
Abstract
All-trans retinoic acid (ATRA) induces clinical remission in most acute promyelocytic leukemia (APL) patients by inducing terminal differentiation of APL cells toward mature granulocytes. Here we report that human umbilical cord-derived mesenchymal stem cells (UC-MSCs) are capable of inducing granulocytic differentiation of the APL-derived NB4 cell line as well as primary APL cells and also cooperate with ATRA in an additive manner. Transwell coculture experiments revealed that UC-MSCs' differentiation-inducing effect was mediated through some soluble factors. Differentiation attenuation by IL-6Ra neutralization and induction by addition of exogenous IL-6 confirmed that IL-6 secreted by UC-MSCs was at least partially responsible for this differentiation induction process. Moreover, we found that UC-MSCs activated the MEK/ERK signaling pathway in promyelocytic cells and pharmacological inhibition of the MEK/ERK pathway reversed UC-MSC-induced differentiation, indicating that UC-MSCs exerted effect through activation of the MEK/ERK signaling pathway. These results demonstrate for the first time a stimulatory effect of MSCs on the differentiation of APL cells and bring a new insight into the interaction between MSCs and leukemic cells. Our data suggest that UC-MSCs/ATRA combination could be used as a novel therapeutic strategy for APL patients.
Introduction
A
Since mesenchymal stem cells (MSCs) constitute a key part of the microenvironment in vivo and could be easily expanded in vitro, the potential clinical value of MSCs is a subject of great interest in recent years. Reports indicate MSCs' therapeutic significance in diseases, including tissue damage [21], autoimmune disorders [22], graft versus host diseases after allogeneic stem cell transplantation [23]. However, there are controversial opinions regarding the role of MSCs in tumorigenesis and antitumor therapy.
For years, a considerable amount of research focused on the influence of MSCs on the growth and apoptosis of tumor cells of hematopoietic and nonhematopoietic origin [24 –29], although presenting controversial results. Nevertheless, little is known about the influence of MSCs on the differentiation of tumor cells. Numerous reports demonstrated that MSCs enhanced the differentiation of normal hematopoietic progenitor cells toward both myeloid and lymphoid lineages [30 –35]. However, whether MSCs also play a role in regulating the differentiation of leukemic stem/progenitor cells remains unknown.
To gain insight into the exact interaction between MSCs and leukemic cells and figure out a new way of differentiation therapy, we sought to find out whether MSCs could affect differentiation of APL cells. In this study, we established umbilical cord MSCs (UC-MSCs) and leukemic cells (APL-derived NB4 cell line [1] as well as primary APL cells) coculture system to fully characterize the possible influence of UC-MSCs on the differentiation of APL cells. We found that UC-MSCs caused G0/G1 cell cycle arrest and granulocytic differentiation of APL cells, and cooperated with ATRA to exert an additive effect. Regarding the underlying mechanism, we identified that UC-MSCs exerted effect at least by secreting IL-6, which led to the activation of the MEK/ERK signaling pathway in APL cells.
Our study revealed a role of MSCs in promoting the differentiation of APL cells and suggested a novel and promising cell-based combinatorial differentiation therapy for APL.
Materials and Methods
Reagents
ATRA, nitroblue tetrazolium (NBT), phorbol myristate acetate, and indomethacin were purchased from Sigma-Aldrich. Stock solutions of ATRA were dissolved in ethanol at 1 mM and stored protected from light at −20°C. Phycoerythrin-conjugated anti-CD11b and anti-CD14 were from BD Biosciences Pharmingen. The human IL-6Ra neutralizing antibody was from R&D Systems. The human granulocyte colony-stimulating factor (G-CSF) neutralizing antibody was from Abcam. Recombinant human IL-6 was from Peprotech. Antibodies against phospho-MEK1/2, phospho-ERK1/2, MEK1/2, ERK1/2, and actin were purchased from Cell Signaling Technology. PD98059, U0126, and SB203580 were from Cell Signaling Technology.
Isolation and culture of UC-MSCs
UC-MSCs were isolated from umbilical cords obtained from local maternity hospitals with donors' informed consent. Human tissue collection for research was approved by the Institutional Review Board of the Chinese Academy of Medical Science and Peking Union Medical College. Informed consent was obtained from patients in accordance with the Declaration of Helsinki. The details of isolation, ex vivo expansion, and identification of UC-MSCs were essentially as previously described [36]. UC-MSCs were maintained in Dulbecco's modified Eagle medium: Nutrient Mixture F-12(Ham) (1:1) supplemented with 10% fetal bovine serum (FBS; Hyclone), 10 nM dexamethasone (Sigma Aldrich), 2 mM
Culture of acute promyelocytic leukemic cells and coculture experiments with UC-MSCs
The NB4 cell line, reserved in our laboratory, is derived from FAB-M3 and is highly representative of the human APL pathophysiology [1]. The HL-60 cell line, reserved in our laboratory, is derived from FAB-M2 [37]. Bone marrow samples were collected from newly diagnosed APL patients at the Chinese Academy of Medical Sciences & Blood Disease Hospital. Informed consent was obtained from patients in accordance with the Declaration of Helsinki. Patients were diagnosed according to the French-American-British classification. Mononuclear cells were aspirated by Ficoll-Paque liquid. Each sample contained >95% leukemic blasts, as verified by morphologic examination. Leukemic cells were maintained in the RPMI 1640 medium (Gibco Life Technologies), supplemented with 10% FBS, 2 mM
Determination of differentiation of NB4 cells
Differentiation status of NB4 cells was determined by Wright–Giemsa staining, NBT reduction test, and cell surface differentiation markers CD11b and CD14 as previously reported [20]. Wright–Giemsa staining was performed for morphological evaluation of granulocytic differentiation. Superoxide generation was assessed quantitatively by the NBT reduction test. Four hundred cells were counted to determine the fraction of NBT-positive cells. Cell surface differentiation markers CD11b and CD14 were quantified via flow cytometry (LSR II, BD) to show if leukemic cells differentiate along granulocytic lineage or monocytic lineage. In some cases, PD98059, U0126, SB203580, human IL-6Ra neutralizing antibody or recombinant human IL-6 was administered.
Analysis of cell cycle distribution by flow cytometry
Tumor cells were plated into six-well culture plates at a density of 1×105/mL and were cultured with or without underlying UC-MSCs or/and ATRA for 48 h before harvesting and quantified. Cells were fixed with 70% cold ethanol at 4°C for more than 12 h, washed with phosphate-buffered saline twice, then incubated with 50 μg/mL RNase A (Sigma Aldrich) for 30 min at 37°C, and stained with 50 μg/mL propidium iodide (Sigma Aldrich) at room temperature for 5 min. The DNA content was analyzed by flow cytometry using MODFIT software (Verity).
