words, FGF1-induced phosphorylation of Erk1/2 is downregulated by LIF, and this downregulation is blocked when the Stat3 inhibitor is introduced. Taken together, these results provide a basis for how mESCs maintain in an undifferentiated stage under the stimulation of either autocrine or exogenous FGFs. Our finding demonstrates, for the first time, that LIF-induced Stat3 phosphorylation plays an important role in promoting the binding of phospho-Erk1/2 and Sprouty2, and thus inhibiting the FGF-induced differentiation.
Conclusion
The cross talk of FGF1-Erk1/2 signaling and LIF-Stat3 signaling was studied. Our results show that LIF-stimulated Stat3 phosphorylation could induce the downregulation of FGF1-induced Erk1/2 signaling through increasing the binding affinity of phospho-Erk1/2 and Sprouty2. This study provides an explanation for how LIF blocks the Erk1/2 signaling that is induced by either autocrine or exogenous FGFs in 46C mESCs. As shown in Fig. 5, mESCs differentiate under the stimulation of FGF1-induced Erk1/2 signaling; however, this differentiation process was blocked by LIF-induced Stat3 signaling due to the increased binding affinity between phospho-Erk1/2 and Sprouty2. The binding between phospho-Erk1/2 and Sprouty2 then resulted in the dephosphorylation of phospho-Erk1/2 and the concomitant inhibition of the differentiation process. Thus, we demonstrated that LIF serves a dual role in 46C mESCs, in not only the maintenance of self-renewal, but also the inhibition of differentiation mediated by FGF (Fig. 5).
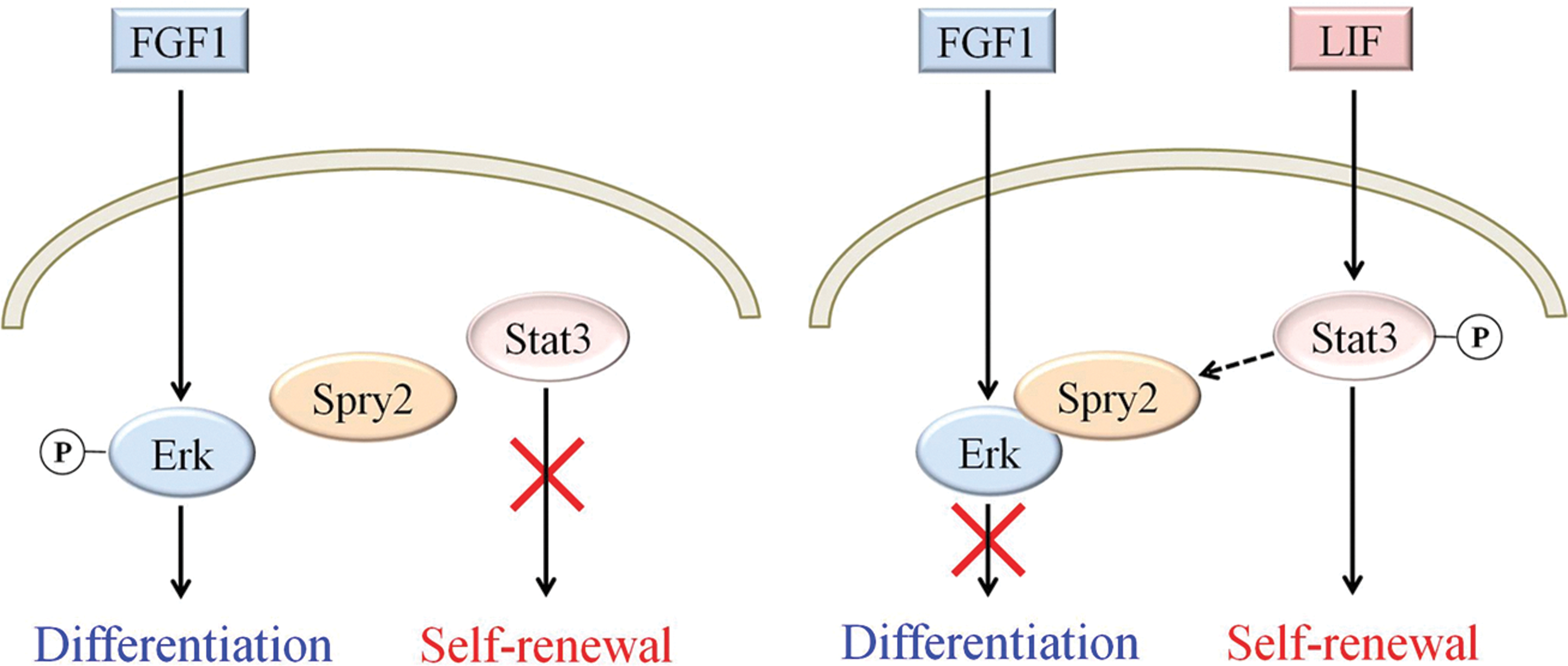
Cross talk of LIF-Stat3 signaling and FGF1-Erk1/2 signaling pathways in mESCs. Under the stimulation of FGF1, Erk1/2 signaling is activated, thus activating the differentiation process. However, this differentiation process would be inhibited through increased binding affinity of phospho-Erk1/2 and Spry2 by LIF-induced Stat3 phosphorylation/activation. In addition, LIF also induced self-renewal process to maintain mESCs pluripotency. Therefore, LIF serves a dual role in mESCs, not only maintaining self-renewal, but also inhibiting differentiation. Color images available online at
Footnotes
Author Disclosure Statement
There are no conflict of interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.