
Abstract
Hypoxic culture has been shown to delay premature senescence occurring during in vitro culture. Human mesenchymal stem cells (hMSCs) cultured under hypoxia have been reported to maintain their stemness properties and delay senescence compared to the cells cultured under normoxia. However, the molecular mechanism by which hypoxia regulates premature senescence has not been fully revealed. In this study, hMSCs were cultured under the conditions of 21% (normoxia) and 1% O2 (hypoxia) tension and analyzed for cell growth, expression of MSC surface markers, multilineage differentiation, and cellular senescence. Our results showed that more cells retained MSC surface markers in hypoxic culture than those in normoxic culture, and hypoxia was able to enhance multilineage differentiation of hMSCs. The hypoxic condition also delayed cellular senescence of hMSCs, increased activation of AKT signaling, and upregulated both intra- and extracellular levels of macrophage migration inhibitory factor (MIF) compared to the normoxic condition. Inhibition of AKT activity in hypoxic culture increased the number of cells with positive staining for senescence-associated β-galactosidase activity, upregulated expression levels of senescence-associated markers p16 and p21 mRNA transcripts, and decreased expression levels of potency-associated markers, including NANOG, OCT3/4, and SOX2. On the other hand, upregulated intra- and extracellular levels of MIF by stable MIF overexpression in normoxic culture increased the activation of AKT while decreasing mRNA expression of senescence-associated markers and increasing expression of potency-associated markers. Taken together, our findings suggest that hMSCs in hypoxic culture produce endogenous MIF to activate AKT signaling to delay the progression of cellular senescence.
Introduction
S
The serine/threonine kinase AKT, also known as protein kinase B, is a critical signaling molecule involved in the regulation of cellular senescence [18 –23]. Previous studies have shown that the activation of AKT suppresses cellular senescence and maintains stem cell properties of human skin-derived precursors and mouse embryonic stem cells [18 –20]. It has also been reported that PI3K/AKT signaling is activated by hypoxia [24,25] and inhibits transcriptional activity of p53 [26]. Given that p53 is a potent senescence-inducing factor, suppression of p53 activity by the activation of the PI3K/AKT signaling pathway may also result in inhibition of cellular senescence. However, several previous studies have shown contradicted findings that AKT activation promotes cellular senescence in mouse embryo fibroblasts (MEFs), primary esophageal epithelial cells, and endothelial cells [21 –23]. These studies suggest that the role of AKT is critical in the regulation of cellular senescence and may be dependent on cell type and/or physiological status.
Macrophage migration inhibitory factor (MIF) is a proinflammatory cytokine [27] and can be induced by hypoxia, a feature of tissue inflammation [28]. MIF was first identified in activated T cells [29,30] and has also been found in other cells, such as endocrine cells, epithelial cells, and fibroblasts [31 –33]. MIF plays a crucial role in innate and acquired immunity systems and is involved in the pathogenesis of acute and chronic inflammation [27,34,35]. In addition, recent studies have shown that MIF activates the mitogen-activated protein kinase signaling pathway [36,37] and regulates p21 and p53 to modulate cell cycle and cell growth [33,38,39]. Particularly, it has been shown that MIF as an antisenescence factor antagonizes the activity of potent senescence factor p53 [40], and overexpressing MIF in MEFs suppresses the progression of cellular senescence [41].
Premature cellular senescence during in vitro culture is one of the major challenges encountered in hMSC research for translational applications. It would be of great benefit that hMSCs expanded in vitro are able to maintain their multipotency without undergoing premature cellular senescence. Several groups have reported that hypoxic culture delays cellular senescence of hMSCs [14,17]. The current knowledge gap is that it is not clear (1) if MIF plays a role in the regulation of cellular senescence of hMSCs under hypoxia and (2) how MIF activity regulates the activation of the AKT signaling pathway and vice versa in hypoxic culture. In this study, we hypothesized that hypoxic culture modulates MIF activity, which in turn regulates AKT signaling to control senescence of hMSCs. To test this hypothesis, we compared cellular senescence of hMSCs cultured under normoxia (pro-senescence condition) and hypoxia (antisenescence condition) and analyzed extracellular levels of MIF and activation of AKT in both normoxic and hypoxic culture conditions.
Materials and Methods
Chemicals, culture medium, and antibodies
Triciribine (TCN) and AKT Inhibitor IV were obtained from EMD Millipore Chemicals (Billerica, MA). Dulbecco's modified Eagle's medium-low glucose (DMEM-LG), DMEM-high glucose (DMEM-HG), and fetal bovine serum (FBS) were obtained from Invitrogen (Carlsbad, CA); antibiotic–antimycotic solution and Hank's balanced salt solution (HBSS), and phosphate-buffered saline (PBS) were purchased from Cellgro (Manassas, VA). Anti-MIF antibody was obtained from R&D Systems (Minneapolis, MN); antibodies for flow cytometric analysis were obtained from BD Biosciences (San Diego, CA); and all other antibodies for western blotting were obtained from Cell Signaling Technology (Boston, MA).
Isolation and maintenance of hMSCs
With the approval from the Institutional Review Board of the University of Wisconsin-Madison, femoral heads of two female patients at the ages of 25 and 65 years undergoing total hip arthroplasty were obtained through the collaboration with Dr. Matthew Squire at the Department of Orthopedics and Rehabilitation. Bone marrow was collected from femoral heads, diluted in 30 mL of DMEM-LG, and centrifuged at 1,200 rpm for 5 min. The collected cell pellet was then resuspended in 25 mL of HBSS, slowly added to 20 mL of Ficoll-Paque PLUS (GE Healthcare Life Science, Pittsburgh, PA) in a 50-mL tube, and centrifuged at 500 g for 30 min. After centrifugation, mononuclear cells were collected, diluted in 50 mL of HBSS, and centrifuged at 1,300 rpm for 10 min. The cell pellet was resuspended in 50 mL of HBSS and centrifuged again at 1,300 rpm for 10 min. Cells were collected, resuspended in DMEM-LG supplemented with 10% FBS and antibiotics, plated in cell culture plates, and maintained at 37°C in a humidified, 5% CO2 incubator. Culture medium was changed every 3 days, and upon 70% confluence, cells were passaged using 0.05% trypsin–EDTA (Invitrogen) and replated at a seeding density of 1,000 cells/cm2. Cells isolated from the different patients were independently cultured and used separately in repeats of an assay. Specifically, each analysis of this study was carried out using one donor's cells and then repeated using the other donor's cells to confirm the reproducibility of experimental data. The results of each assay presented in this study are representative results based on a donor's cells from reproducible data of assay repeats.