Real-time polymerase chain reaction analysis of MPO, c-Myc, C/EBPβ, C/EBPɛ, and IL-6 mRNA
Total RNA was extracted by using Trizol (Invitrogen). cDNA synthesis was done using the MLV RT kit (Invitrogen) according to the manufacturer's instructions. cDNAs and an internal housekeeping gene (GAPDH) were quantified using an SYBR Green-based real-time detection method (Applied Biosystems 7500 Real-Time Polymerase Chain Reaction [PCR] System). The primers used were as follows: MPO forward 5′-CGGGAGCGATTGTTTGAGC-3′, reverse 5′-TGTTGGGCGTGCCATACTG-3′; c-Myc forward 5′-CCACGTCTCCACACATCAG-3′, reverse 5′-GCTGGTGCATTTTCGGTTG-3′; C/EBPβ forward 5′-GGGCCCTGAATCGCTTAA-3′, reverse 5′-ATCAACAGCAACAAGCCCG-3′; C/EBPɛ forward 5′-GCTGGCTGGTGGATTGTG-3′, reverse 5′-CTGGGTCCT GCCCTC TTT-3′; IL-6 forward 5′-CCAGTACCCCCAGGAGAAGAT-3′, reverse 5′-TTGCCTTTTTCTGCAGGAAC-3′; GAPDH forward 5′-CTCCTCCACCTTTGACGCTG-3′, reverse 5′-TCCTCTTGTGCTCTTGCTGG-3′. PCR cycling conditions were 95°C for 10 min, 95°C for 15 s, and 60°C for 1 min for 40 cycles, followed by the final single peak-melting curve program. Each sample was done in triplicate, and mean values were used for quantification. PCR products of IL-6 were resolved on 1% agarose gels.
Transwell experiments
The transwell experiment was conducted in a six-well plate with lower and upper chambers separating by a membrane with 0.4-μm-diameter pores (Corning Costar). UC-MSCs were plated into the lower chamber and irradiated by 30 Gy before NB4 cells (5×105 per well) were added into the upper compartment. Tumor cells were collected after 48 h culture and the cell surface marker CD11b was analyzed by flow cytometry.
Determination of cytokine secretion by enzyme-linked immunosorbent assay
Cell-free supernatants were collected and kept frozen at −80°C until assayed for cytokine concentrations by enzyme-linked immunosorbent assay (ELISA). ELISA assay kits for IL-6 and G-CSF were used following the supplier's instruction (Neobioscience Biotech Co, Ltd.). The PGE2 ELISA kit was purchased from Cayman Chemicals and used according to the protocol of the manufacturer.
Western blot analysis
After 24 and 48 h incubations, tumor cells were harvested and lysed in the RIPA buffer [50 mM Tris-HCl (pH 7.6), 150 mM sodium chloride, 1% NP40, 0.25% deoxycholic acid, 1 mM ethylene glycol bis(2-aminoethyl) tetraacetic acid, 1 mM ethylene diamine tetraacetic acid, 1 mM sodium fluoride, 1 mM sodium orthovanadate] with protease inhibitor cocktail (Sigma Aldrich). The protein concentration of the lysates was quantified by the BCA protein assay kit (Pierce). Equal amounts of protein (20 μg) were electrophoresed in 12% or 15% sodium dodecyl sulfate-polyacrylamide gel and transferred to polyvinyldifluoride membranes (Millipore). The membranes were blocked for 1 h with 5% bovine serum albumin (BSA), and incubated with primary antibodies diluted 1:2,000 for phospho-ERK1/2 and actin, and 1:1,000 for ERK1/2, phospho-MEK1/2, as well as MEK1/2 in 5% BSA at 4°C overnight. After washing, membranes were incubated with the corresponding horseradish peroxidase-conjugated secondary antibody (Pierce) diluted 1:2,000 in 5% BSA. The immunocomplexes were visualized with an enhanced chemiluminescence detection kit according to the manufacturer's instructions (Pierce). When necessary, membranes were stripped by incubation in 62.5 mM Tris-HCl (pH 6.8), 2% sodium dodecyl sulfate, and 100 mM 2-mercaptoethanol for 30 min at 50°C. After washing, membranes were reprobed with other antibodies.
Statistical analysis
Data are presented as mean±standard deviation. Comparisons of the data of each group were performed by the one-way analysis of variation test. The level of significance was set at a P value of <0.05, and when positive indicated by asterisks (*P<0.05; **P<0.01). The SPSS 16.0 software package was used for the statistical analysis.
Results
UC-MSCs promote the differentiation of NB4 cells toward mature granulocytes and present additive differentiation-promoting activity with ATRA
For the evaluation of myeloid cell maturation, we monitored NB4 cell differentiation using three classical assays: cell morphology, the NBT reduction test, and flow cytometric analysis of CD11b and CD14 expression [20]. Since ATRA, although having therapeutic limits is the main differentiation inducer used in clinical settings against APL, we analyzed the capacity of UC-MSCs combined with suboptimal ATRA in the mean time. In our experiment, UC-MSCs and ATRA (10 nM) were applied individually or in combination to NB4 cells. As shown in Fig. 1A, NB4 cells treated with UC-MSCs for 72 h showed morphological evidence indicative of granulocytic maturation. Undifferentiated control NB4 cells were found to be predominantly promyelocytes with round and large nuclei, but cells cultured with UC-MSCs had a more condensed and smaller nucleus pattern, resembling myelocytes and metamyelocytes. Compared with cells treated with ATRA alone, those treated with both ATRA and UC-MSCs appeared more mature, presenting more band-form nuclei and accidental segmented nuclei. These morphological data were further corroborated by the results of NBT test as well as the immunophenotyping of CD11b, a myeloid differentiation marker that is expressed from myelocyte stage onward, and CD14, a monocytic differentiation marker. After 72 h treatment, a significant increase in NBT reduction (Fig. 1B, C) and CD11b expression (Fig. 1D) were triggered in the leukemic cells treated either with UC-MSCs or ATRA compared with control cells. When used together, UC-MSCs and ATRA cooperated to enhance NBT reduction and CD11b cell surface expression in an additive manner. This upregulation was shown to be time dependent as UC-MSCs upregulated CD11b positivity to 18.37%±2.53% at 24 h, 30.7%±1.97% at 48 h, and 39.83%±2.53% at 72 h. However, CD14 expression consistently stayed at a level lower than 3% (data not shown). Previous work has demonstrated that NB4 cells have the potential to differentiate into granulocytes or monocytes [38]. We herein confirmed that UC-MSCs promote granulocytic rather than monocytic differentiation of NB4 leukemic cells. Further, we also examined the differentiation-inducing effects of UC-MSCs or/and 10 nM ATRA on primary blast cells from APL patients by detecting CD11b positivity after 48 h culture. UC-MSCs and 10 nM ATRA individually increased CD11b positivity to more than 60% and more than 80%, and when combined together, to 95.1% (Fig. 2A). Therefore, we draw conclusion that UC-MSCs induce granulocytic differentiation of APL cells.
