
Abstract
Amniotic fluid stem cells (AFSs) are interesting mesenchymal stem cells (MSCs) that are characterized by their great potential for cell proliferation and differentiation compared with other types of MSCs identified to date. However, MSCs in prolonged culture have been found to exhibit defects in genetic stability and differentiation capacity. Epigenetic anomalies have been hypothesized to be a cause of these defects. Here, we investigated the genomic methylation and genetic imprinting in AFSs during prolonged in vitro culture. Four human imprinted genes, insulin-like growth factor 2 (IGF2), H19, small nuclear ribonucleoprotein polypeptide N gene (SNRPN), and mesoderm-specific transcript (MEST), were evaluated for their expression levels and methylation statuses in AFS lines. The data revealed epigenetic instability in high passage number AFS cultures. The real-time polymerase chain reaction analysis showed that the expression levels of the imprinted genes gradually increased with increased time in culture. The loss of parental allele-specific imprinting for at least 1 gene among IGF2, H19, and SNRPN was observed in every AFS line after passage 8 using allelic expression analysis. The imprinting control regions (ICRs) of the IGF2 and H19 genes were assayed for site-specific methylation using bisulfite sequencing. This assay revealed a variable level of methylated CpG sites in the ICRs of IGF2 and H19. This variable level of CpG methylation is related to the aberrant expression of the IGF2 and H19 genes in late-passage AFSs. Our results did not reveal any irregularity in the epigenetic control system in the early-passage AFSs, indicating that the standard in vitro culturing of AFSs used in medical treatments should be limited to 8 passages.
Introduction
M
Taken together, epigenetics comprise many mechanisms that work together to regulate gene expression and cellular phenotype by modifying the genome without changing the nucleotide sequence. These regulatory mechanisms have been implicated in the maintenance of stem cell-specific characteristics. DNA methylation, one of the most well-characterized epigenetic mechanisms, plays an important role in controlling cellular activity and development. In addition, DNA methylation regulates the function of imprinted genes by contributing the bias of parental allele expression by adding methyl groups to the imprinting control region (ICR) of the silent parental (maternal or paternal) allele. This silencing of 1 allele results in monoallelic expression. Improper gene imprinting is involved in many developmental disorders [9]. In mammalian species, 80% of all imprinted genes are clustered [10]. Three notable clusters of human imprinted genes are located at the chromosome positions 11p15, 15q11-13, and 7q32. Insulin-like growth factor 2 (IGF2), a maternally imprinted gene, and H19, a paternally imprinted gene, are located in the same cluster at 11p15. The methylation in the ICR of H19 results in IGF2 promoter activation. The small nuclear ribonucleoprotein polypeptide N gene (SNRPN), a maternally imprinted gene, is located in the 15q11-13 region, and the paternally imprinted mesoderm-specific transcript (MEST) gene was the first imprinted gene found in the 7q32 region. Abnormalities in the expression of these imprinted genes is correlated with many disorders involving cell growth and development [11 –13].
To evaluate the suitability of AFSs for use in medical applications, the present work investigates the alteration of global DNA methylation and genomic imprinting characteristics in terms of expression levels, allelic expression patterns, and site-specific methylation on the control regions of 4 imprinted genes: IGF2, H19, SNRPN, and MEST. These characteristics were analyzed temporally during the in vitro expansion of AFS lines.
Materials and Methods
Establishment and expansion of AFS lines
Amniotic fluid collected from 12 pregnant women, who provided informed consent according to the regulations of the Ethics Committee of Siriraj Hospital, Mahidol University, Thailand, was cultured in minimal essential medium alpha (α-MEM; Gibco, Invitrogen) supplemented with 10% fetal bovine serum (FBS; PAA) at 37°C and 5% CO2. On day 4 of culture, the medium was replaced with AFS medium containing α-MEM (Gibco), 15% embryonic stem cell grade-FBS (ES-FBS), 20% Chang medium (Irvine Scientific), 1% (v/v) 100 U/mL penicillin and 100 mg/mL streptomycin (Biochrom), and 1% (v/v) 2 mM
Characterization of the AFS lines
For phenotypic analysis, AFSs from early (P2) and late (P17) passages were detached and stained with fluorescein isothiocyanate- and phycoerythrin-conjugated monoclonal antibodies against CD29, CD44, CD90, CD105 (eBioscience), CD34, CD45, and CD73 (Becton Dickinson). The cells were subsequently fixed in 1% paraformaldehyde and analyzed using an FACSort flow cytometer (Becton Dickinson).