Normoxic and hypoxic cell culture
Isolated hMSCs were first maintained in culture with 21% O2, and when reaching 70% confluence, the cells were then divided into two groups for different culture: one group of cells was plated at a high cell density of 1,000 cells/cm2 in culture with 21% O2 (normoxia) and the other group of cells at the same seeding density in culture with 1% O2 (hypoxia). A commercial hypoxic culture chamber (BioSpherix, Lacona, NY) was used to create a hypoxic condition for hMSC culture. The oxygen tension in the chamber was detected by a sensor and precisely regulated by a computerized controller to pump N2 and CO2 mixtures into the chamber. Both groups were maintained either for two passages or until cultured cells reach senescence before various analyses were performed. The procedure of passaging cells during the study was conducted in the ambient environment within the least operation time to minimize exposure to normoxia.
Cell growth and bromodeoxyuridine assay
Cell growth in normoxic and hypoxic culture was evaluated by the analysis of accumulated population doublings (PDs). Cell numbers at each passage were determined by cell counting with a hemacytometer, and the number of PDs was calculated based on the formula PD=ln(C1/C0)/ln(2), where C0=the initial number of seeded cells; C1=the number of cells at the time of passaging. To analyze cell proliferation, the assay of bromodeoxyuridine (BrdU) incorporation was performed using the BrdU Flow kit following the manufacturer's instructions (BD Biosciences). Briefly, hMSCs in normoxic or hypoxic culture were incubated with 10 μM BrdU for 2 h, and then trypsinized, fixed, and permeabilized on ice. The cells were then incubated with DNAse for 1 h at 37°C before treated with anti-BrdU antibody and analyzed by flow cytometry (BD Biosciences). The data were analyzed using the FloJo software (Tree Star, Ashland, OR).
Analysis of cell surface markers by flow cytometry
To analyze the expression of cell surface markers, hMSCs in normoxic or hypoxic culture were harvested and washed with an ice-cold PBS buffer containing 1% bovine serum albumin and 0.1% sodium azide (Sigma-Aldrich, St. Louis, MO). The cells were then incubated with PE-conjugated anti-CD73 antibody, APC-conjugated anti-CD90 antibody, FITC-conjugated anti-CD105 antibody, and PerCP-conjugated anti-CD45 antibody at 4°C for 30 min. After washed with the buffer to remove unbound antibodies, the cells were analyzed by flow cytometry with the FloJo software to determine the expression of cell surface markers.
Senescence-associated β-galactosidase assay
A commercial staining kit (Cell Signaling) was used to detect the activity of senescence-associated β-galactosidase. Briefly, hMSCs were fixed in 1×fixative solution (2% formaldehyde and 0.2% glutaraldehyde in PBS) for 10 min at room temperature, rinsed twice in PBS, and incubated in a β-galactosidase staining solution (40 mM citric acid/sodium phosphate, 150 mM NaCl, 2 mM MgCl2, 50 mM potassium ferrocyanide, and 50 mM potassium ferricyanide) adjusted to a pH of 6.0 for 16 h at 37°C. To quantify the number of cells with positive staining for senescence-associated β-galactosidase, 100 cells in each of three independent wells of hMSC culture were counted, and the ratio of the number of stained cells to that of total cells was calculated.
Evaluation of multilineage differentiation of hMSCs
Human MSCs maintained under the normoxic or hypoxic condition for two passages were harvested and then seeded in cell culture plates at a density of 5,000 or 10,000 cells/cm2 for induction of osteogenesis or adipogenesis, respectively. The osteogenic medium was composed of DMEM-LG, 10% FBS, 0.1 μM dexamethasone, 10 mM β-glycerophosphate, 50 μg/mL ascorbic acid-2-phosphate (Sigma-Aldrich), and antibiotics. The adipogenic medium was composed of DMEM-HG, 10% FBS, 1 μM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine, 1 μg/mL insulin (Sigma-Aldrich), and antibiotics. To induce chondrogenesis, 250,000 hMSCs previously cultured under the normoxic or hypoxic condition for two passages were centrifuged at 600 g for 5 min to form a high-density cell pellet following a previous published method with modifications [42]. The chondrogenic medium was composed of DMEM-HG, 0.1 μM dexamethasone, 50 μg/mL ascorbic acid-2-phosphate, 1% ITS+(BD Biosciences), 0.9 mM sodium pyruvate, 40 μg/mL
To analyze osteogenic differentiation, osteogenic-induced hMSCs were stained for alkaline phosphatase (ALP) activity using the ALP staining kit following the manufacturer's instructions (Sigma-Aldrich). Furthermore, to quantify ALP activity, the cells were first treated with a digestion buffer containing 2% Triton X-100, 0.15 mM Tris base, 0.1 mM ZnCl2, 0.1 mM MgCl2·6H2O at pH 9.0 for 1 h at 37°C and then overnight at 4°C. The digestion buffer was then analyzed by measuring the reaction kinetics with p-nitrophenyl phosphate (Sigma-Aldrich). The results were normalized to double-stranded DNA (dsDNA) contents that were separately determined by the PicoGreen assay (Invitrogen).
To evaluate adipogenic differentiation, adipogenic-induced hMSCs were stained with Oil Red O (Sigma-Aldrich). After microscopic analysis, the stain of Oil Red O was extracted using 2-propanol and quantified by a spectrometer at the absorbance wavelength of 540 nm.