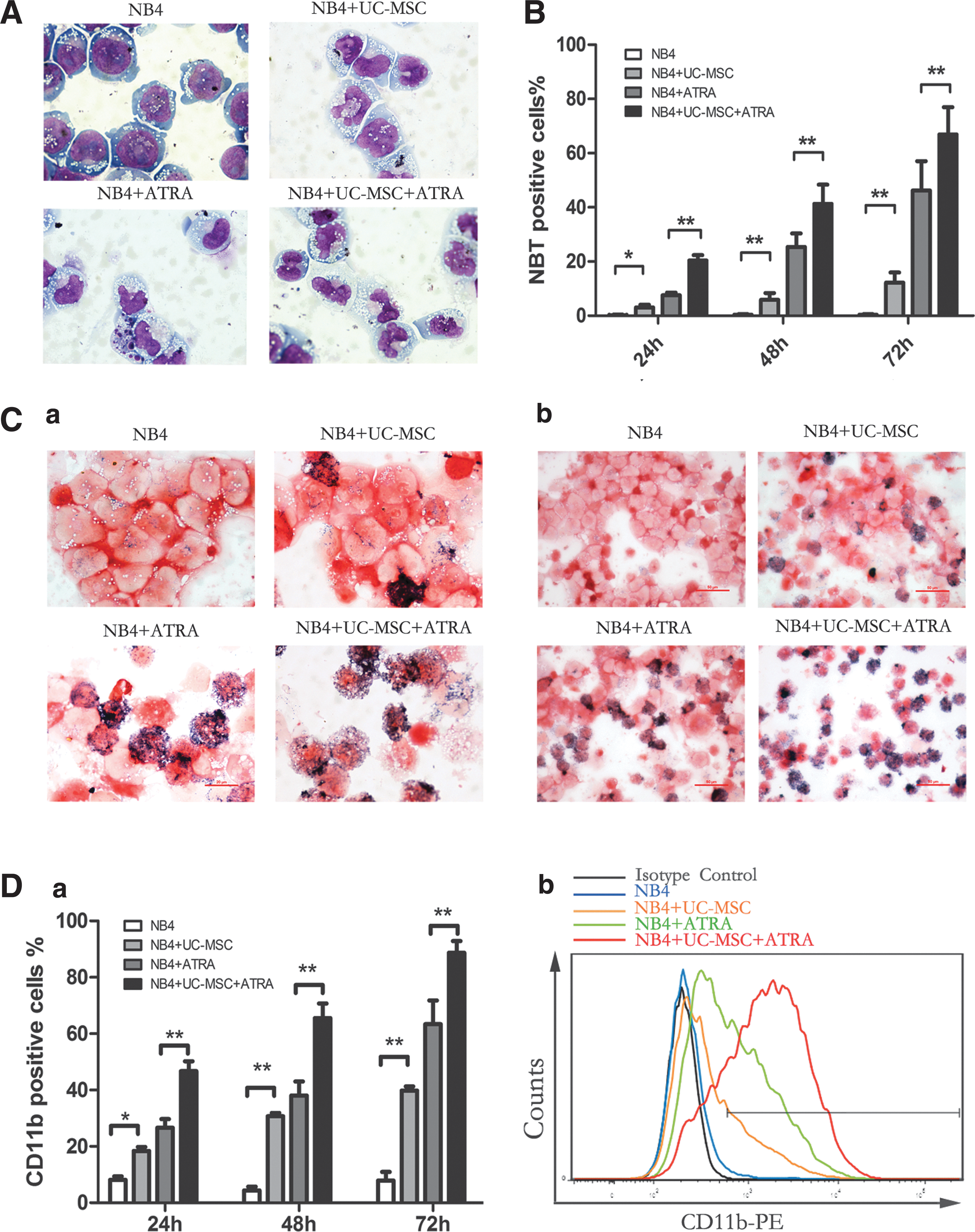
Umbilical cord-derived mesenchymal stem cells (UC-MSCs) induce granulocytic differentiation of NB4 cells and present additive effect with all-trans retinoic acid (ATRA). Differentiation of NB4 cells were assessed by Wright–Giemsa staining, the nitroblue tetrazolium (NBT) reduction test, and CD11b positivity after incubation with UC-MSCs or/and 10 nM ATRA for 72 h.
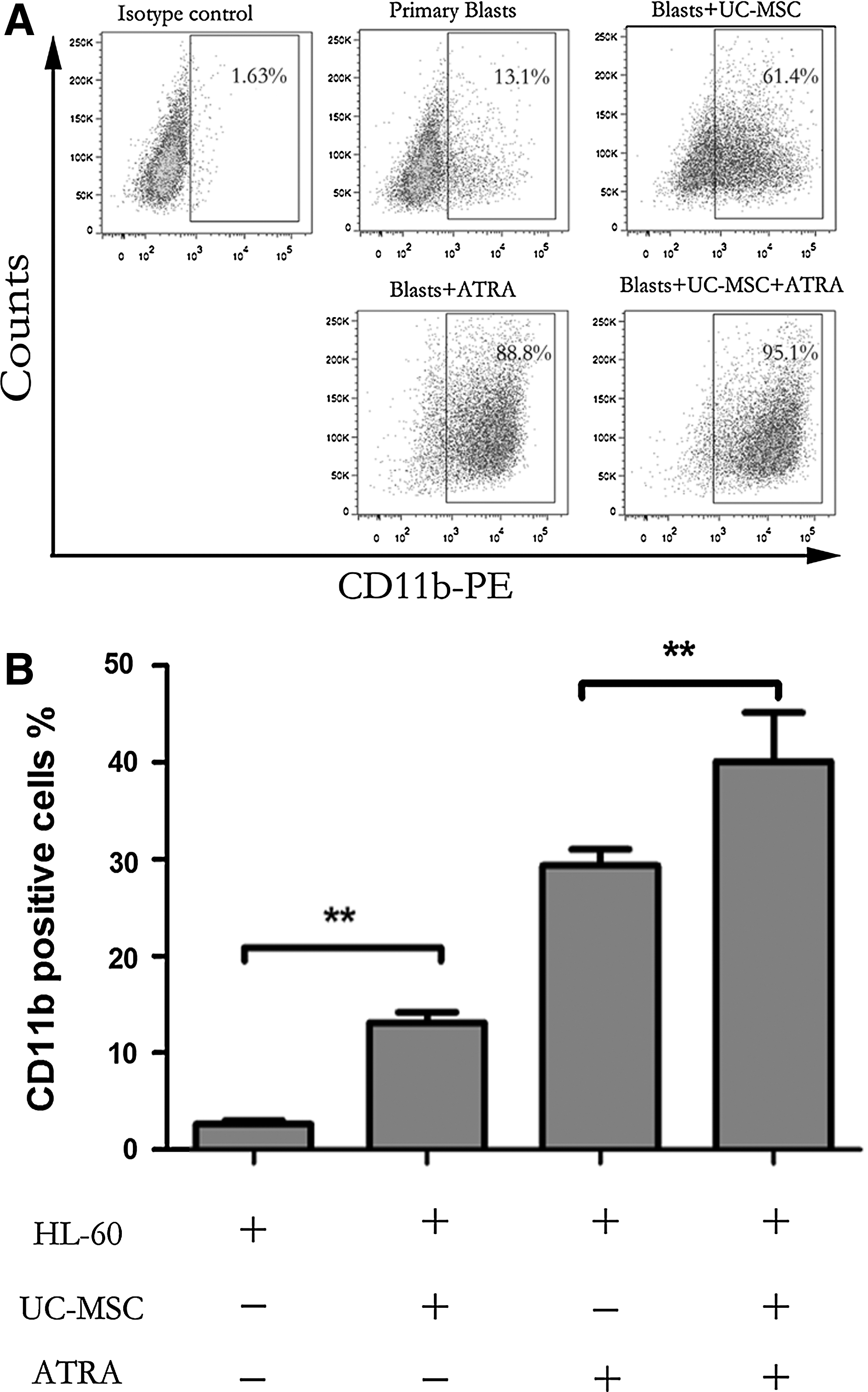
UC-MSCs induce granulocytic differentiation of primary blast cells from acute promyelocytic leukemia (APL) patients and HL-60 cells.