For differentiation capacity analysis, the cells from early and late passages were cultured to 70% confluence and then exposed to either adipogenic or osteogenic induction medium at 37°C in 5% CO2 and 5% O2. The medium was changed twice per week. The adipogenic induction medium contained α-MEM (Gibco), 10% ES-FBS (PAA), 1 μM dexamethasone (Sigma), 5 μg/mL insulin (Sigma), 0.5 mM 3-isobutyl-1-methylxanthine (Sigma), and 60 μM indomethacin (Sigma). The osteogenic induction medium contained α-MEM (Gibco) supplemented with 10% ES-FBS (PAA), 0.1 μM dexamethasone (Sigma), 10 mM glycerol-2-phosphate (Sigma), and 50 μM ascorbic acid (Sigma). After 3 weeks of growth in the induction medium, the size of the intracellular lipid droplets was investigated in the adipogenic-induced cells, and the alkaline phosphatase activity and calcium accumulation were assayed in the osteogenic-induced cells.
Global methylation analysis using reverse-phase HPLC
Genomic DNA was extracted from each AFS line using the phenol-chloroform-isoamyl alcohol (50-49-1) method. The gDNA suspension was boiled to obtain single-stranded DNA. Contaminating RNA was removed with RNA ribonuclease H and ribonuclease A (Ambion). Single nucleotides (sNTs) were prepared by adding 10 U nuclease P1 (US Biology) and 150 U of calf intestinal phosphatase (New England Biolab). The sNT suspension was analyzed by HPLC (Waters HPLC Corporation) using a gradient method with 2.5% (v/v) and 8.0% (v/v) methanol in 0.05 M KH2PO4, pH 4.0. The level of global DNA methylation was determined based on the concentration of methylated cytosine against a standard curve.
Imprinted gene expression analysis using quantitative real-time reverse transcriptase–polymerase chain reaction
Total RNA was extracted from each AFS line using phenol-chloroform. cDNA was synthesized using a RevertAid First-Strand cDNA Synthesis Kit (Fermentas). Selective primer validation of the amplification efficiency of the genes was studied, and the endogenous reference gene was completed before use. The real-time reverse transcriptase–polymerase chain reaction (RT-PCR) mixture contained 5 ng of cDNA in 5, 12.5 μL of SYBR Green Master Mix (Brilliant II SYBR Green Master Mix; Applied Biosystems), and 10 pmol of forward and reverse primers (Table 1). The specificity of the real-time RT-PCR product was determined by melting curve analysis. The relative amounts of each transcript were calculated using the comparative cycle (CT) method (the 2_DDCT method). The quantities of IGF2, H19, SNRPN, and MEST were determined relative to the quantity of beta-actin. All AFS lines were analyzed in triplicate for every gene.
IGF, insulin-like growth factor; PCR, polymerase chain reaction; SNRPN, small nuclear ribonucleoprotein polypeptide N gene; MEST, mesoderm-specific transcript; SNP, single-nucleotide polymorphism.
Allelic expression analysis
Primers were designed to amplify segments of the IGF2, H19, SNRPN, and MEST genes containing single-nucleotide polymorphisms (SNPs). Each primer contained a restriction site for a specific restriction enzyme. The primers and the enzymes used for IGF2, H19, SNRPN, and MEST allelic expression analysis are listed in Table 1. Genomic DNA and cDNA were subjected to PCR for 35 cycles with the following conditions: 94°C for 30 s, specific melting temperature for 30 s and 72°C for 45 s. After PCR, allelic expression was analyzed using restriction fragment length polymorphism analysis. The PCR products were digested with the specific restriction enzymes, and the 2 parental alleles were resolved on 10% polyacrylamide gels.
