
Abstract
Neural stem cells (NSCs) play essential roles in nervous system development and postnatal neuroregeneration and their deregulation underlies the development of neurodegenerative disorders. Yet how NSC proliferation and differentiation are controlled is not fully understood. Here we present evidence that tumor suppressor p53 regulates NSC proliferation and differentiation via the bone morphogenetic proteins (BMP)-Smad1 pathway and its target gene inhibitor of DNA binding 1 (Id1). p53 deficiency led to increased neurogenesis in vivo, and biased neuronal differentiation and augmented NSC proliferation of ex vivo NSCs. This is accompanied by elevated Smad1 expression/activation in the brain and NSC, which contributes to accelerated neuronal differentiation of p53−/− NSCs. p53 deficiency also leads to upregulation of Id1, whose expression is repressed by p53 in BMP-Smad1-dependent and -independent manners. Elevated Id1 expression contributes to augmented proliferation and, unexpectedly, accelerated neuronal differentiation of p53−/− NSCs as well. This study reveals a molecular mechanism by which tumor suppressor p53 controls NSC proliferation and differentiation and establishes a connection between p53 and Id1.
Introduction
P53
Neural stem cells (NSCs) are responsible for early nervous system development and postnatal nervous tissue regeneration and repair [4 –6]. They are located in several regions of the adult brain—in particular, the subventricular zone and the hippocampus. Upon the stimulation of unknown cues, NSCs differentiate into neurons or glia cells and thus provide new parts for the brain [6]. Defects in NSC compartments, including maintenance/renewal, migration, differentiation, and death, are believed to underlie various diseases of the nervous system, including neurodegeneration and psychiatric disorders [7].
One of the major pathways controlling neurogenesis is the bone morphogenetic proteins (BMP)-Smad1/5/8 pathway [8,9]. They execute their functions by binding to and activating BMP receptors I and II, with the former phosphorylating Smad1, 5, and 8 at the C′ terminal SXS motif. Smad1/5/8 then associate with Smad4, move into the nucleus, and turn on BMP target genes. BMPs play various and sometimes distinct roles throughout the development of the nervous system, often in a context and stage dependent manner [10]. In mouse embryonic stem (ES) cells, BMPs act to inhibit neuronal differentiation and help self-renewal [11]. In early embryogenesis, BMPs inhibit neuroectoderm formation [12,13], whereas in later neural differentiation, BMPs promote the differentiation of both neural cell types and astroglial cells [14 –16]. Recent studies show that BMPs help to keep NSC cells at quiescent status in adult mice. Blockade of BMPs action with Noggin- or tissue-specific inactivation of BMPRIA led to eventual decrease of the NSCs in hippocampus compartment [17]. However, little is known about the BMP target genes that control neurogenesis, with the exception of inhibitor of DNA binding 1 (Id1). Id1 binds to the pro-neuronal transcription factors such as Mash1 to inhibit their functions. Thus, Id1 appears to mediate the inhibitory effects of BMPs on neuronal differentiation, at least in mouse ES cells [11].
p53 has been implicated in the nervous development [18,19]. In vivo studies have shown that p53 mRNA levels and promoter activity are increased during neuronal progenitor cell differentiation [20,21]. p53 null mice (about 16%) develop exencephaly, which is caused by overproliferation of neurons or lack of apoptosis of neurons [22]. It is possible that other members of the p53 family, p63 and p73, provide compensatory functions in the unaffected p53 null mice. Recently, p53 has been shown to negatively control the proliferation of NSCs and p53 deficiency facilitates glioblastoma development via Myc and p21 [23]. However, it is still uncertain how p53 regulates neural differentiation as conflicting results have been reported (reviewed in [18,24,25]). While several studies show that p53 inhibits neuronal lineage, there are studies showing that p53 plays no effect on neuronal differentiation [23,26 –30].
Here we further analyzed neurogenesis in p53−/− embryo/mouse and NSC proliferation and differentiation and found that p53 is highly expressed during neuronal differentiation, and that p53 appears to inhibit NSC proliferation and NSC neural differentiation. p53 deficiency results in an increase in 2 critical neurogenesis regulators, Smad1 and Id1, which contribute to the effects of p53 deficiency on NSC proliferation and differentiation. These findings demonstrate that p53 is a critical regulator of neural differentiation in vivo and uncover a potential mechanism by which p53 regulates neurogenesis.
Materials and Methods
Mice, primary NSC isolation, and culture
p53−/−
mice (on 129S genetic background) were obtained from The Jackson Laboratory. p53−/−
mice on C57BL/6 genetic background were generated by successive crosses to normal C57BL/6 mice. NSCs were isolated from E13.5 embryos following a standard protocol. Briefly, the brains were taken out from the embryos and the bodies were used for genotyping. The telencephalon was separated from the rest and was trypsinized for 5 min at 37°C. After 3 times washing in 5 mL Dulbecco's modified Eagle's medium (DMEM)/F12 (1:1) (Invitrogen) supplemented with 2 mM
NSC siRNA transfection
Cell transfection with specific siRNAs (ON-TARGETplus Smart pool, SiGenome) was performed 24 h after seeding; for this purpose, cells were changed to antibiotic-free medium and transfected using DharmaFECT transfection reagent, following the manufacturer's instructions. DharmaFECT transfection reagent was removed 48 h later and cells were maintained for 1–3 additional days before being counted, fixed for immunocytochemistry or western blot, or used for further experiments.