To evaluate chondrogenic differentiation of hMSCs, chondrogenic cell pellets were digested with a buffer containing 20 μg/mL papain (Sigma-Aldrich), 0.1 M sodium acetate, and 0.05 M EDTA at pH 5.53 for 20 h at 60°C. The amount of sulfated glycosaminoglycan (GAG) was determined using the 1,9-dimethylmethylene blue-based Blyscan Assay kit following the manufacturer's instructions (Biocolor, Carrickfergus, United Kingdom). The GAG results were normalized to dsDNA contents that were separately determined by the PicoGreen assay.
Inhibition of AKT signaling
To investigate if the regulation of AKT signaling affects senescence and potency of hMSCs, hMSCs at passage 4 or 5 in hypoxic culture were treated with AKT inhibitors: TCN at the final concentration of 0, 0.1, and 1.0 μM or AKT Inhibitor IV at the final concentration of 0, 0.01, and 0.1 μM for two passages before analysis. To investigate if the regulation of AKT signaling modulates MIF expression, hMSCs in hypoxic culture treated with or without 1.0 μM TCN were collected after 3, 6, 12, 24, 48, or 72 h for analysis.
Enzyme-linked immunosorbent assay for quantification of MIF
The amount of MIF in hMSC culture medium was quantified by a commercial ELISA kit (R&D Systems, Minneapolis, MN). Briefly, the cultured medium was collected from hMSC culture, centrifuged at 1,000 rpm for 5 min to remove any particles in the medium, and stored at −80°C before analysis. Enzyme-linked immunosorbent assay (ELISA) was performed to analyze the collected medium samples following the manufacturer's instructions.
Plasmid construction
To construct a plasmid for MIF overexpression, referred to as MIFover, the coding sequence of MIF gene of the pCMV6-Entry vector (Origene, Rockville, MD) was isolated by digesting the vector with EcoRI and FseI. The EcoRI and FseI fragment was inserted into the EcoRI and XhoI sites of the pMCs-Puro retroviral vector (Cell biolabs, San Diego, CA) through multiple ligation reactions. Briefly, the MIF fragment and viral vector were first ligated at the EcoRI site and then ligated at the FseI and XhoI sites after blunting the digestion sites using Klenow DNA polymerase. To prepare a control construct, referred to as MIFctl, pMCs-Puro was digested with EcoRI and XhoI and the linearized vector was re-ligated after blunting the digestion sites using Klenow DNA polymerase.
The retroviral vector-mediated shRNA against MIF (shMIF) was constructed by inserting double-stranded oligonucleotides (sense: 5′- gatc
Transfection and transduction for MIF misexpression
The MIFover or shMIF plasmid was co-transfected with the pCMV-VSV-G envelope vector (Cell biolabs) into the packaging cell Plat-GP (Cell biolabs) by calcium–phosphate precipitation. Briefly, 5 μg of the MIFover or shMIF plasmid and 5 μg pCMV-VSV-G were diluted in 250 mM CaCl2 solution with a final volume of 250 μL and then slowly mixed with 250 μL of 2×HEPES-buffered saline [2×HBS, 50 mM HEPES, 280 mM NaCl, 1.5 mM Na2HPO4 (pH 7.0)]. The plasmid/vector mixture was incubated for 20 min at room temperature and slowly added to Plat-GP cell culture treated with chloroquine at 50 μM for 15 min. Five hours after transfection, the medium was replaced with fresh medium, and 48 h after transfection, virus-containing medium was collected from the transfected packaging cell culture and filtered through a 0.45-μm PES filter to eliminate packaging cell contamination from the collected supernatant. The viral supernatant was then concentrated using Retro-X concentrator (Clontech, Mountain View, CA) by following the manufacturer's instructions and added into hMSC culture at 70%–75% confluence. To enhance transduction efficiency, hMSCs with virus were spun at 1,200 g for 5 min at room temperature before incubating the cells in a culture incubator. Successfully transduced cells were selected after the cells treated with 1 μM puromycin for 3 to 4 weeks reached ∼70% confluence. The transduction efficiency was ∼40%. After the selection, cells were expanded for six to eight doublings to obtain a sufficient amount of cells for experiments.
Total RNA isolation and real-time PCR analysis
Since senescence-associated activities are possibly affected by cell–cell contact, cells before reaching 30% density confluence were harvested for real-time PCR analysis to avoid potential effects by cell–cell contact. Total RNA was isolated using the NucleoSpin RNAII kit (Clontech) and converted to cDNA by the high-capacity cDNA reverse transcription kit (Invitrogen). Real-time PCR was performed using the iQ™ SYBR® Green Supermix (BioRad, Hercules, CA) with the primers listed in Table 1. Ubiquitin C (UBC) was used as a reference gene, and the relative expression levels of target genes to UBC were calculated using the ΔΔCT method.
Forward and reverse primers are indicated as “F” and “R,” respectively.
Protein extraction and western blot analysis
To prepare whole cell lysate, cells were harvested, washed with PBS, and lysed in 1×RIPA (50 mM Tris-HCl (pH 7.5), 1% NP-40 substitute, 0.25% sodium deoxycholate, 150 mM sodium chloride, 1 mM EDTA) supplemented with 1×Complete EDTA-free protease inhibitor cocktail, and 1×PhosSTOP (Roche, Indianapolis, IN). After the centrifugation to remove cell debris, the supernatant containing whole cell lysate was collected and subjected to the BCA assay using the Pierce BCA protein assay kit (Thermo scientific, Waltham, MA) to determine protein concentration of the lysate. Fifteen to 25 μg of protein samples were electrophoresed on a 4%–20% gradient gel and then transferred to PVDF membranes. The membrane was incubated with antibodies detecting MIF, AKT, phospho-AKT (Ser473) (pAKT), or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in a blocking solution [5% nonfat milk in 1×Tris-buffered saline containing 0.1% Tween 20 (TBS/T)] overnight at 4°C, washed three times in TBS/T, and then incubated with horseradish peroxidase-linked secondary antibody in the blocking solution at room temperature for 1 h. Western blot images were obtained using the Kodak Image Station 4000R Pro system (Carestream Health, Rochester, NY).
Statistical analysis
All quantitative assays were performed in triplicate. Data were expressed as the mean±SD, and a Student's t-test was performed to determine levels of statistical significance. A p-value of<0.05 was considered significant.