Site-specific methylation analysis by bisulfite sequencing
Genomic DNA was modified by bisulfite treatment using the EZ DNA Methylation Kit (Zymo research Corp.) according to the manufacturer's instructions. The primers for IGF2 and H19 were designed using the online software MethPrimer (
Statistical analyses
The differences in the relative mRNA levels for each gene and the difference in the global DNA methylation between AFSs of different passage numbers were analyzed by a one-way ANOVA using the general linear model procedure in SPSS version 17.0 (SPSS, Inc.). A probability value of P<0.05 was considered significant.
Results
Morphologic appearance and proliferation capacity of AFS
On day 4 of the primary culture of fresh amniotic fluid, P0 AFSs were small in size and exhibited a fibroblastic morphology. The cells had a high rate of proliferation; the population-doubling time (PDT) was 0.3–0.5 days. AFS colonies containing 300–600 cells were observed on day 5 of culture. One of these colonies was chosen and expanded to generate a clonal AFS line, and the remaining colonies were pooled and used as the P0 sample. The clonal AFS line of each donor was repeatedly subcultured, and cells were collected at early (P2), middle (P8 and P12), and late (P17) passages. The morphologies of these cells are shown in Fig. 1. AFSs from the early passage (P2) showed rapid growth, with a PDT of 0.3–0.7 days. After repeated expansion to P8, the rate of cell propagation declined to a PDT of 0.9–1.2 days. Seven of the 12 AFS lines were subcultured for more than 25 passages (105 population doublings) with a PDT of 1.1–1.9 days, whereas the other 5 entered into senescence and became larger before passage 18 (84 population doublings). The cells at P17 were used as the late-passage sample. Chromosomal analysis was performed for each line and showed a normal diploid karyotype of 46 XX or 46 XY.
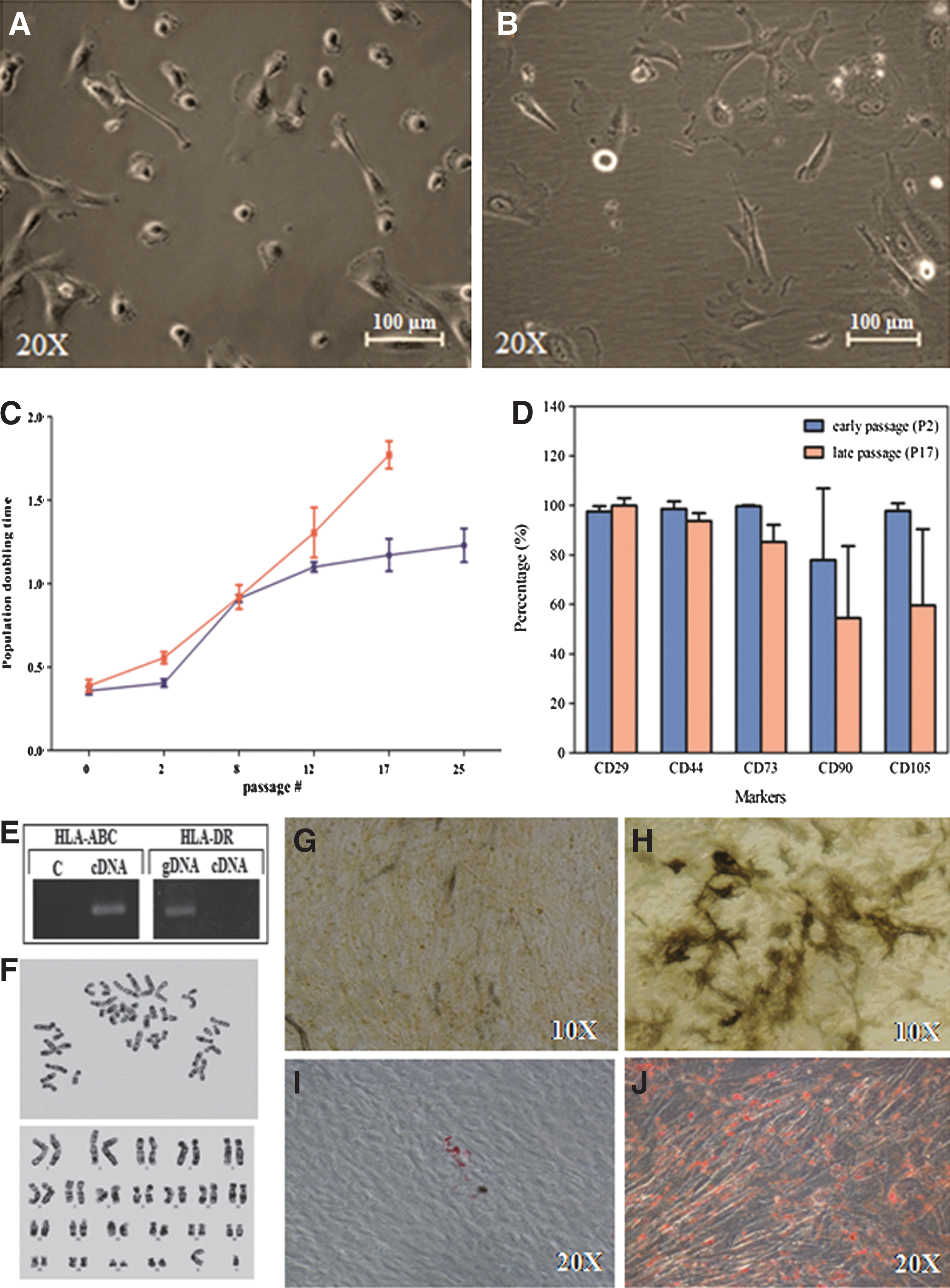
Morphologic appearances of an amniotic fluid stem cell (AFS) line at early and late passages. AFSs at passage 2 (9 days in culture) were small, and many were in metaphase