Tissue and cell immunocytochemistry
E11.5, E13.5, and E17.5 embryos and day 3 pups were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) overnight, and then equilibrated in 30% sucrose. The tissues were then frozen and coronally sectioned (8 μm) using a cryostat microtome. Sections were dried on poly-l-lysine-coated slides and processed immediately for immunocytochemistry. These slides as well as cultured NSCs were blocked in 5% bovine serum albumin in PBS with 0.5% Triton X-100 for 30 min. Primary antibody were diluted in the same blocking solution and applied overnight at 4°C. Slides were then washed 3 times in PBS for 10 min and incubated with secondary antibodies at 1:100 in PBS for 1 h at room temperature. After washing 3 times in PBS for 10 min, the slides were incubated with 3,3-diaminobenzidin at room temperature without light for 10 min. Slides were then dehydrated and mounted with neutral gums. For cell culture slides, secondary antibodies conjugated with fluorescein isothiocyanate or tetramethyl rhodamine isothiocyanate (Sigma–Aldrich) were used. The following antibodies were used: anti-beta-III Tubulin (Tuj1) (ab78078, 1:100, Abcam), anti-microtubule-associated protein 2 (MAP2) (05-346, 1:200, Millipore), anti-glial fibrillary acidic protein (GFAP) (mAB3402, 1:500, Millipore), anti-Nestin (ab11306, 1:100, Abcam), anti-NeuN (ABN78, 1:50, Millipore), and anti-O4 (mAB345, 1:200, Millipore).
Western blot analysis
Cells or mouse brains were lysed in TNEN buffer (50 mM Tris, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, and 0.1% Triton X-100) supplemented with 1 mM NaF, Na2VO3, 1 mM phenylmethanesulfonyl fluoride, and 1 μg/mL of aprotonin, leupeptin, and pepstatin. Protein concentration was determined using an assay (Bio-Rad Laboratories). Proteins were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore). The antibodies against the following proteins were used for western blot: GFAP (mAB3402, 1:1000, Millipore), MAP2 (05-346, 1:1000, Millipore), Nestin (mAB5326, 1:1000, Millipore), p53 (2524, 1:1000, Cell Signaling,), Id1 (sc-488, 1:500, Santa Cruz), p21 (556430, 1:500, BD), Actin (sc-81178, 1:1000, Santa Cruz), p-Smad1 (9511, 1:1000, Cell Signaling), Smad1 (9743, 1:1000, Cell signaling), and Tuj1 (ab78078, 1:500, Abcam).
Luciferase assay
The Smad1 promoter (fragments 1.0 or 2.0 kb from the start of transcription) was cloned into pGL3 (luciferase basic vector; Promega). P53 expression constructs, the promoter plasmid, and renilla plasmid (internal control) were co-transfected into HCT116 or MC3T3-E1 cells. Cells were harvested 24 h later, washed with PBS, and lysed with reporter lysis buffer (Promega). The luciferase activities were measured following the manufacturer's procedures and were normalized against the renilla activity. All transient expression in this assay was performed in triplicate.
RNA isolation and real-time PCR
Total RNA was isolated from cells growing on 60 mm dishes using TRIzol reagent (GIBCO BRL). Total RNA was subjected to DNase treatment (Ambion) and quantitated. Five μg of total mRNA was reverse transcribed into complementary DNA (cDNA) using AMV (Roche) reverse transcriptase. The total reaction was used in the real-time polymerase chain reaction (PCR) to assay. The primers were designed following the instructions from Roche.
Quantitation of western blot results
Western blot results were scanned with a Molecular Dynamics scanning densitometer. The relative levels of protein of interest were then determined by measuring the intensity of the corresponding bands. All values were averages of cell cultures isolated from at least 3 p53−/− mice and their control littermates and were normalized to the constitutive expression of Actin. In all the quantitation data, the values of wild type (WT) were set as 1.0.
Statistical analysis
Each experiment was repeated with 3 or more mutant and control mice. Statistical analysis was performed using an unpaired t test (STATISTICA Software; StatSoft, Inc.). P values were provided for all in vivo results. Significant association was defined when *P<0.05 compared with control.
Results
p53 plays an active role in neurogenesis
To reexamine a possible role for p53 in neural development, we first analyzed the expression of neuron and astrocyte-specific markers in the brain homogenates of 13.5-, 15.5-, and 17.5-day embryos of p53−/− and WT (of the same litter) on C57BL/6 background. The initiation of embryonic neurogenesis and gliogenesis is controlled by multiple developmental cues [4 –6]. We found that in WT embryos, neuron markers Tuj1 and MAP2 and astrocyte marker GFAP were expressed at low levels at day 13.5, and the expression of Tuj1 and MAP2 was increased at day 15.5, while the expression of GFAP was increased at day 17.5 (Fig. 1A), confirming that neuron differentiation is earlier than that of glia. p53−/− brains showed much higher levels of Tuj1 and MAP2 but lower levels of GFAP than WT embryos (Fig. 1A), suggesting that p53 deficiency promotes neuron differentiation but inhibits astrocyte differentiation at this stage of development.

p53 deficiency led to an increase in neurogenesis at the sacrifice of gliogenesis during embryonic development.
To substantiate these findings, we carried out immunohistochemical staining of neuronal and glial markers on brain sections. Although only 16% of the p53−/−
embryos were reported to develop exencephaly, we found that all the 9 p53−/−
brain sections showed more Tuj1 at day 11.5 embryonic cerebral cortex region (MAP2 undetectable) and more NeuN (another neuron marker) at the day 13.5 embryonic cerebral cortex region than WT littermates (Fig. 1B and 1C). On the other hand, a decrease in GFAP staining was observed on brain sections of E17.5 embryos compared to control littermates (Fig. 1D). Moreover, a 30% decrease in the number of GFAP-positive cells in the corpus callosum region of day 3 p53−/−
pups and the CA3 region of the hippocampus of 2-month-old p53−/−
mice were also observed compared to WT controls (Supplementary Fig. S1 and S2; Supplementary Data are available online at
We also found that p53 protein levels went down from day 15.5 and were almost undetectable at day 17.5 (Fig. 1A), consistent with the expression pattern of p53 mRNA [21]. This p53 expression pattern, together with our observation that p53−/− embryos showed an increase in neurogenesis, suggests that p53 might help to curtail neurogenesis before day 15.5 but permit neurogenesis afterward. Alternatively, the p53 action after day 15.5 may be taken over by other members of the p53 superfamily such as p63 and p73 [24]. The observations that the start of GFAP expression at day 17.5 was associated with a decline of p53 and that p53 deficiency impeded gliogenesis suggest that the p53 action may be taken over by other members of the p53 superfamily, or the decline of p53 may serve other purposes at this stage, for example, promoting cell expansion.