Results
Hypoxia delays cellular senescence of hMSCs
Previous studies have reported that hypoxic culture conditions prolong lifespan of many types of cells, including MSCs [14,15,17,43]. To confirm the findings, we cultured hMSCs under 21% O2 (normoxia) and 1% O2 (hypoxia) to determine the effect of oxygen tension on cell growth and cellular senescence. The growth curve of cells maintained in normoxic and hypoxic culture showed that the total number of PDs of the cells increased under the hypoxic condition (22 PD), compared to that under the normoxic condition (14 PD) (Fig. 1A). Despite the difference in the numbers of total accumulated PDs between the two groups of cells, growth rates based on slopes of the growth curves during the early culture period (the first 30 days of culture) were comparable between the groups (Fig. 1A). We selected hMSCs at passage 4 (p4) and passage 7 (p7) as representative cells at different stages of growth for the analysis of morphology, stem cell surface markers, proliferation, and senescence-associated markers. Cell morphology of p4 hMSCs in both hypoxic and normoxic culture was similar in size and shape but became substantially different with increased cell passages (Fig. 1B). Specifically, p7 hMSCs in normoxic culture displayed an enlarged flattened morphology, a characteristic of senescent cells, whereas those at the same passage maintained in hypoxic culture maintained spindle-shaped morphology. We further analyzed the expression of hMSC surface markers using flow cytometry to determine the percentage of cells expressing CD73+/90+/105+/45− in the whole cell population [44]. The results showed that 84.7% and 92.9% p4 hMSCs in normoxic and hypoxic culture, respectively, were CD73+/90+/105+/45− cells, whereas 45.3% and 75.0% p7 hMSCs in normoxic and hypoxic culture, respectively, were this cell population (Fig. 1C), suggesting that hypoxic culture is more capable of retaining the population of CD73+/90+/105+/45− cells than normoxic culture. In addition, the results of cell proliferation by the BrdU assay showed that BrdU was incorporated in ∼22% p4 hMSCs in normoxic or hypoxic culture (Fig. 1D). At cell passage 7, BrdU was incorporated in less than 1% hMSCs in normoxic culture, whereas BrdU was incorporated in 5.15% hMSCs in hypoxic culture (Fig. 1D). Hypoxia also reduced the number of p7 hMSCs with positive staining for the activity of senescence-associated β-galactosidase. The ratio of senescence-associated β-galactosidase-positive cells to total cells in hypoxic culture was less than that in normoxic culture (Fig. 1E). Real-time PCR assay was carried out to analyze the mRNA expression of p16 and p21, which are considered senescence-associated markers for the detection of cellular senescence [45 –47]. The real-time PCR results showed similar trends in the expression of p16 and p21 between hMSCs cultured under hypoxia and normoxia: the expression levels of p16 and p21 mRNA transcripts increased with the number of cell passage. However, when comparing the cells at the same passage number between hypoxic and normoxic culture, the expression levels of p16 mRNA transcripts of p7 hMSCs in hypoxic culture were lower than those of the cells in normoxic culture. Likewise, the expression levels of p21 mRNA transcripts of p7 hMSCs under hypoxia were significantly lower (P<0.005) compared to those of the cells under normoxia (Fig. 1F). Collectively, our results suggest that hypoxic culture is able to delay the progression of premature cellular senescence of hMSCs in vitro.
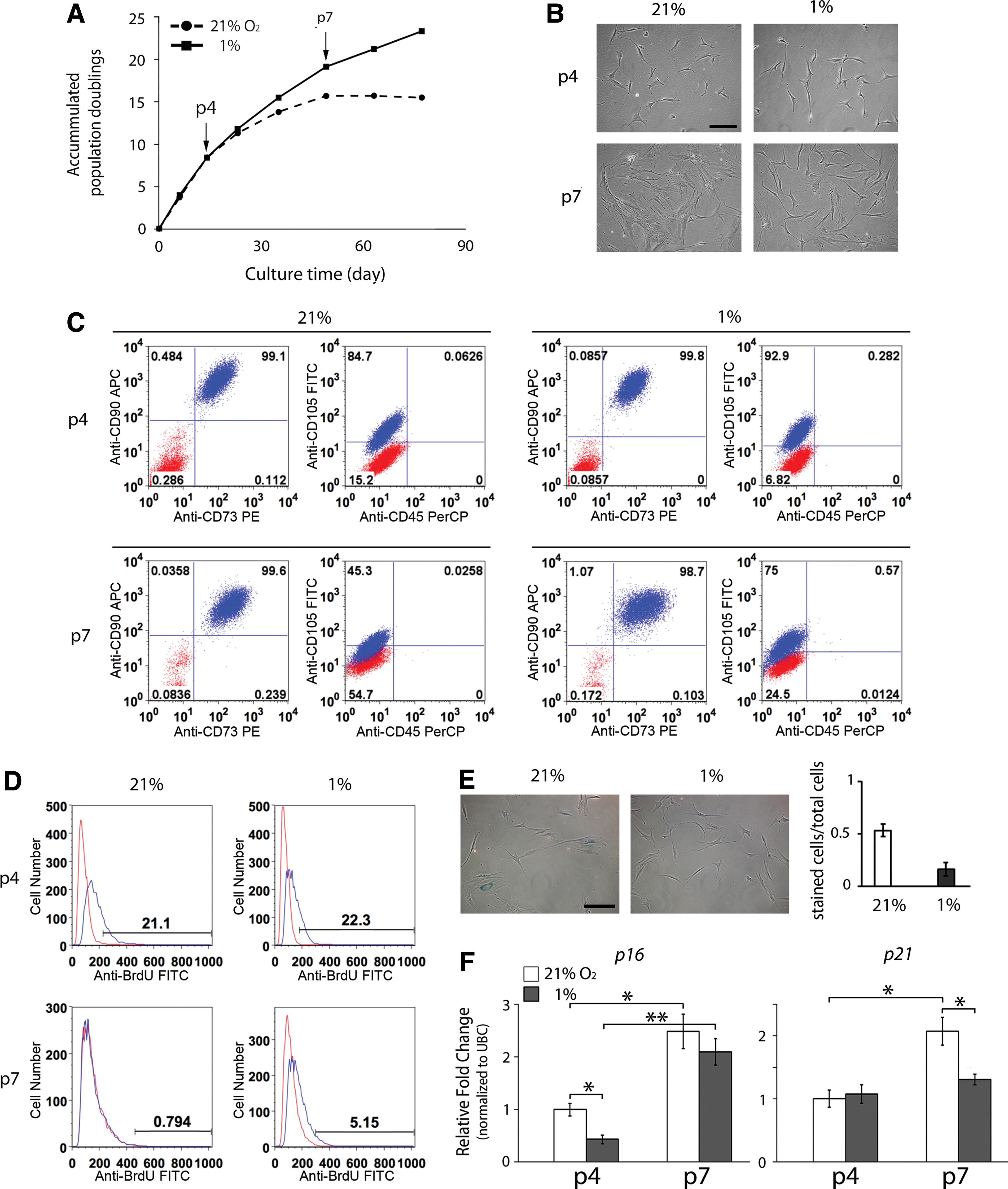
Cell response of human mesenchymal stem cells (hMSCs) in hypoxic and normoxic culture.