Phenotypic characteristics and cell differentiation
To assess the antigenic characteristics, specific MSC surface markers were investigated using flow cytometry analysis in every AFS line at early and late passages. All AFS lines were positive for CD29, CD44, CD73, CD90, and CD105 but negative for CD34 and CD45. The mean±standard deviation values of the signals observed in the AFSs at the early passage versus the late passage were as follows: CD29, 97.56%±2.1% versus 99.95%±3.03%; CD44, 98.57%±3.1% versus 93.76%±3.1%; CD73, 99.66%±0.4% versus 85.26%±6.9%; CD90, 77.93%±28.96.% versus 54.50%±29.03%; and CD105, 97.8%±3.1% versus 59.55%±30.9%. The major histocompatibility complex (MHC) was detected by RT-PCR. mRNA for HLA-ABC, but not for HLA-DR, was found in all AFS lines at the early and late passages.
In the analysis of the differentiation ability of AFS lines, alkaline phosphatase and oil red O staining were investigated. Early-passage AFS were found to be positive after culturing in the appropriate induction medium for 3 weeks, indicating that these cells had the potential to differentiate into osteogenic and adipogenic lineages, respectively. In contrast, most of the AFSs from the late passage yielded low or no signal for alkaline phosphatase or oil red O staining (Fig. 1). At passage 25, 3 of 7 lines could differentiate into both adipogenic and osteogenic lineages, but with low yields. One of these 7 cell lines could only be differentiated into the adipogenic lineage, and one could only be differentiated into the osteogenic lineage. The remaining 2 AFS lines did not exhibit either adipogenic or osteogenic differentiation.
Epigenetic variation in early versus late passages of AFSs
The epigenetic status of AFSs was determined by DNA methylation analysis of imprinted genes. The analyses were performed using AFSs from the primary culture (P0), an early passage (P2), middle passages (P8 and P12), and a late passage (P17).