An active role for p53 in NSC proliferation and cell fate determination
A role for p53 in NSC differentiation is still debatable as some studies showed that p53 inhibits neural differentiation, whereas the others did not find such a role [26 –28]. To further clarify this issue, we isolated NSCs from brains of day 13.5 p53−/− and WT embryos (C57BL/6 genetic background) of the same litter with a standard protocol [23,26,28,30]. NSCs from individual embryos formed spheres and were further expanded in proliferation medium containing bFGFs and EGFs. These cells stained positive for NSC marker Nestin in spheres or monolayers, regardless of the p53 status (Fig. 2A); once cultured in the differentiation medium, they started to express markers for neuron or glia (Fig. 2B), suggesting that these freshly isolated cells are undifferentiated NSCs. Judged from the spheres size, it is obvious that p53 deficiency led to increased NSC proliferation (see later results). Immunocytochemistry staining of NSC cultured in the differentiation medium showed that all 4 p53−/− NSC cultures analyzed differentiated into more MAP2-positive cells but fewer GFAP- or O4-positive cells than WT NSC cultures (Fig. 2B, and 2C). However, the percentage of differentiated cells was similar (49%) in WT and p53−/− NSC cultures, suggesting that p53 deficiency led to a shift in differentiation from glia to neuron (Fig. 2C). We did not find significant difference in apoptotic cells between p53−/− and WT NSC cultures under our experimental settings (Supplementary Fig. S3). We also cultured NSCs in medium containing only B27 to exclude possible influence of growth difference on differentiation as cell proliferation is limited in B27 medium. Similarly, we found that p53−/− NSC tend to differentiate into neurons, justified by western blot results of NSC differentiation markers, especially at 72 h (Fig. 2D), confirming an inhibitory role for p53 in neuronal differentiation, which might involve compensatory mechanisms due to the loss of p53. Note that, in general, NSC differentiation was analyzed after placing the cells in the differentiation medium for 3 or more days. We included early time points in our experiments to analyze Smad1 expression and phosphorylation and Id1 expression. Surprisingly, we detected low levels of Tuj1 in WT NSCs (but not in p53−/− NSCs) at early time points. One explanation for this is that GFAP expression might transiently respond to the components of B27 medium in a p53-dependent manner, which warrants further investigation. Although day 13.5 embryo rarely express GFAP in the brain, justified by western blot analysis of the brain homogenate (Fig. 1A), GFAP expression in the NSC cultures isolated from day 13.5 embryos is readily detectable. One possible explanation is that NSC themselves and the radial glia cells that exist in the in vitro NSC cultures express GFAP [31,32], whereas these cells are rather rare in day 13.5 brains. Alternatively, B27 medium (used in Fig. 2D) may affect gliogenesis and/or GFAP expression. The GFAP difference between p53−/− and WT NSC cultures appears to be greater when cultured in B27 than that with serum (Fig. 2C and 2D). These results, taken together, indicate that p53−/− NSCs differentiated into neurons at the sacrifice of astrocytes, supporting the conclusion drawn by Armesilla-Diaz et al. that p53 plays a negative role in NSC neural differentiation but a positive role in gliogenesis [26,28,30], but are inconsistent with 2 reports showing that p53 deficiency did not alter neural or glia differentiation of NSCs [23,27]. This discrepancy is not less likely to be caused by genetic backgrounds, as similar results were obtained from NSCs isolated from p53−/− mice on 129 and C57BL/6 background (Supplementary Fig. S4 and Fig. 3).
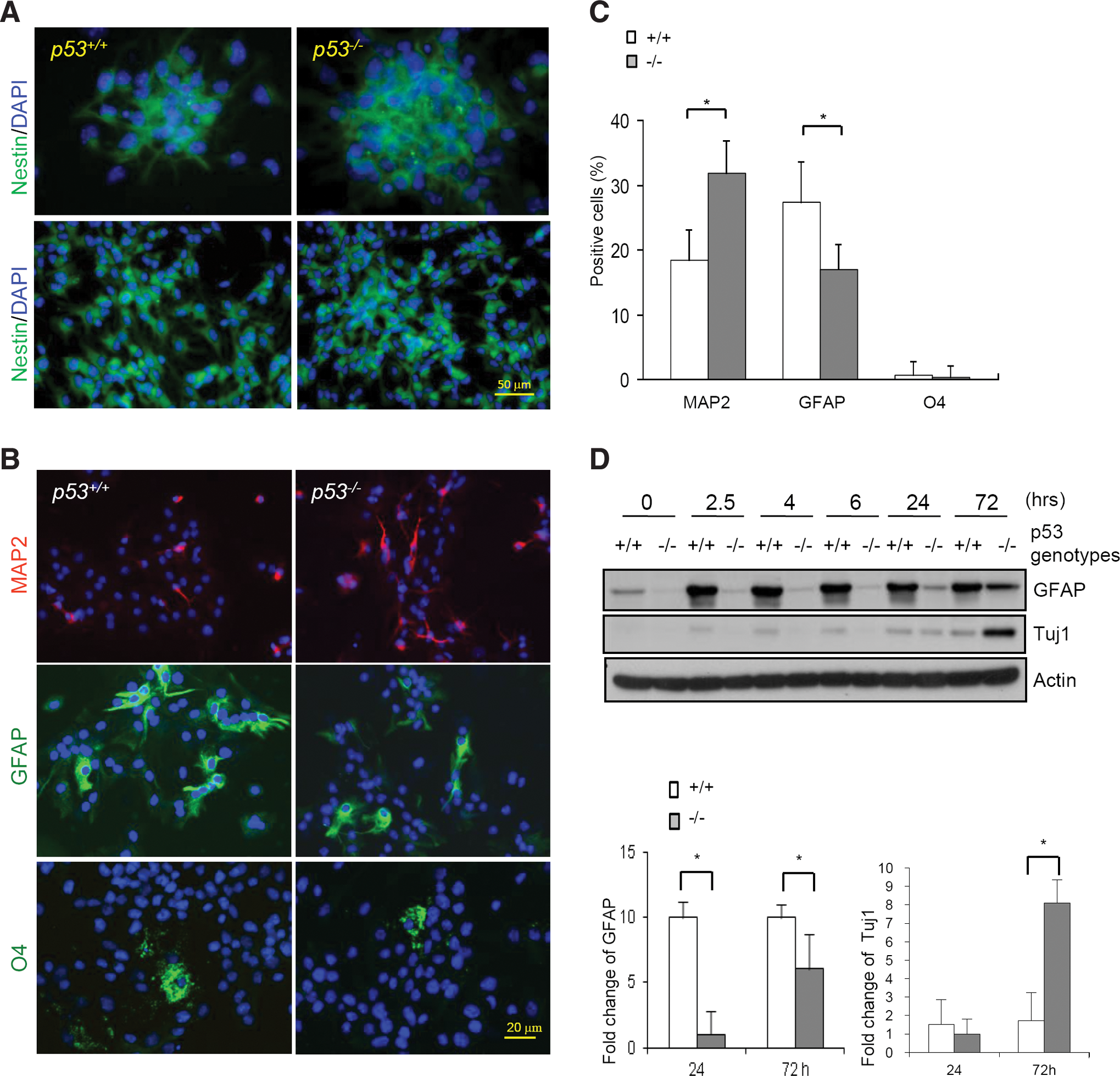
p53 deficiency promoted neural differentiation of neural stem cells (NSCs).