We next investigated the effect of hypoxia on the regulation of hMSC multipotency. The mRNA expression of potency-associated markers NANOG, OCT3/4, and SOX2 was analyzed to determine hMSC properties [48 –50]. The expression levels of potency-associated markers of hMSCs under both hypoxic and normoxic conditions were downregulated with increased cell passage numbers. When comparing the cells at the same passage number between hypoxic and normoxic culture, the expression levels of potency-associated markers NANOG, OCT3/4, and SOX2 were upregulated in hMSCs under hypoxia compared to those in the cells under normoxia (Fig. 2A). We further induced p4 hMSCs harvested from normoxic or hypoxic culture for osteogenesis, adipogenesis, and chondrogenesis under normoxia to determine the effect of normoxia and hypoxia during hMSC expansion/maintenance culture on the capacity of multilineage differentiation. The results showed that after 7 days of osteogenic induction, the mRNA levels of CBFA1 and ALP in hypoxia-cultured hMSCs were significantly higher than those in normoxia-cultured hMSCs (Fig. 2B). Hypoxia-cultured hMSCs also showed significantly upregulated ALP activity compared to normoxia-cultured cells after 14 days of induction (Fig. 2C). For adipogenesis, hypoxia-cultured hMSCs expressed higher mRNA levels of peroxisome proliferator-activated receptor gamma 2 (PPARG2) and lipoprotein lipase (LPL) than normoxia-cultured cells after 7 days of induction (Fig. 2D). The results of Oil Red O staining also revealed that hypoxia-cultured hMSCs produced more lipid droplets than normoxia-cultured hMSCs after 14 days of induction (Fig. 2E). Finally, our results showed that after 7 days of chondrogenic induction, the mRNA levels of SOX9 and aggrecan (AGN) in hypoxia-cultured hMSCs were significantly higher than those in normoxia-cultured hMSCs (Fig. 2F), whereas the GAG production was comparable between the two culture after chondrogenic induction for 14 days (Fig. 2G). Taken together, these results suggest that compared to normoxic culture, hypoxic culture is able to more effectively retain multipotency of hMSCs.
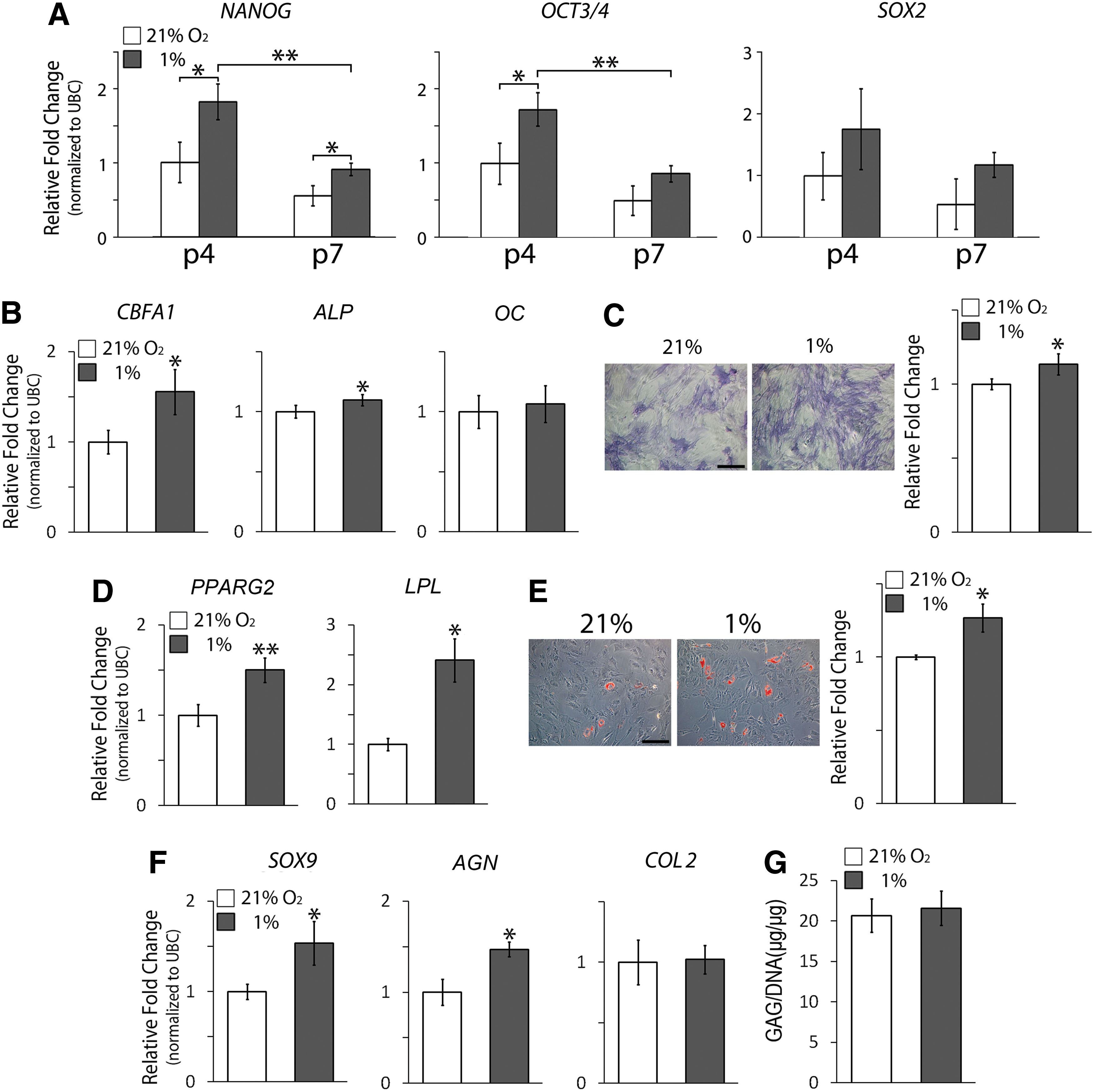
Potency and multilineage differentiation of hMSCs under hypoxia and normoxia.