Gene imprinting
To analyze the imprinted gene expression level using real-time RT-PCR, 4 imprinted genes from 4 different ICRs—IGF2 and H19 from domain 11p15.5, SNRPN from domain 15q11, and MEST from domain 7q32—were utilized for mRNA expression analysis using quantitative real-time RT-PCR in 12 AFS lines. The expression levels of the IGF2, H19, SNRPN, and MEST genes were comparable among the cells from the early passage (P2) and from P0 (Fig. 2). With repeated passage in in vitro culture, all AFS lines exhibited a slight variation in H19 expression and a gradual increase in the IGF2, SNRPN, and MEST expression levels (Fig. 2). The increase in IGF2 expression was first observed after the seventh passage, whereas the SNRPN and MEST gene expression levels did not increase until after the tenth passage. With regard to the middle passages (P8 and P12), the P8 AFS lines did not differ from the P12 AFS lines (P value>0.05) with regard to the IGF2, H19, SNRPN, and MEST expression levels. The IGF2 expression level of AFSs from the middle passages was significantly higher (P value<0.05) than the expression levels in P0 and P2 AFSs. The expression levels of SNRPN and MEST increased slightly relative to the levels in the P2 cells, but this increase was not significant (P value>0.05). The level of H19 expression was observed to be stable. The high levels of IGF2, SNRPN, and MEST expression were maintained in the late passages. The levels of transcription of IGF2 and MEST were significantly higher (P value<0.05) than early passage cells, but they did not differ significantly from the middle passages. SNRPN expression was strongly up-regulated, and this gene was expressed at a significantly higher level in late-passage cells (P value<0.05) than in cells from early and middle passages (Fig. 2).

The differences in the relative IGF2, H19, small nuclear ribonucleoprotein polypeptide N gene (SNRPN), and mesoderm-specific transcript (MEST) expression levels in early, middle, and late passage. AFSs were determined using quantitative real-time reverse transcriptase–polymerase chain reaction. The different letters (a, b, and c) within a gene indicate a significant difference (P<0.05) in the mean and standard deviation of the expression level among cells from different passages.
For the allelic expression analysis of imprinted genes, the presence of SNPs in the IGF2, H19, SNRPN, and MEST genes was analyzed in 12 AFS lines. These polymorphisms were used to distinguish the maternal and paternal alleles. Eight AFS lines carried SNPs in the IGF2 and H19 genes, 9 cell lines contained an SNP in SNRPN, and 7 lines had an SNP in MEST. The cell lines harboring SNPs were used for the allelic expression analysis. At the early passage, all of the studied lines showed typical genetic imprinting, with monoallelic expression of IGF2, H19, and SNRPN; whereas all lines containing an SNP in MEST displayed the same biallelic expression found in the cells at P0 (Fig. 3). In the middle passages, 3 of 8 P8 AFS lines exhibited biallelic expression of the previously silent IGF2 allele, whereas the biallelic expression of the H19 gene was found in only one P8 AFS line and in 2 P12 lines. Both alleles of MEST were expressed, and all 9 AFS lines that carried an SNP in SNRPN showed biallelic expression of this gene after passage 12. Among the late-passage (P17) cells, 7 of 8 AFS lines showed biallelic expression for IGF2. One of 8 AFS lines exhibited biallelic expression of the H19 gene, and 2 AFS line did not express either allele of H19.