It should be pointed out that, in fact, we initially performed the experiment with MSCs not irradiated. However, MSCs proliferated so fast that they were detached from the six-well plate at the third day. Hence, we had MSCs irradiated when they were 80%–90% confluence to stop them from proliferating. We compared the CD11b expression of NB4 cells after 48 h coculture with MSCs irradiated and not irradiated, and the values were similar without statistical difference. Overall, irradiation does not impact the effect of MSCs in our experiments.
To know whether the differentiation-inducing effect of UC-MSCs is generalizable or unique to promyelocytic leukemic cells, we next investigated UC-MSCs' effect on HL-60 cells. We measured the expression level of CD11b on HL-60 cells after 48 h culture, and found that UC-MSCs also enhanced CD11b expression (Fig. 2B), although to an extent much less compared with NB4 cells, probably because HL-60 cells are more premature than NB4 cells.
The effect of UC-MSCs on NB4 cell cycle and mRNA expression of MPO, c-Myc, C/EBPβ, and C/EBPɛ
As previously reported, differentiation of promyelocytic leukemic cells is accompanied with cell cycle arrest, mostly G0/G1 block [39]. We then analyzed the cell cycle distribution of NB4 cells treated with or without UC-MSCs or/and 10 nM ATRA for 48 h. As shown in Fig. 3, UC-MSCs and ATRA caused G0/G1 block, respectively, and an even more obvious effect when they are applied together, with the proportion of S phase conversely regulated and no obvious change of the proportion of the G2/M phase.
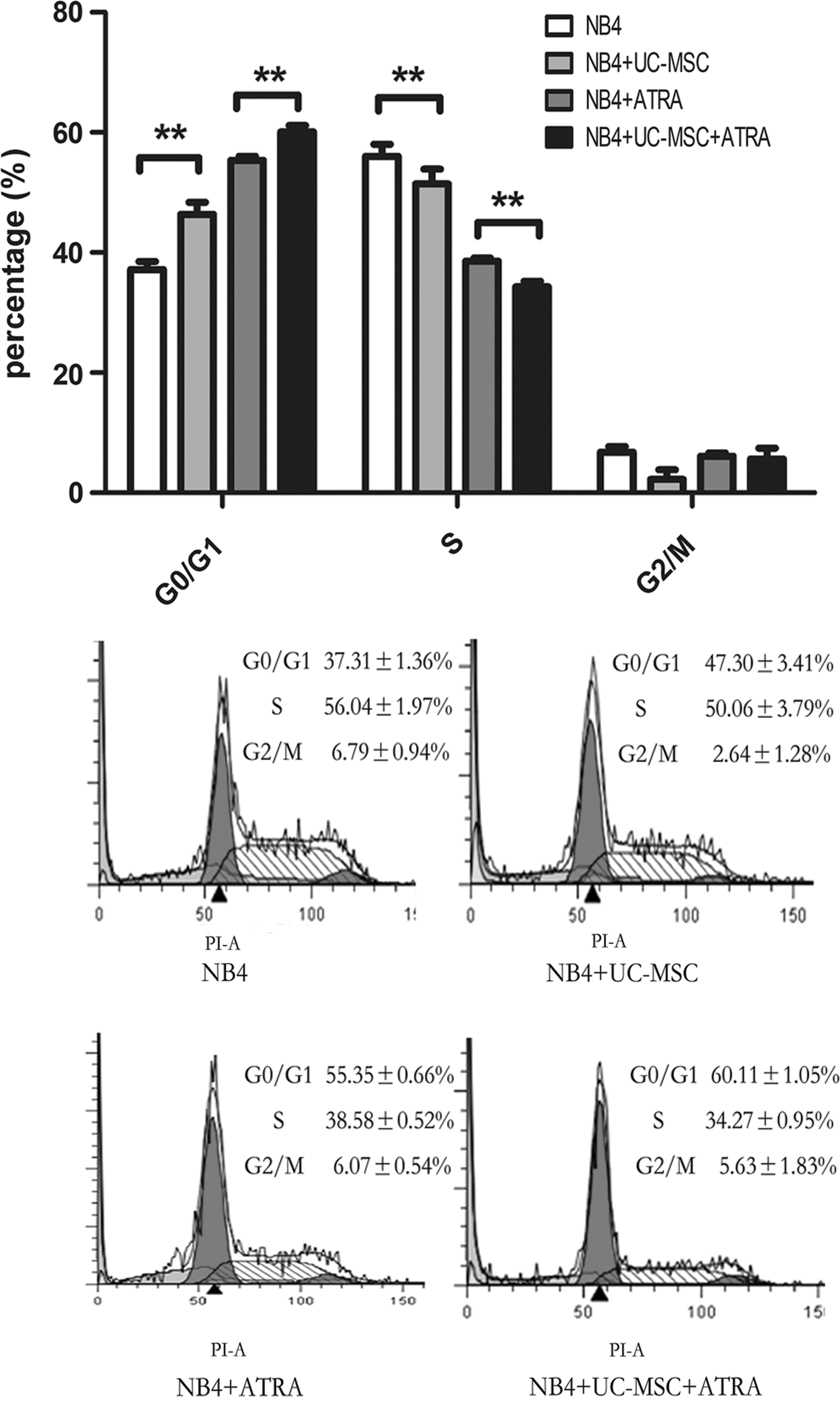
UC-MSCs' differentiation-promoting effect is accompanied by G0/G1 cell cycle arrest cell cycle distribution of NB4 cells was analyzed as indicated in Materials and Methods section after being treated with UC-MSCs or/and 10 nM ATRA for 48 h. Data represent the mean±standard deviation of at least three independent experiments. **p<0.01.