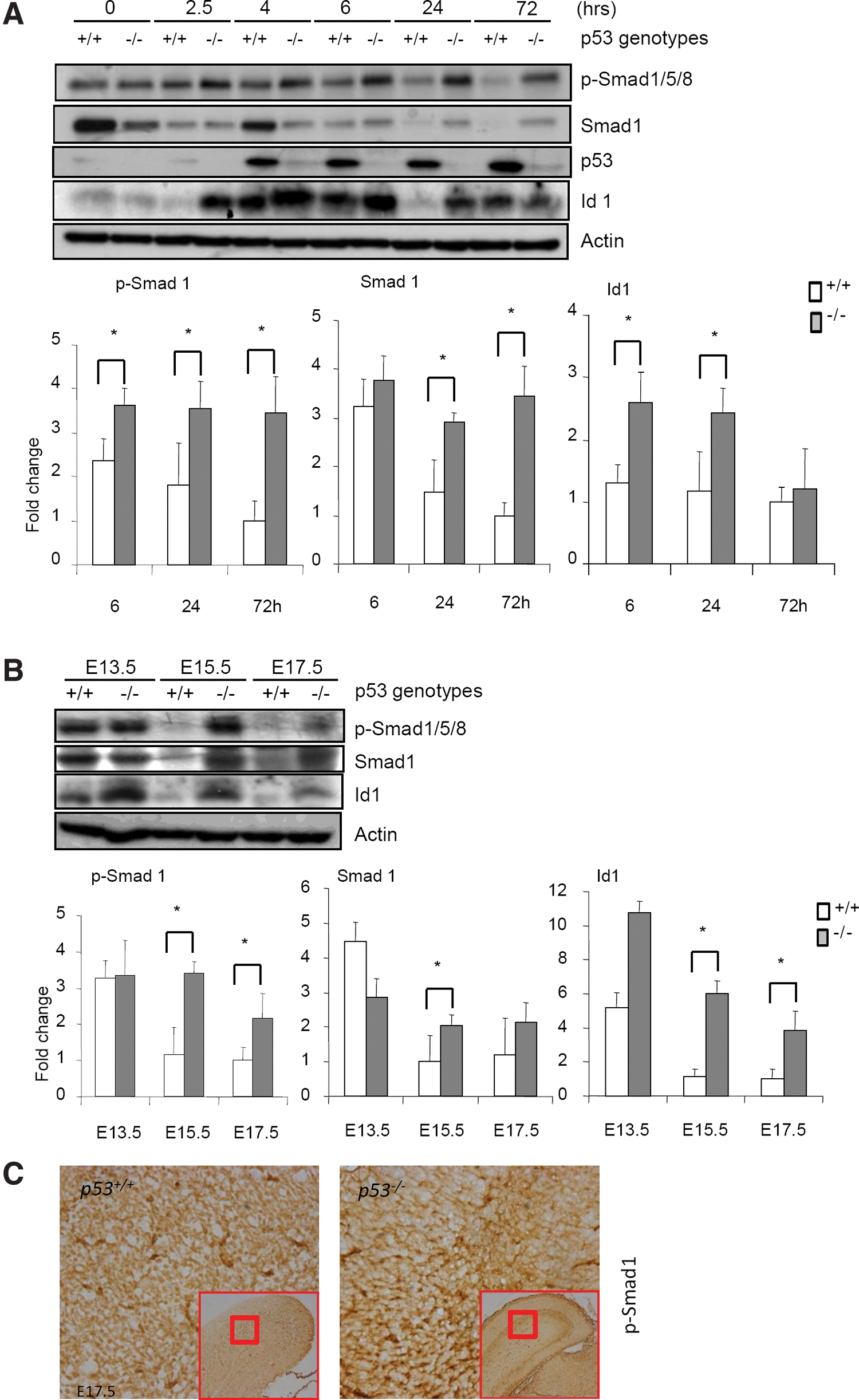
p53 deficiency led to upregulation of Smad1, hyperactivation of Smad1, and upregulation of inhibitor of DNA binding 1 (Id1) in NSCs and mouse brains.