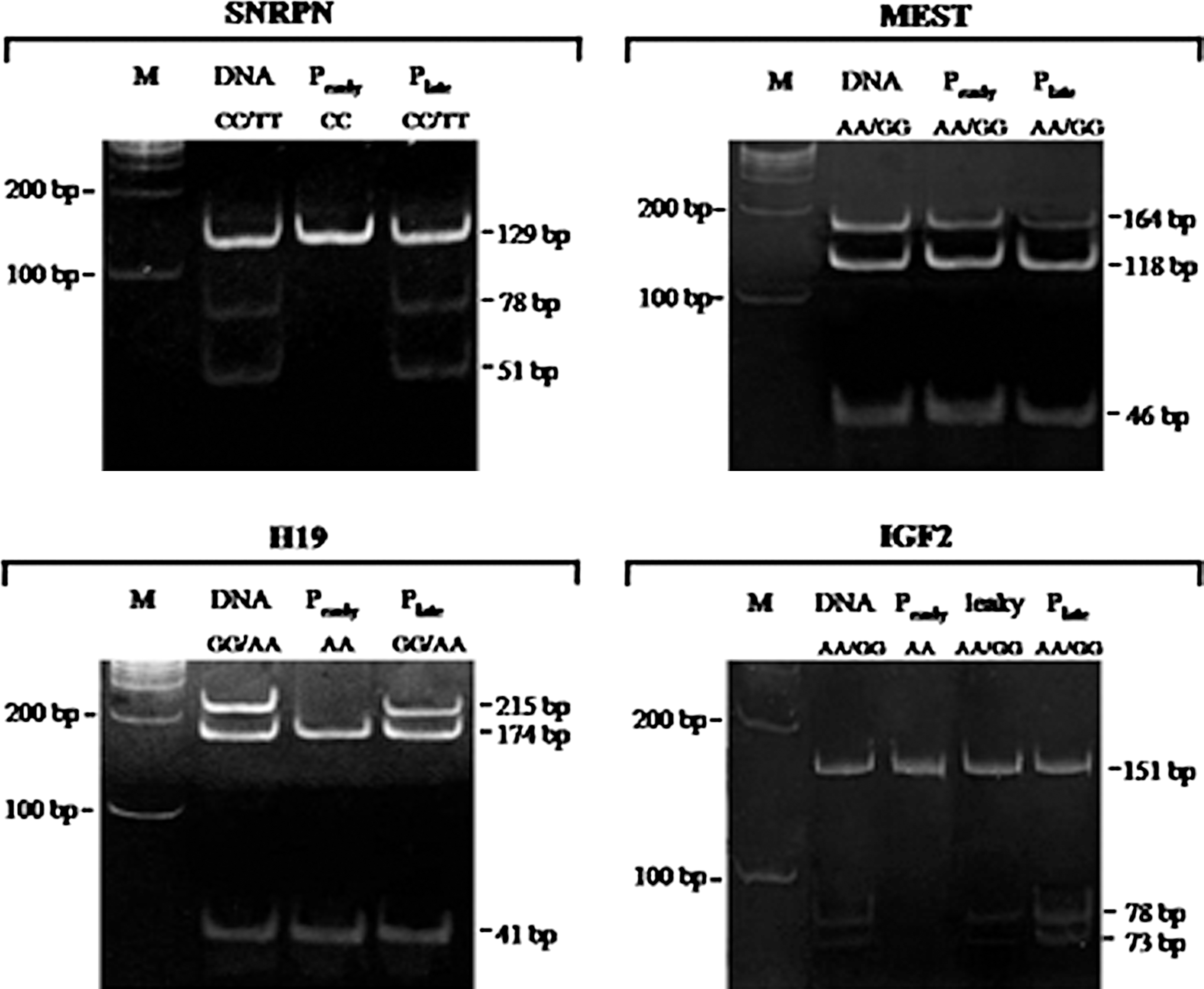
Allelic expression patterns of 4 imprinted genes in the early- and late-passage AFSs. A biallelic expression analysis was used to determine the bias of parental allele expression in AFS lines carrying SNPs in IGF2 (n=8), H19 (n=8), SNRPN (n=9), and MEST (n=7). The DNA lane shows the heterogeneous alleles after restriction enzyme digestion at the site of the SNP. The allelic expression patterns of AFS line at passage 2 and passage 17 were, respectively, shown in Pearly and Plate lanes. The leaky expression pattern of IGF2 was observed in AFS at passage 4, indicating the loss of gene imprinting.
DNA methylation
The AFS lines at the early and late passages were analyzed to determine the global change in the level of the DNA methylation using reverse-phase HPLC. The results revealed DNA hypomethylation in the genome of late-passage AFSs (7.6%±2.5%) compared with the cells at early passages (9.3%±2.1%). Although no significant difference (P>0.05) was observed in genomic methylation levels of AFSs between early and late cultures, these data demonstrate that global methylation of the genome of undifferentiated AFSs was lost during long-term culture.