Hypoxia increases AKT activation and MIF production
Our results shown in Fig. 1 suggest that hypoxic culture delays the progression of cellular senescence. Based on the finding, we further identified the role of MIF and AKT regulated by oxygen tension in the modulation of cellular senescence in hMSCs. We chose AKT and MIF for this study because both the molecules have been reported to be involved in the regulation of cellular senescence [18 –23]. The results of western blotting showed that the expression levels of total AKT in hMSCs under the normoxic condition were slightly higher than those under the hypoxic condition, but the levels of pAKT were increased in hypoxic culture compared to those in normoxic culture (Fig. 3A). Likewise, the expression levels of total MIF were upregulated in hypoxic culture than those in normoxic culture. Moreover, the amounts of soluble MIF produced by hMSCs in the culture medium determined by ELISA showed that a significantly higher (P<0.005) amount of MIF was detected in the medium of the culture under hypoxia than that in the culture under normoxia (Fig. 3B), suggesting that hypoxic culture induces hMSCs to secret more soluble MIF than normoxic culture in the medium. Similarly, the expression levels of MIF mRNA transcripts were also significantly upregulated (P<0.05) in hMSCs cultured under hypoxia, compared to those in the cells under normoxia (Fig. 3C), suggesting that hypoxia increases the production of MIF in hMSCs, which in turn enhances the release of soluble MIF from the cells.
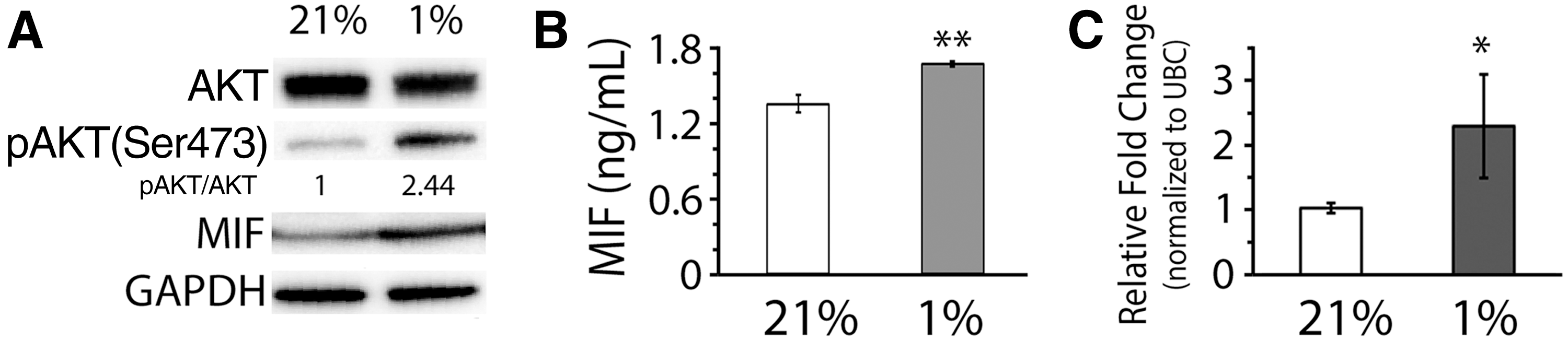
AKT and migration inhibitory factor (MIF) activities in hMSCs modulated by hypoxia or normoxia.