We also observed that the levels of p53 were increased up to day 3 once cultured in the differentiation medium (Fig. 3A). This result, together with the p53 expression pattern during embryonic neurogenesis (Fig. 1A), suggests that one function of elevated p53 expression is to curb neuronal differentiation but promote gliogenesis. However, this finding is not consistent with a recent study showing that the level of p53 is not altered during NSC in vitro differentiation [33]. Further studies are needed to determine what causes this discrepancy.
p53−/− NSCs showed an increase in Smad1 expression and activation
How does p53 deficiency promote neuronal differentiation? A clue came from our mouse genetic studies of p53 in osteoblastogenesis and bone formation [34,35]. p53−/− mice show osteosclerotic phenotypes, with accelerated osteoblast differentiation due to upregulation of Runx2 and Osterix, 2 transcription factors essential for osteoblastogenesis [34,35]. Both Runx2 and Osterix were recently shown to be target genes of BMPs [36], which are critical regulators of the development of bone as well as the nervous system. This link encouraged us to test whether p53−/− NSCs have any alteration in BMP-Smad1 signaling during differentiation. We indeed found that WT NSCs display considerable levels of Smad1/5/8 phosphorylation when cultured in the differentiation medium, which quickly goes down, accompanied by a downregulation of Smad1 (Fig. 3A). The cues that elicit this change, as well as the change in p53 levels, are currently unclear. However, p53−/− NSCs maintained high levels of Smad1 protein and Smad1/5/8 activation at 6, 24, and 72 h in the differentiation medium, without showing alterations in the protein levels of Smad5 or Smad8 (Fig. 3A and Supplementary Fig. S5). Moreover, p53−/− embryos also displayed an upregulation of Smad1 and hyperactivation of Smad1 compared to WT littermates, justified by western blot analysis of the brain homogenates and immunohistochemical staining of p-Smad1 on brain sections (Fig. 3B and 3C). Comparison of the levels of p-Smad1 and the levels of Smad1 suggests that elevated expression of Smad1 contributes to the enhanced Smad1 activation (Fig. 3B).
Pervious studies showed BMP2 treatment did not alter the protein levels of p53 or the protein levels of p53 target gene p21 in several cell types [37]. However, we showed that the loss of p53 leads to Smad1 upregulation (Fig. 3A and 3B), suggesting that p53 represses the expression of Smad1 under normal conditions. We found that in osteoblasts, p53 represses Smad1 expression via the well-established p53-p21-CDK-E2F1 pathway, and that Smad1 promoter has multiple binding sites for E2F1 (G. Ma et al., unpublished results). It is likely that p53 represses Smad1 expression with the same mechanism in NSCs and this needs further investigation.
Enhanced BMP-Smad1 signaling contributes to accelerated neuronal differentiation of p53−/− NSCs
The enhanced activation of Smad1 signaling is predicted to participate in neuronal differentiation of p53−/− NSCs, as BMPs (the main activator of Smad1/5/8) are known to regulate neurogenesis in cell context, localization, and stage-dependent manners [9,38 –40]. To determine the function of BMPs in NSCs, we treated WT and p53−/− NSCs with 50 ng/mL of BMP2, which led to an increase in Smad1/5/8 phosphorylation (Supplementary Fig. S6), and followed the differentiation process. Immunocytochemistry staining showed that BMP2 treatment increased the number of Tuj1-positive cells and decreased the number of GFAP-positive cells, in the presence or absence of p53 (Fig. 4A). The greater effect of BMP2 on GFAP-positive cells in WT than in p53−/− NSCs could be due to the pre-existing hyperactivation of Smad1/5/8 in p53−/− NSCs. To corroborate these findings, we inhibited BMPs action with both Noggin and Chordin in the differentiation assays. Noggin and Chordin are protein antagonists specific for BMPs and the combinational use is intended to efficiently block BMPs action. Immunocytochemistry staining showed that Noggin and Chordin treatment led to a decrease in the number of Tuj1-positive cells but an increase in the number of GFAP-positive cells (Fig. 4B). Western blot analysis showed that Noggin and Chordin decreased Smad1/5/8 activation although they somehow led to an upregulation of Smad1 (Supplementary Fig. S7). However, the effects of Noggin/Chordin treatment on GFAP were minimal in p53−/− NSCs (Fig. 4B). Although inconsistent results regarding BMPs' role in neuronal differentiation has been reported, our findings support the conclusion that hyperactivated BMP-Smad1 signaling contributes to accelerated neuronal differentiation of p53−/− NSCs. The differential effects of Noggin and Chordin on p53−/− and WT NSCs suggest that factors other than BMP-Smad1 may also play an important role in p53−/− NSCs.

Bone morphogenetic protein 2 (BMP2) treatment promoted, while Noggin and Chordin inhibited, neural differentiation of NSC.
p53−/− NSCs showed an upregulation of Id1
Id1 is a BMPs target gene and has been shown to inhibit neuronal differentiation but promote NSC self-renewal [41,42]. As a helix-loop-helix protein, Id1 binds to helix-loop-helix transcription factors such as Mash1 to inhibit their activities [11,43]. Since BMP-Smad1 pathway is hyperactivated in p53−/− NSCs, we determined the protein levels of Id1 in p53−/− and WT NSCs during differentiation and found that Id1 levels were initially very low, went up at 2, 4, 6 h, and went down at 72 h in WT NSCs, and that p53−/− NSCs showed an increase in Id1, except at 72 h after cultured in differentiation medium (Fig. 3A). A decline of Id1 levels from day 1 to day 8 during NSC differentiation was previously reported [33]. Moreover, the brain tissue of p53−/− embryos also showed an upregulation of Id1 compared to WT littermates, justified by western blot analysis (immunohistochemical staining did not work with the available antibodies) (Fig. 3B). Realtime PCR analysis showed that the mRNA levels of Id1 are also much higher in p53−/− NSCs than in WT cells (Fig. 5A), suggesting that p53 might repress Id1 transcription.