Myeloid differentiation is characterized by downregulation of MPO [40], and the proto-oncogene c-Myc is rapidly downregulated during differentiation of myeloid leukemia [41]. We then evaluated the mRNA levels of MPO and c-Myc by real-time PCR after 24 h culture. UC-MSCs had the trend to downregulate MPO transcription, yet with no statistical significance, while they could significantly enhance ATRA's effect on the attenuation of MPO transcription (Fig. 4A). Both UC-MSCs and ATRA reduced the c-Myc mRNA level on their own, and this downregulation effect was further enhanced with the combination of UC-MSCs and ATRA (Fig. 4B). The downregulation effect of MSCs on MPO and c-Myc is consistent with the UC-MSCs' differentiation-promoting activity. Previous studies also hinted at a role for the CCAAT/enhancer-binding protein (C/EBP) family in myeloid differentiation [42]. Among them, C/EBPβ [43] and C/EBPɛ [44] were most commonly reported to be implicated in ATRA-induced granulocytic differentiation of APL cells. Of note, C/EBPɛ was indicated to be upregulated during granulocytic, but not monocytic differentiation [45]. To address the regulation of the C/EBP family members during UC-MSC-induced granulopoiesis, we measured the mRNA level of C/EBPβ and C/EBPɛ by real-time PCR and found that UC-MSCs increased transcription of C/EBPβ, and produced an additive effect with ATRA (Fig. 4C). Regarding C/EBPɛ, UC-MSCs alone were inclined to upregulate it yet with no statistical significance. However, UC-MSCs together with ATRA significantly increased C/EBPɛ mRNA to a much higher level compared with ATRA alone (Fig. 4D). These results suggest the upregulation of C/EBPβ or C/EBPɛ might be responsible for the differentiation induction by UC-MSCs.
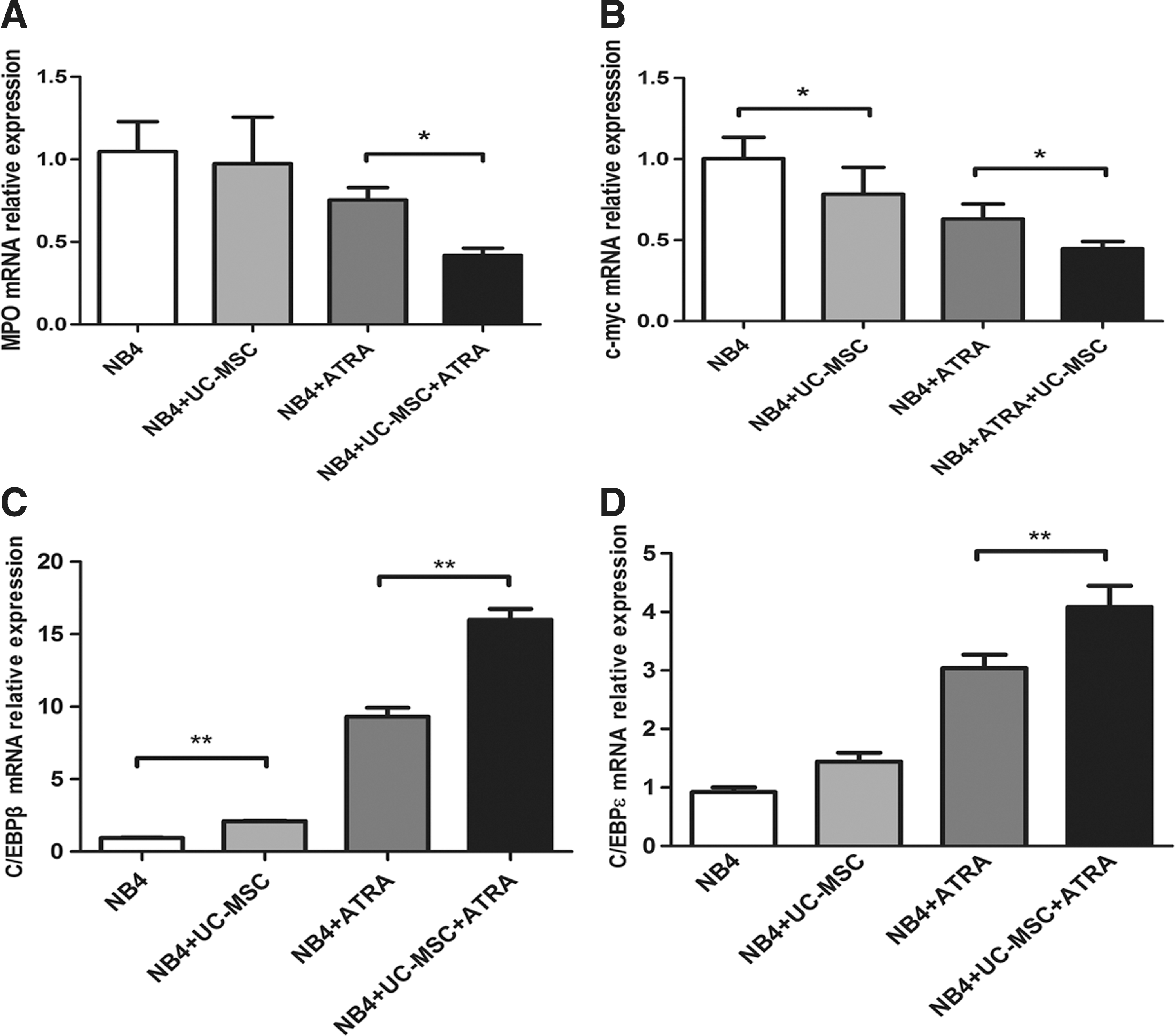
UC-MSCs downregulate mRNA level of MPO and c-Myc, and upregulate mRNA level of C/EBPβ and C/EBPɛ in NB4 cells. Real-time polymerase chain reaction was conducted to measure the mRNA level of MPO
IL-6 is the major soluble mediator of the differentiation induction by UC-MSCs
To elucidate whether UC-MSCs exert effect through soluble factors or direct cell–cell contact, we performed coculture experiments using the transwell system in which leukemic cells and UC-MSCs were physically separated by a membrane permeable only for soluble factors, but not cells. As shown in Fig. 5A, even in the absence of direct cell–cell contact, UC-MSCs were still able to effectively augment CD11b expression of NB4 cells, with no statistical difference between the cell–cell contact group and the transwell group. Cells treated with UC-MSCs for 48 h were 29.35%±2.21% positive without transwell and 26.35%±6.57% positive with transwell. Meanwhile, CD11b positivity of cells treated with UC-MSCs plus ATRA were 67.18%±10.77% without transwell and 65.23%±10.18% with transwell. Results here indicate that the effect of UC-MSCs was almost completely mediated by some soluble factors. Previous studies have suggested the contribution of G-CSF [46,47] and PGE2 [48] to granulocytic maturation of promyelocytic leukemic cells, or to the enhancement of ATRA-induced differentiation. Therefore, we analyzed the possible role these factors might play in the differentiation process. By ELISA, both the G-CSF and PGE2 concentration dramatically increased in the coculture supernatants (data not shown). However, when either the G-CSF neutralizing