For the site-specific analysis of methylation, we studied differentially methylated regions in the ICRs of IGF2 and H19 in 8 AFS lines containing SNPs in IGF2 and H19 using bisulfite sequencing. At the early passage, all cell lines contained an equal proportion of fully methylated and fully unmethylated DNA in both genes, demonstrating the fidelity of the monoallelic imprinting of IGF2 and H19 during short periods of in vitro culture. These results are consistent with the expression levels and allelic expression analysis results. In the analysis of the late-passage cell lines, 4 of 8 AFS lines were found to exhibit variations in or loss of methylation at a specific CpG site in the imprinted allele at DMR0 of IGF2 (Fig. 4). In addition, 3 of 8 AFS lines exhibited variations or loss of methylation at H19 (Fig. 5). We found that 3 late-passage AFS lines had aberrant site-specific methylation patterns in the ICRs of both IGF2 and H19.

The methylation of the imprinting control region (ICR) of IGF2. The methylation of the ICR of IGF2 was analyzed using bisulfite sequencing.

The methylation of the ICR of H19. The methylation of the ICR of H19 was analyzed using bisulfite sequencing.
Discussion
AFSs are interesting alternative MSC source that have potential uses in regenerative medicine. Based on the loss of differentiation capacity that has been observed in long-term cultured stem cells, we investigated the changes in genetic control mechanisms in AFSs related to stem cell development during early, middle, and late periods of in vitro culture. Gene imprinting analysis was performed for 4 imprinted genes (IGF2, H19, SNRPN, and MEST) in 3 different imprinting clusters. The results indicate that IGF2, SNRPN, and MEST became overexpressed after long-term culture. Increases in the expression of these imprinted genes in mid- and late-passage AFSs were associated with aberrant allele-specific imprinting, variations, and loss of methylated CpG sites in the ICR. Genomic methylation analysis also revealed global changes in hypomethylation in late-passage AFSs. Based on our results, we suggest that MSC lines cultured for extended passages harbor genomic epigenetic anomalies and should not be used for therapeutic purposes.
Previous studies have reported the loss of differentiation potential in stem cells cultured for extended passages derived from various sources, such as embryonic stem cells [4], MSCs obtained from adipose [5], and bone marrow [6]. AFSs exhibit an intermediate potency between embryonic and adult stem cells. AFSs maintain some embryonic stem cell characteristics, giving these cells superior proliferation potential compared with MSCs from other sources. In this study, 7 of the 12 AFS lines could be expanded in vitro for more than 105 population doublings. However, despite the fact that phenotypic antigenic surface markers P25 are not different from P2 and P17, we found that the cell differentiation potential was lower in the higher passage cells. This finding supports that analysis limited to phenotypic characteristics of cell surface markers is not enough to indicate the suitable use of stem cells for study or medical applications. IGF2, an imprinted gene, was studied in AFSs, and this analysis revealed the precise coordination of the epigenetic mechanisms of genomic imprinting and methylation. Variations in site-specific methylation in the ICR of IGF2 (started in mid-culture AFS) affect the imprinting of parental allele expression and are related to changes in the level of expression of imprinted genes in AFSs during long-term culture.
Our work presents evidence for the association between epigenetic modifications and the differentiation capacity of stem cells. One AFS line, AF 2F, displayed a high proliferation potential, with expansion for more than 32 passages. In the early passages, this cell line exhibited monoallelic expression of the IGF2 gene and expressed IGF2 at the same level as P0 AFSs. In addition, this cell line was able to differentiate into osteogenic and adipogenic lineages at the early passages. However, after P8, the IGF2 expression level of AF 2F gradually increased concomitant with biallelic expression, indicating the loss of parent specific imprinting of the IGF2 gene. The cell line at P8 had a lower potential to differentiate into both osteogenic and adipogenic lineages when compared with P2. At late passages, the AF 2F line sustained a high proliferation rate. These cells exhibited biallelic expression and overexpression of IGF2, whereas methylation was found on previously unmethylated CpGs, resulting in a loss of expression of H19. The cell line lost the ability to differentiate into the osteogenic lineage, and the yield of adipogenic differentiation was low. In contrast, another 2 AFS lines, AF T21 and AF T12, showed high proliferation potential in passage 31 with a PDT of 1.3 days. These cell lines displayed normal expression levels and monoallelic patterns of IGF2 and H19 genes at passages 2, 17, and 25. These 2 AFS lines were capable of differentiating into adipogenic and osteogenic lineages at passage 25. These observations support the relationship between epigenetic instability and differentiation capacity observed in late-passage stem cells.