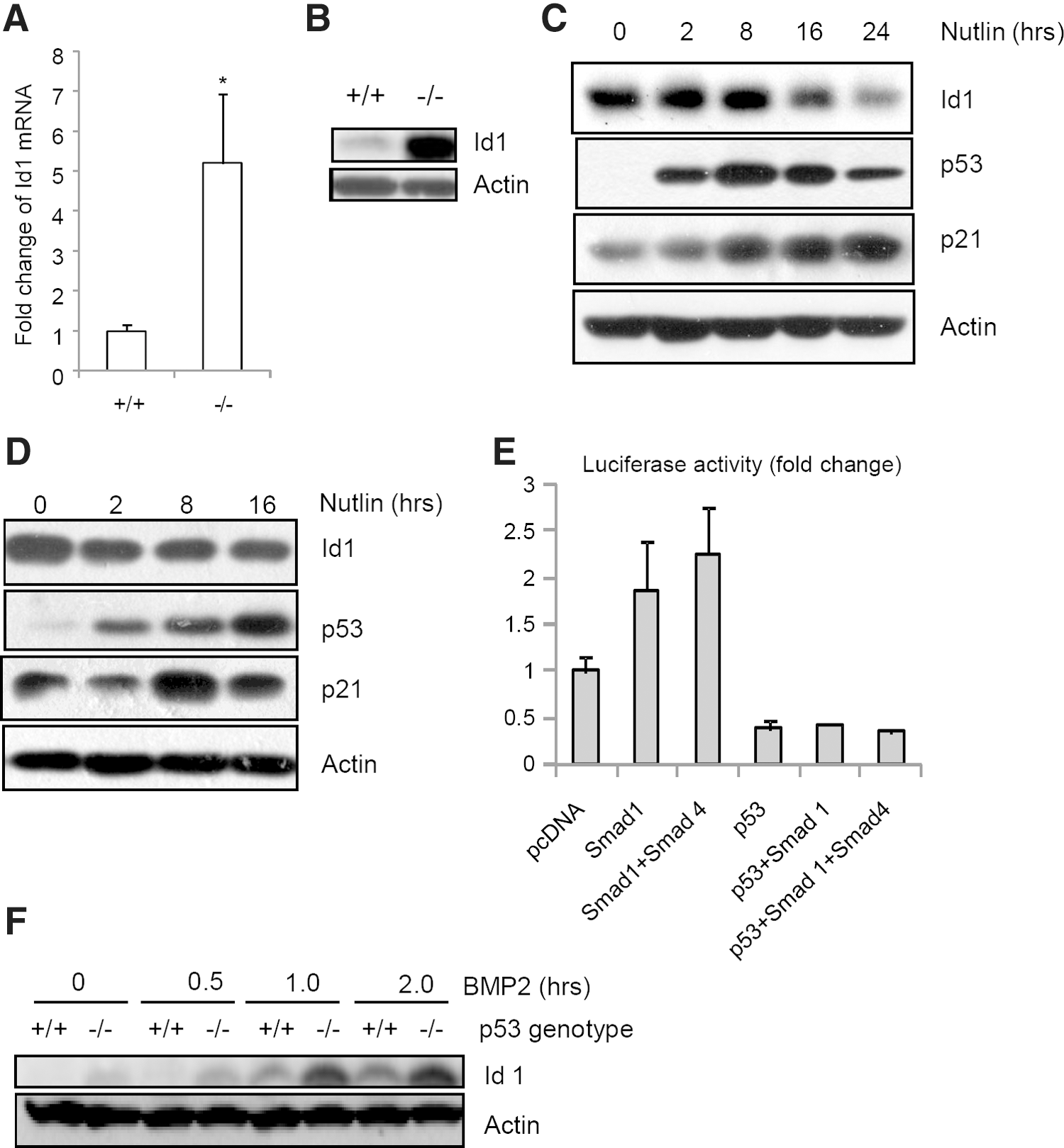
p53 represses Id1 expression in BMP-Smad1-dependent and independent manners.
Moreover, p53−/− mouse embryonic fibroblast also showed an upregulation of Id1 at the protein levels (Fig. 5B). On the other hand, Id1 was downregulated when p53 is transiently upregulated by nutlin-3 in PC-12 and NSC cells (Fig. 5C, 5D), a small molecule compound that disrupts Mdm2-p53 interaction and stabilizes p53 [44]. These results, taken together, suggest that p53 is a repressor of Id1 transcription.
p53 represses Id1 expression in BMP-Smad1-dependent and -independent manners
It has been established that Id1 is a target gene of BMP-Smad1 pathway [42,45,46]. We found that p53−/− cells show enhanced Smad1 activation and increased Id1 expression (Fig. 3A). In a reporter assay using Id1 promoter fused to luciferase, p53 was able to inhibit the promoter activity of Id1, while co-expression of Smad1 or Smad1 and Smad4 was able to activate Smad1 promoter (Fig. 5E). However, Id1 promoter has no typical p53-binding site, suggesting that p53-medaited repression of Id1 transcription does not involve direct binding to the promoter. Alternatively, p53 may interfere with transactivators of Id1 gene such as Smad1. We indeed found that co-expression of p53 abolished Smad1-mediated Id1 promoter activation (Fig. 5E). This result, together with the finding that loss of p53 leads to an increase in Smad1 expression and activation, suggests that p53 is a transcription repressor of Id1.
However, regulation of Id1 expression during neural differentiation seems to be rather complex. We noticed that Id1 expression pattern was not always positively correlated to that of BMP-Smad1 activation (Fig. 3A), suggesting that there exist other signals that control Id1 transcription. Moreover, Id1 is expressed at higher levels in p53−/− NSCs, especially 2.5 and 4 h after being placed in the differentiation medium, when the BMP-Smad1 signaling is comparable between WT and p53−/− NSCs (Fig. 3A), suggesting that p53 may also regulate Id1 independently of the BMP-Smad1 pathway. This is further supported by our observation that p53−/− cells still showed a much higher induction of Id1 than WT cells in response to 50 ng/mL of BMP2 (Fig. 5F). Id1 promoter has binding sites for YY1, Egr1, Sp1, ATF/CREB, and other transcription factors [47]. It is possible that p53 might interfere with the transactivation activity of 1 or more of these transcription factors to repress Id1 transcription.