antibody (50 μg/mL) or indomethacin (10 μM) was applied to the coculture system to block G-CSF function or prevent PGE2 synthesis, the CD11b augmentation by UC-MSCs was not diminished at all (Fig. 5B, C). Further, when exogenous G-CSF (100 ng/mL) was applied to the coculture system, the level of CD11b was barely changed (data not shown). Based on these results, we ruled out the possibility of participation of G-CSF and PGE2 in UC-MSCs' differentiation induction. Recently, IL-6 was identified to mediate human bone marrow MSCs' (BM-MSCs) contribution to in vitro differentiation of hematopoietic progenitor cells toward myeloid and lymphoid cells [35]. Xie et al. reported that IL-6 dramatically enhanced retinoic acid-induced granulocytic differentiation of HL-60 cells (a leukemic cell line derived from M2) [49]. Then, we focused our attention on IL-6, a factor abundantly produced by UC-MSCs [50]. The concentration of IL-6 was significantly upregulated in the coculture supernatant (Fig. 6A). To determine the role of IL-6, we performed neutralizing experiments with the IL-6Ra neutralizing antibody (50 μg/mL) be added to the coculture system to prevent IL-6 from binding to its receptor, the isotype antibody used as a control simultaneously. After incubation for 48 h, CD11b was detected by flow cytometry. As shown in Fig. 6B, IL6Ra neutralization attenuated CD11b upregulation by UC-MSCs from 28.7%±6.49% to 14.0%±4.26%, and decreased the value in UC-MSCs plus the ATRA group from 71.5%±8.19% to 54.8%±9.4%, a level slightly higher compared with the ATRA group, which was 47.8%±9.02%. The reversion was not complete, with the possibility that some other soluble factors are involved in this differentiation process or the IL-6Ra neutralizing antibody does not fully block the function of IL-6. To further address IL-6's function in promoting differentiation, exogenous IL-6 (50 ng/mL) was applied and CD11b positivity was evaluated after 48 h treatment. As shown in Fig. 6C-a, IL-6 and ATRA could, respectively, elevate the CD11b expression to be 39.1%±2.91% and 63.4%±2.34% positive, while IL-6 plus ATRA could result in a greater effect to 81.27%±2.9%. This differentiation-inducing effect of IL-6 was also corroborated by morphological change shown by Wright–Giemsa staining (Fig. 6C-b). To define the origin cells of IL-6 production in the coculture system, we conducted real-time PCR to quantify IL-6 mRNA in UC-MSCs or NB4 cells after 24 h incubation. We observed that IL-6 mRNA expression in UC-MSCs was significantly upregulated to more than sevenfold when it was cocultured with NB4, and further increased to more than 13-fold when ATRA was added (Fig. 6D-a). Conversely, cDNA reverse transcribed from IL-6 mRNA in NB4 cells either untreated or treated with UC-MSCs or/and ATRA could not be amplified with the same PCR condition. Electrophoresis of the PCR products was conducted afterward, and the photographed image in Fig. 6D-b verified the results of real-time PCR. These results suggested that IL-6 was derived from UC-MSCs in the culture. Taken together, we concluded that UC-MSCs promote granulocytic differentiation of NB4 leukemic cells at least, in part, through IL-6 secretion.
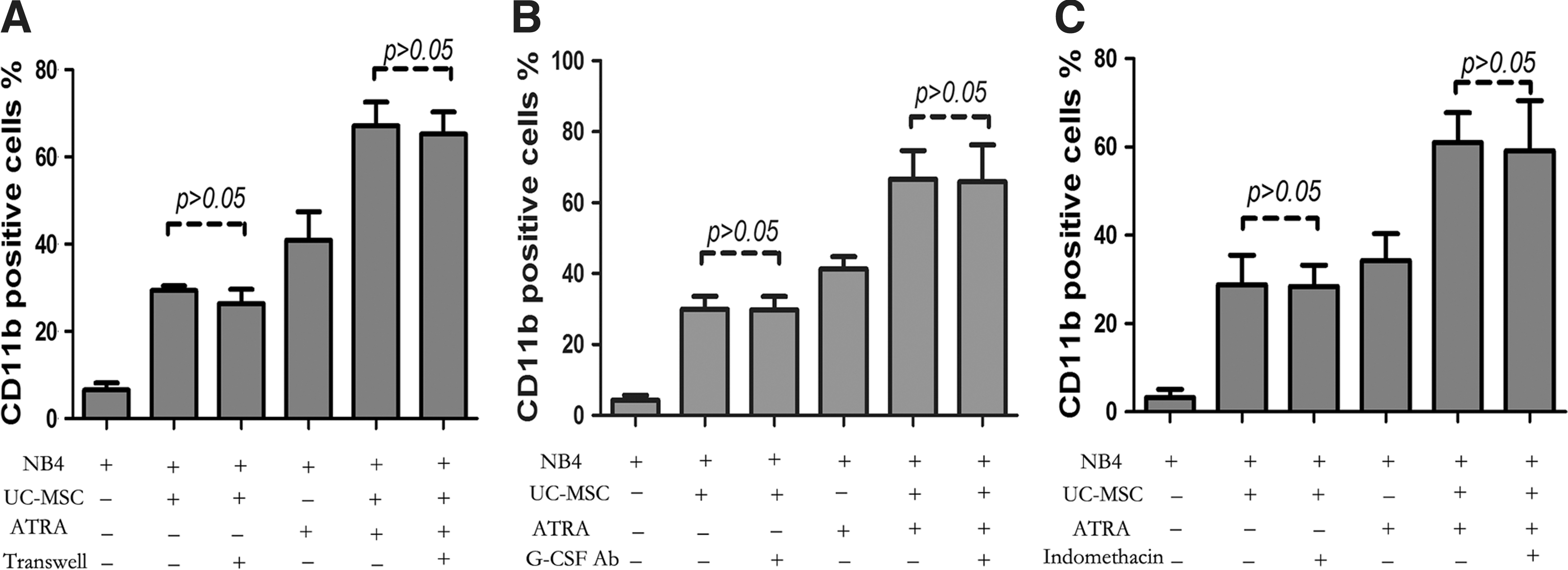
UC-MSCs' differentiation-promoting effect is achieved through some soluble factors and granulocyte colony-stimulating factor (G-CSF) and PGE2 are not the mediator.