Reduction of AKT activation in hypoxic culture increases cellular senescence of hMSCs
To study if AKT activation is critical to the regulation of cellular senescence of hMSCs under hypoxia, we attenuated the activation of AKT in hMSCs under the hypoxic condition by treating the cells with the potent AKT inhibitor TCN for two passages and then analyzed the effect of AKT-attenuation on cell proliferation, cellular senescence, and potency. We confirmed that TCN treatment was able to effectively reduce the activation of AKT by demonstrating that the levels of pAKT decreased with increased doses of TCN without affecting the levels of total AKT (Supplementary Fig. S1A; Supplementary Data are available online at
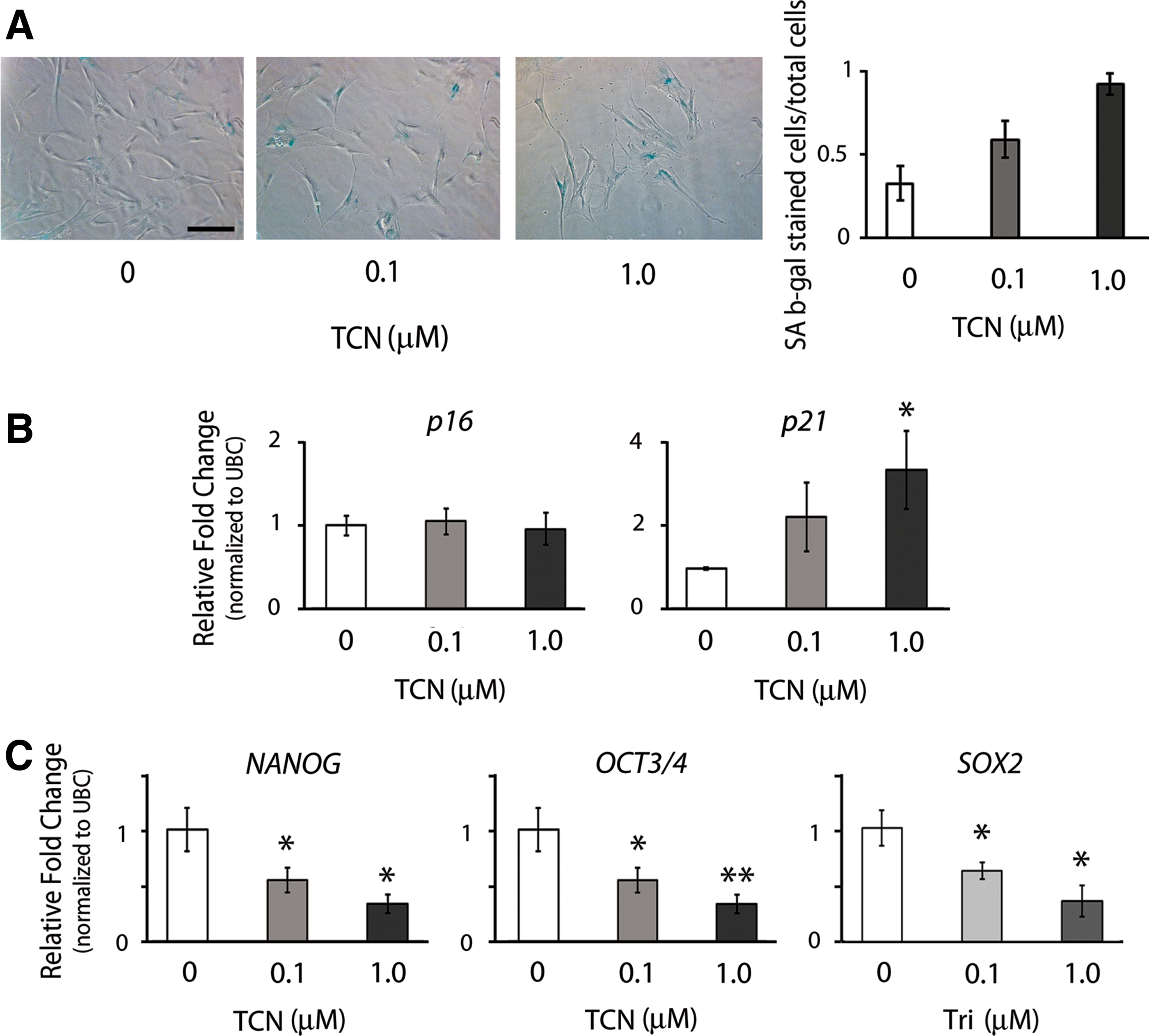
Cell response of hMSCs to the regulation of AKT activity in hypoxic culture.
Inhibition of AKT activity in hypoxic culture does not affect MIF expression
With our results showing hypoxic culture increased the activation of AKT and expression of MIF, we would like to further determine the regulatory mechanism between AKT and MIF. To this end, we regulated AKT activity by treating hMSCs in hypoxic culture with TCN to analyze the expression of MIF. Our western blot results showed that TCN treatment continued to decrease phosphorylation of AKT in hMSCs under hypoxia during the first 24 h of culture, and compared to the control culture without TCN treatment, the culture treated with TCN showed decreased levels of pAKT in hypoxic culture (Fig. 5A). On the other hand, attenuation of AKT activation seemed not to affect expression levels of MIF in hMSCs (Fig. 5A). We also measured the amounts of soluble MIF in the medium of culture treated with TCN, and the ELISA results showed that the levels of soluble MIF in the culture medium were comparable among the groups detected 24, 48, and 72 h after TCN treatment and between the groups treated with and without TCN (Fig. 5B). Similarly, the treatment of AKT Inhibitor IV also did not alter the expression levels of MIF in hMSCs under the hypoxic condition (Supplementary Fig. S2A). Taken together, our results suggest that the production of MIF by hMSCs under hypoxia is unlikely regulated by the activation of AKT signaling.
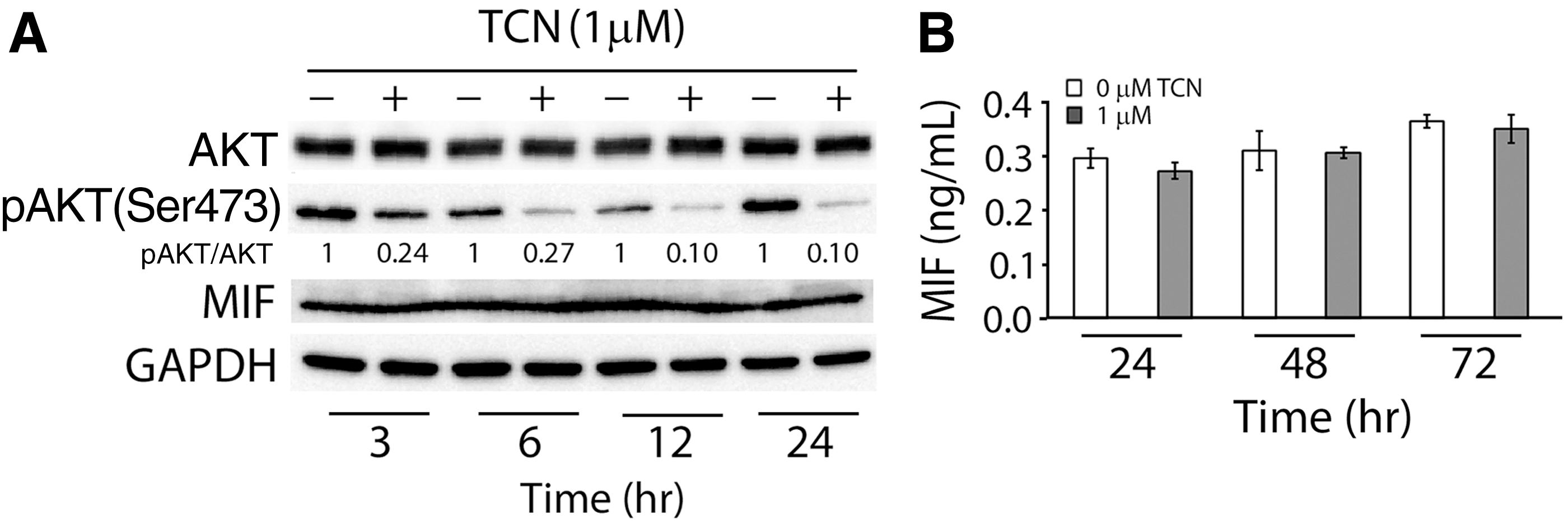
Regulation between AKT activation and MIF expression in hMSCs under hypoxia.