Elevated Id1 expression contributes to increased self-renewal, proliferation, and differentiation of p53−/− NSC
To determine the function of elevated Id1 in p53−/− NSCs, we tried to knock down Id1 with siRNA. Pooled siRNA could efficiently lower the protein levels of Id1 (to the levels similar to that of WT NSCs) when transiently transfected into WT and p53−/− NSCs for 3 days. We then tested a possible role of Id1 in neuronal differentiation of WT and p53−/− NSCs and found that knockdown of Id1 enhanced neuronal differentiation of WT NSCs, evidenced by an increase in Tuj1 and MAP2 expression (Fig. 6A), consistent with previous studies showing that Id1 is a negative regulator of neural differentiation [41,42]. Unexpectedly, Id1 knockdown resulted in a decrease in the expression of neuronal markers in p53−/− NSCs (Fig. 6A). This finding was further validated by immunocytochemistry experiments that show that Id1 knockdown increased the number of Tuj1-positive cells in WT cultures but decreased the number of Tuj1-positive cells in p53−/− cultures (Fig. 6B). Thus, loss of p53 might have altered the cell context and as such, highly elevated Id1 seems to promote neural differentiation under this condition. Further studies are needed to understand how p53 status makes such a switch in determining the function of Id1. These data, taken together, suggest that Id1 elevation contributes to the increase in neuronal differentiation of p53−/− NSCs.
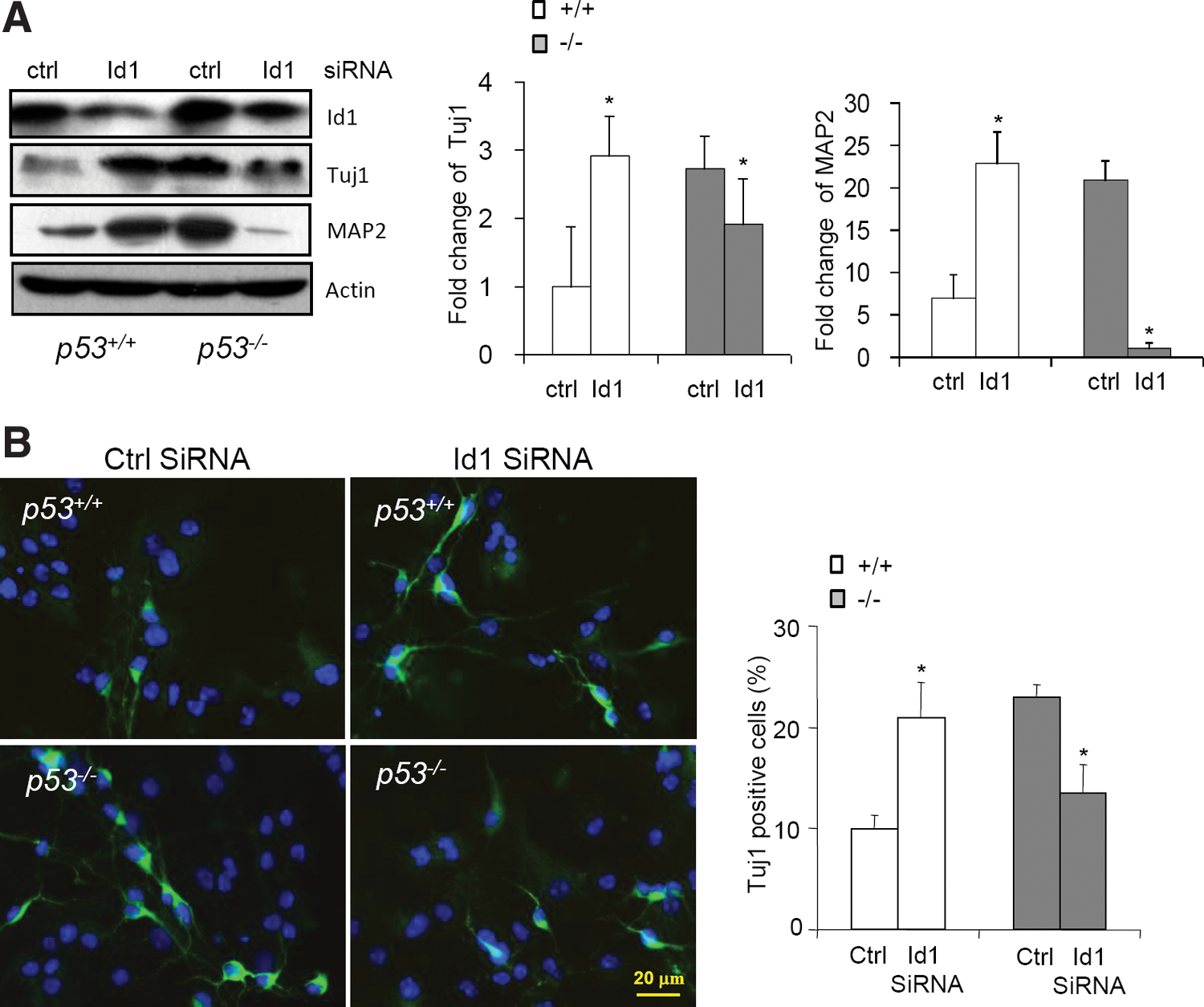
Knock-down of Id1 impeded neural differentiation of p53−/−
NSCs, but not WT NSCs.
There is evidence that the growth advantage of p53−/− cells is attributable to decreased expression of p53 target gene p21, a CDK inhibitor [23]. Consistent with previous studies, we found that p53−/− NSCs showed enhanced proliferation (Fig. 7A). To test the potential effect of elevated expression of Id1 in NSC proliferation, we knocked down Id1 with siRNA and found that Id1 knockdown led to a decrease in the sphere size of p53−/− NSCs, whereas it has a minimal effect on WT NSCs (Fig. 7A). This could be because Id1 knockdown was transient and less than complete in our experimental settings. Counting the cell numbers in the NSC spheres transfected with Id1 or control siRNA also confirmed that Id1 promoted the proliferation of p53−/− NSCs, as the cell numbers were decreased by 29% in the presence of Id1 siRNA (Fig. 7B). BrdU labeling experiments also showed that p53−/− NSC cultures had more proliferating cells, which was reduced by Id1 knockdown (Fig. 7C). Moreover, we found that p53−/− NSC had increased self-renewal ability, which requires elevated expression of Id1 as Id1 knockdown decreased the self-renewal of p53−/− NSCs (Fig. 7D and Supplementary Fig. S8). These results, taken together, suggest that elevated Id1 protein mediates the role of p53 deficiency on NSC self-renewal and proliferation.