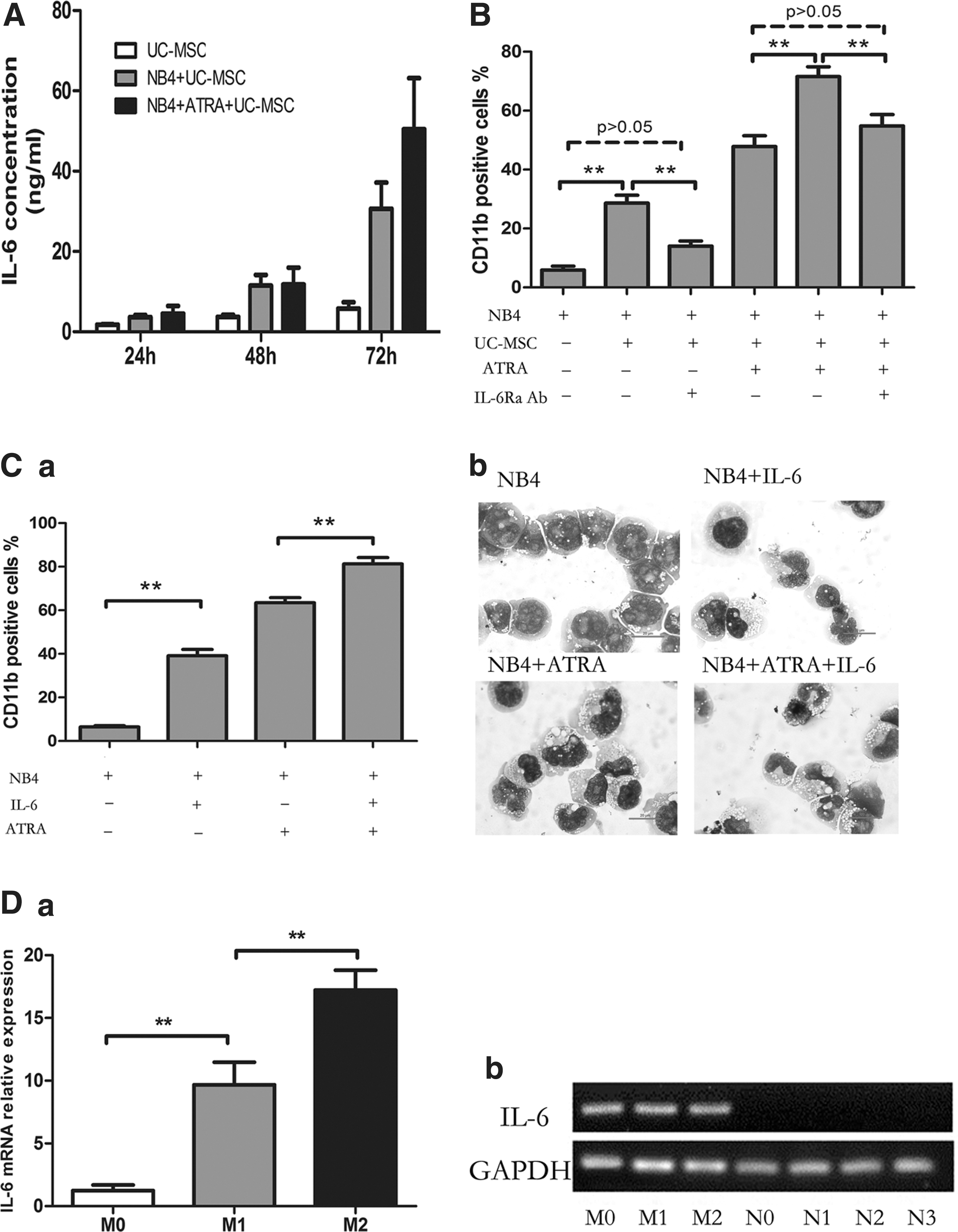
UC-MSCs promote granulocytic differentiation of NB4 cells by way of secreting IL-6.
Role of MEK/ERK-signaling pathway in differentiation of NB4 cells triggered by UC-MSCs
Next, we explored the signaling pathway elicited in NB4 cells during the UC-MSC-induced differentiation process. Previously, MEK/ERK signaling has been widely reported to play a vital role in differentiation of various kinds of cells, including normal cells and tumor cells [51,52]. In the hematopoietic system, the MEK/ERK pathway has been shown to be required for granulocytic [53], megakaryocytic [54], and erythrocytic differentiation [55]. MEK/ERK has also been reported to play a major role in ATRA-induced granulocytic differentiation of HL-60 cells [56]. In recent years, the MEK/ERK pathway was reported to be involved in the differentiation of NB4 cells induced by ATRA [57] or genistein [19]. Hence, we examined the activation status of MEK and ERK by western blotting after treatment with UC-MSCs and/or ATRA for 24 and 48 h. Our data showed that UC-MSCs and ATRA enhanced phosphorylation of MEK1/2 and ERK1/2, respectively, and cooperated to exert an even more obvious effect (Fig. 7A). To further determine whether activation of MEK/ERK signaling is responsible for NB4 cell differentiation induced by UC-MSCs, we used PD98059 (a specific inhibitor of MEK1) and U0126 (a nonselective inhibitor of MEK1 and MEK2) to see the influence of MEK/ERK inhibition on the expression of CD11b. For comparison, we also examined the effect of the p38MAPK inhibitor SB203580, since p38MAPK [58] was earlier reported to regulate myeloid cell differentiation. NB4 cells were pretreated with PD98059 (30 μM), U0126 (10 μM), or SB203580 (10 μM) for 1 h before treatment by UC-MSCs or/and ATRA for additional 48 h. As shown in Fig. 7B, PD98059 could partially reverse UC-MSCs' effect, while U0126 almost totally reversed it with no significant difference between NB4 cells treated by UC-MSCs plus U0126 and control cells. At the same time, we observed that PD98059 reduced CD11b expression of tumor cells treated with ATRA plus MSCs to a level even lower than ATRA-treated cells, and U0126 reduced CD11b positivity to a basal level, which is consistent with previous reports that MEK/ERK plays a critical role in ATRA-induced NB4 cell differentiation [57]. The reversion of the differentiation marker by PD98059 and U0126 suggested that UC-MSCs exert effect almost totally by activation of the MEK/ERK pathway. It is worth noting that SB203580 further raised the CD11b level of NB4 cells treated with UC-MSCs from 28.97%±6.49% to 46.1%±4.12%, and the positivity of cells treated with UC-MSCs plus ATRA from 71.07%±5.11% to 87.57%±3.51%, indicating that p38MAPK plays a negative role in regulating NB4 differentiation. Since IL-6 was proved to be the soluble mediator of this differentiation-promoting effect, and was previously shown to activate the MEK/ERK-signaling pathway in various kinds of cells, such as phagocytes [59], epithelial cells [60], and neurons [61], we wonder whether IL-6 exerts effect via MEK/ERK activation under this circumstance. Herein, we carried out experiments with PD98059 or U0126 added to the culture system of NB4 cells stimulated by IL-6. Data in Fig. 7C displayed that PD98059 partially reversed and U0126 completely abolished its upregulation of CD11b. Based on these facts, we conclude that UC-MSCs regulate differentiation partly by secreting IL-6 to activate the MEK/ERK-signaling pathway in NB4 cells.

UC-MSCs induce granulocytic differentiation of NB4 cells by activation of its intracellular MEK/ERK signaling pathway and IL-6 also functions through this pathway.
Discussion
MSCs have been demonstrated to have great potential for treating various diseases, especially those related to tissue damage as well as immune disorders. However, there are discrepancies on its application in malignancies. Whether MSCs are pro- or antitumorigenic is a subject of controversial reports. The possible role that MSCs might play in influencing tumor cells' behavior has been extensively investigated by a number of groups in recent years, mostly focusing on proliferation and apoptosis [25,26,28]. Our previous study has also shown that UC-MSCs inhibit the growth of HL-60 and K562 leukemic cells [62]. Nevertheless, little is known about the role of MSCs in modulating differentiation of leukemic cells.