The differentially methylation domain of H19 governs 7 binding sites for the insulator proteins CTCF1-CTCF7. The methylation of CTCF blocks enhancer-initiated transcription, resulting in silencing of IGF2 [15]. Our study investigated site-specific methylation of the CTCF6 domain known to be a key regulatory domain for switching of H19 and IGF2 expression [16]. However, we found no correlation between site-specific methylation of CTCF6 and H19 expression levels in 3 of 8 AFS lines. It is possible that other CTCF binding sites have a strong effect on H19 expression in AFS lines. This observation may be explained by the study of Murphy et al. [17], which revealed that hypermethylation of CTCF1 correlates to the higher levels of IGF2 expression.
MEST has been found to promote cell enlargement [18] and to induce the loss of the undifferentiated state [19] in early developmental cells. The overexpression of MEST observed during repeated passaging of AFSs may be associated with instability of the undifferentiated state and senescence of AFSs at a high passage. Our work found the up-regulation of MEST in AFSs at passage 10 under a standard culture medium. This is contradictory to our finding that AFSs generated under xeno-free culture medium supplemented with human umbilical cord serum can sustain normal expression of MEST and IGF2 compared with subculture passage 17 (unpublished data). Taken together, this implies that improvement of the stem cell culture system is needed to provide high-quality MSCs for clinical use.
Our data showed biallelic expression of MEST in AFSs throughout the culture period. There are 2 possible reasons to explain these data. First, MEST may not be imprinted in fetal stem cells derived from amniotic fluid, as a double dose of MEST expression is needed for fetal cell development during gestation [20]. Second, some isoforms of MEST are not differentially imprinted, and this could have biased the amplification of non-methylated alleles during the PCR. As described by McMinn et al. [21], MEST can produce mRNA in many isoforms. Isoform1 of MEST is maternal imprinted in many human organs, including the placenta, and initiates from exon1c. Transcription of MEST Isoform2 initiates from exon1a, is nonimprinted, and can be found in many nonplacental organs. Our study investigated allelic expression using an SNP at a site in exon 14. To distinguish this possibility, further work should investigate allelic expression of MEST using an SNP at a site on exon1 of MEST.
DNA methylation is important for regulating gene activity and cell development. DNA methyltransferases (DNMTs), including DNMT1, DNMT3a, and DNMT3b, play important roles in the methylation of genomic DNA. The loss of methylation in specific regions and genomic hypomethylation in late-passage AFSs may be caused by the dysregulation of these enzymes. Future work should investigate the expression and function of DNMTs in long-term cultured stem cells to determine whether DNMTs activity can be assayed as bio-markers to define stem cell quality in large-scale MSC production for research and therapeutic applications.
In summary, the present work demonstrates that epigenetic instability correlates to the loss of differentiation potential in late-passage AFSs. Our findings suggest that the appropriate timelines for the in vitro culture of MSCs from other sources should be determined. Our results also demonstrate that long-term cultured AFSs are not suitable for research and in vivo applications where imprinted gene expression and differentiation potential can influence the outcomes.
Footnotes
Acknowledgments
The authors are grateful to Peter A. Mcguin for editing this article. This work was funded by the Faculty of Medicine of Siriraj Hospital, Mahidol University. Partial funding was provided by Thailand Research Funds, Thailand.
Author Disclosure Statement
The authors have no conflict of interests.