Knock-down of Id1 inhibited proliferation of p53−/−
NSCs.
Discussion
There is increasing evidence that p53 regulates cell differentiation in addition to cell proliferation, apoptosis, and senescence. For instance, p53 has been reported to promote the differentiation of myogenic and hematopoietic cells but inhibit the differentiation of osteogenic and adipogenic cells, suggesting that the function of p53 in regulating differentiation is cell type specific [24,48]. Our in vivo results and ex vivo NSC culture results, by detecting cell marker with immunocytochemistry and western blot, show p53 deficiency led to enhanced proliferation and accelerated neuronal differentiation of NSCs, at the sacrifice of glia. This is similar to mesenchymal stem cells, in which p53 deficiency leads to an increase in both proliferation and differentiation [49]. However, there are studies showing that p53 does not regulate NSC differentiation [18,23,27]. This discrepancy is not likely caused by the genetic background of the p53−/−
mice as in most studies C57BL/6 or FvB/B6 mice were used [23,26
–28]. Moreover, we found that NSC isolated from 129 background behaved like that isolated from C57BL/6 background. We suspect that the culture conditions might be a major factor causing the discrepancy. In various studies, NSC differentiation was assayed in the presence of serum on coated slides. Different coating materials (polylysine/laminin, GFR-Matrigel™, poly-ornithine-coated
More importantly, our study revealed 1 molecular mechanism by which p53 regulates NSC proliferation and differentiation (Fig. 7E). We show that p53 deficiency altered the expression of 2 critical regulators of NSC homeostasis, Id1 and Smad1, which seem to mediate the effects of p53 deficiency on NSC proliferation and/or neural differentiation. The link of p53 deficiency to Id1 and Smad1 expression/activation is also observed in osteoblasts (Ma G et al., unpublished results). Previous studies have shown that p53 regulates the stemness of NSCs via altering the expression of Oct4 [54,55], and NSC proliferation via p21, Id2, and c-myc [23,56]. Thus, p53 might act on multiple pathways to control NSC proliferation and differentiation. The coordinated action of these pathways may help to fine-tune the output from p53 in cell proliferation and differentiation. To add to the complexity of p53 in neurogenesis regulation, we found that p53 status also acts like a switch in the action of Id1 in neuronal differentiation.
This study shows that the expression of Id1 is repressed by p53 in NSC, and more importantly, Id1 mediates, at least partially, the effects of p53 deficiency on NSC proliferation and neural differentiation. Lack of p53 led to an elevation of Id1, while upregulation of p53 led to a decrease in Id1. p53 seems to repress the promoter activity of Id1 in a p53-DNA binding independent manner. A recent study suggested that p53 might represses Id1 expression via DEC1, a p53 target gene in DNA damage response [57]. In addition, we show that p53 inhibits the Smad1's activity on Id1 promoter. Thus, p53 might repress Id1 expression via multiple mechanisms, including BMP-Smad1-dependent and -independent manners. The conclusion that Id1 contributes to the enhanced proliferation of p53−/− NSCs is consistent with previous studies showing that Id1 promotes self-renewal of mouse ES cells, extends the lifespan of mouse embryonic fibroblasts [47], and augments proliferation of p53−/− cancer cells [58]. In addition, Id1 elevation is shown here to promote neuronal differentiation in p53−/− NSCs, whereas Id1 inhibits neuronal differentiation in normal NSCs. One possible explanation for this switch is that p53 deficiency may have altered the cell context so that neuronal differentiation process differently responds to Id1, which warrants further investigation. Together with the findings that p53 also represses Id2, which also participates in NSC self-renewal [25,59], our results suggest an important link between p53 and Id proteins.
This study also establishes a connection between p53 and the BMP-Smad1 pathway in NSC. Previous studies have shown that p53 interacts with Smad2/3 to regulate several cellular and developmental processes [60], this study shows that p53 could repress Smad1 expression in NSCs and mouse brain. Furthermore, p53 seems to be able to inhibit the transcription activity of Smad1 on Id1. Recent studies revealed that p53 might inhibit Smad1 expression via the p21-CDK-Rb-E2F1 pathway (Ma G. et al., unpublished results), a well established route that is used by p53 to repress the transcription of some target genes. The link between p53 and BMP-Smad1 signaling might provide an explanation why p53−/− cells are more sensitive to BMPs-induced differentiation of mesenchymal stem cells. In NSCs, the function of Smad1 elevation seems to promote neuronal differentiation and this effect is partially mediated by BMP target gene Id1. While these results suggest that p53 has an impact on BMP-Smad1 signaling (Fig. 7E), we found that BMPs do not activate p53 [37]. Thus, p53 is a unilateral regulator of the BMP-Smad1 pathway.
In summary, this study provides in vivo evidence that p53 plays roles in NSC differentiation and proliferation and uncovered an important mechanism by which p53 executes its functions. p53 expression is altered during NSC differentiation and in vivo neurogenesis, and p53 deficiency leads to increased expression of Smad1 and Id1, both of which contribute to accelerated neuronal differentiation of p53−/− NSCs. Elevated Id1 expression also contributes to the increased proliferation of p53−/− NSC. The link to BMP-Smad1 signaling and Id1 expression may also underlie other aspects of development regulated by p53.
Footnotes
Acknowledgments
We would like to thank Qiang Yu and Huiyi Kua for helpful discussions, and Hang In Ian, Deyu Cai, and Zhongxin Wang for technical assistance. The work was supported by grants from the Ministry of Science and Technology of China (the National Key Scientific Program [2012CB966901, to B. L.]), the National Natural Science Foundation of China (81130039, 31071229, and 81121001), Shanghai Pujiang Program (10PJ1405000), and the Changjiang Scholars Program of the Ministry of Education.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.