Due to abundant supply, painless collection procedure, less chance of microbial contamination, and better expandability [63], UC-MSCs possess advantages in research and great potential in future clinical application. In this work, we observed for the first time that UC-MSCs could promote granulocytic differentiation of NB4 cells as well as primary APL blast cells, and when used in combination with ATRA, could elicit an additive effect. As far as the underlying mechanism is concerned, we established that UC-MSCs enhance differentiation at least partly by secreting IL-6 to activate the MEK/ERK signaling pathway in NB4 leukemic cells. Our proposal was supported by several evidences. First, UC-MSCs secreted a larger amount of IL-6 upon coculture with NB4 in comparison with the basal secretion by UC-MSCs. The IL-6Ra neutralizing antibody could partially reverse the differentiation induction by UC-MSCs, and exogenous IL-6 also promoted granulocytic differentiation of NB4 cells. Second, UC-MSCs enhanced the phosphorylation of MEK/ERK, and inhibition of the MEK/ERK-signaling pathway almost totally abrogated the UC-MSCs' differentiation-promoting effect. Finally, inhibition of the MEK/ERK signaling pathway also abolished IL-6-mediated induction of granulocytic differentiation.
The capability of ATRA to induce the differentiation of APL cells has been well documented. Our findings show that mechanism underlying UC-MSC-induced differentiation overlaps with that of ATRA in G0/G1 cell cycle arrest [39], downregulation of c-Myc [64], upregulation of C/EBPβ [43] and C/EBPɛ [44], and activation of the MEK/ERK-signaling pathway [57] in NB4 cells. The importance of c-Myc in leukemogenesis has been revealed in recent years. Ectopically expressed c-Myc in murine bone marrow progenitors induced acute myelogenous leukemia [65], while abrogation or inhibition of it induced cell cycle arrest and terminal differentiation of leukemic cell lines and primary acute myelogenous leukemic cells [66,67]. Consistent with these previous studies, UC-MSCs significantly reduced the mRNA level of c-Myc in APL cells. Upregulation of C/EBPβ and C/EBPɛ were reported to be closely related with granulocytic differentiation. Transfection of leukemic cell lines with C/EBPβ [43] or C/EBPɛ [45] could drive leukemic cells to differentiate into mature granulocytes. UC-MSCs significantly increased mRNA transcription of C/EBPβ by itself, and dramatically enhanced the mRNA level of C/EBPɛ combined with ATRA, implying that regulation of c-Myc, C/EBPβ, and C/EBPɛ may also contribute to the differentiation-promoting activity of UC-MSCs.
Recently, IL-6 was identified to mediate human BM-MSCs' contribution to in vitro differentiation of hematopoietic progenitor cells toward myeloid and lymphoid cells [35]. Xie et al. reported that IL-6 enhanced retinoic acid-induced granulocytic differentiation of APL cells [49]. However, the cell line used in their experiments was HL-60, which was actually not derived from APL (FAB-M3), but derived from the French-American-British classification M2 (FAB-M2) [37]. In our study, we show that IL-6 induced differentiation of NB4 cells derived from APL, to a much greater extent compared with HL-60 cells, highlighting its role in promoting granulocytic differentiation of promyelocytic leukemic cells.
Previous research compared UC-MSCs and BM-MSCs in cytokine secretion, and discovered that UC-MSCs are more abundant in hematopoietic growth factors, including G-CSF, GM-CSF, IL-6, IL-8, and IL-11 [36,50]. Of note, IL-6 produced by UC-MSCs was 1571.0±617.2 pg/mL, significantly higher than that produced by BM-MSCs which was 704.0±51.5 pg/mL [50]. Since our data highlight the critical role of IL-6 in mediating UC-MSCs' function, we doubt BM-MSCs' capability to compete with UC-MSCs in differentiation-inducing efficiency and whether BM-MSCs from APL patients have defects in promoting myeloid cell differentiation, which needs further exploration.
In our experiment, neutralization of IL6Ra partially, but not completely, reversed the UC-MSC effect with the possible reason that some other soluble factors exerted effect simultaneously, or the neutralizing antibody was not effective enough to totally block IL-6 from binding its receptor. Several lines of investigation have suggested that G-CSF augments ATRA-induced granulocytic differentiation in APL [46,47]. PGE2 was also reported to contribute to granulocytic differentiation of NB4 cells [48], although COX genes were not listed among the ones modulated by ATRA in NB4 using microarray analysis [68]. Discrepant with previous reports, results detailed in the current study indicate that G-CSF and PGE2 did not participate in UC-MSC-mediated differentiation of APL cells.
IL-6 was previously shown to activate the MEK/ERK pathway in a variety of cell types, such as phagocytes [59], epithelial cells [60], and neurons [61], but not in APL cells. In this study, we found that IL-6 induce MEK/ERK signaling in APL cells. MEK/ERK signaling has already been widely reported to be involved in differentiation of various kinds of cells, and verified to be activated during differentiation of NB4 cells induced by ATRA [57] and genistein [19] in recent years. Consistent with those reports, our data show that the MEK/ERK pathway was also activated by UC-MSCs and contributed to the differentiation of NB4 cells.
While MEK/ERK plays a positive role in regulating granulocytic differentiation, opinions conflict regarding the role of p38MAPK. Actually, both the promoting effect [58] and inhibitory effect [69] of p38 on promyelocytic differentiation have been previously reported. Alsayed et al. found that p38MAPK was activated during ATRA-induced differentiation, but exhibited negative regulatory effects on the induction of differentiation [69]. Consistently, inhibition of p38 using SB203580 augmented the expression level of the cell surface differentiation marker CD11b in APL cells in our experiment, supporting that p38 activation is a suppressive factor.
In summary, this is the first report demonstrating UC-MSCs' capability to induce terminal granulocytic differentiation of APL leukemic cells and to elicit an additive effect when combined with a suboptimal dosage of ATRA. Despite the fact that ATRA alone or in combination with chemotherapy results in high rates of complete remission in APL patients [2], its clinical application is still frustrated by the potentially fatal side effects [4] and acquired resistance [12]. Our data here presented a promising cell-based combinatory therapeutic strategy that could lower the dosage of ATRA without reducing effectiveness, somehow alleviating the problems incurred by a therapeutic dose of ATRA. Our study demonstrates for the first time, a role for MSCs in promoting the differentiation of APL cells and brings a new insight into the interaction between MSCs and leukemia.
Footnotes
Acknowledgments
This study was supported by the 863 project (grant no. 2011AA020118 and no. 2012AA02A211), the 973 program of China 2011 CB964800 (grant no. 2011CB964802) from the Ministry of Science & Technology of China, the National Key Scientific Instrument and Equipment Development Projects (grant no. 2011YQ03013404), and the National Natural Science Foundation of China (grant No. 30900557, 81270595, and 81000196).
Author Disclosure Statement
No competing financial interests exist.