Overexpression of MIF in normoxic culture increases the activation of AKT and reduces cellular senescence of hMSCs
To investigate the role of MIF in the regulation of cellular senescence of hMSCs, we transduced hMSCs with a retroviral vector encoding MIF to stably overexpress MIF in hMSCs. After transduction, MIF-overexpressing hMSCs were cultured under normoxia to determine if the regulation of MIF expression leads to alteration of AKT activity, which in turn modulates cellular senescence and potency of hMSCs. Our results showed that intra- and extracellular levels of MIF were upregulated in MIF-overexpressing hMSCs compared to those in the control cells (Fig. 6A, B). In addition to increased expression of MIF, our western blot results also showed that the levels of total and phosphorylated AKT were increased in MIF-overexpressing hMSCs than those in the control cells (Fig. 6A). Moreover, when comparing cell morphology between MIF-overexpressing and the control cells, the control hMSCs displayed enlarged and flattened morphology, whereas the MIF-overexpressing hMSCs maintained spindle morphology (Fig. 6C). In addition, our real-time PCR results showed that the expression levels of p16 and p21 mRNA transcripts were significantly downregulated (P<0.05) in MIF-overexpressing hMSCs compared to those in the control hMSCs (Fig. 6D). The expression levels of NANOG, OCT3/4, and SOX2 mRNA transcripts were significantly upregulated (P<0.05) in MIF-overexpressing cells than those in the control cells (Fig. 6E). Collectively, these results suggest that the regulation of MIF increases both expression and activation of AKT and suppresses the progression of cellular senescence of hMSCs in the normoxic condition.
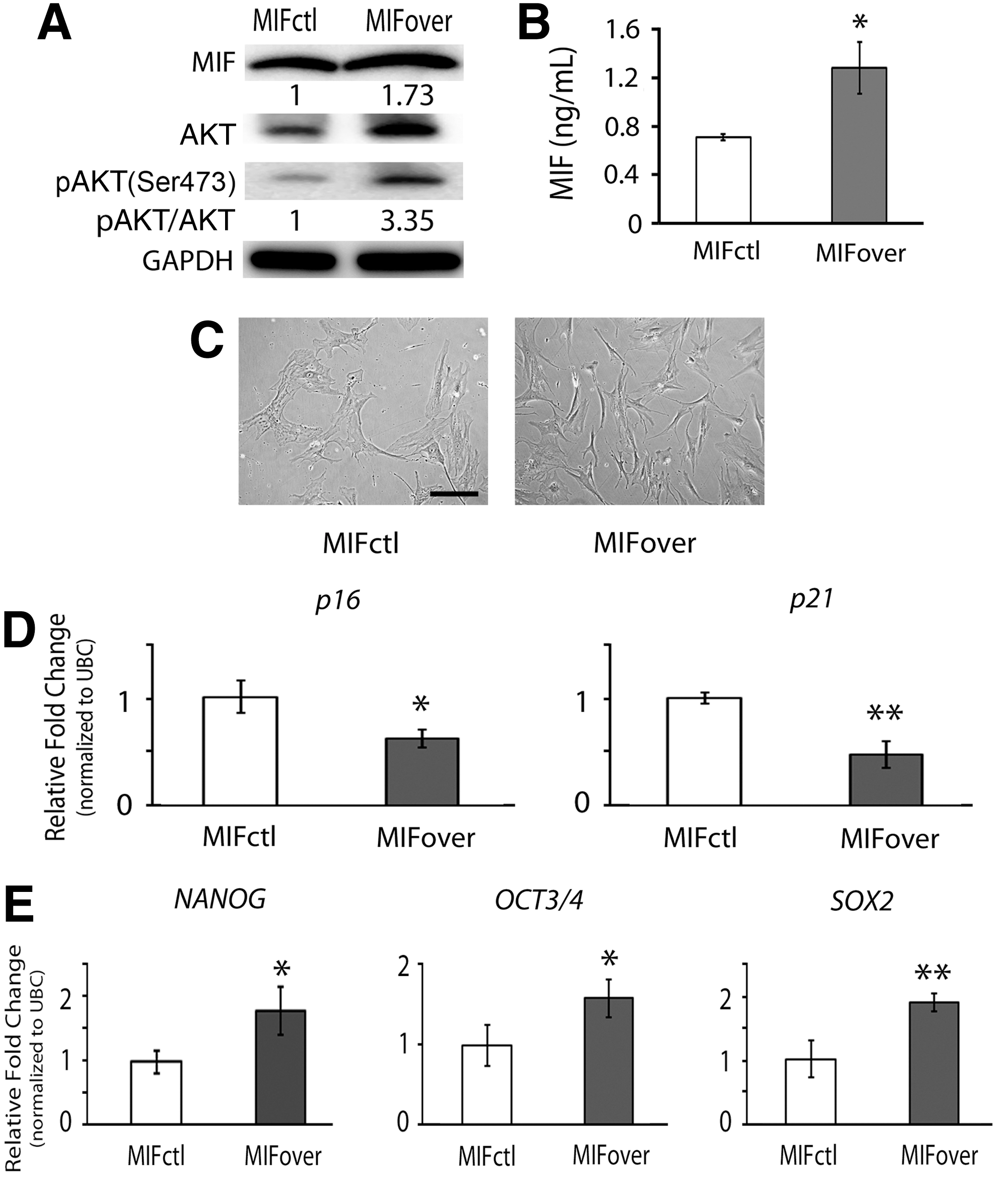
Cell response of MIF-overexpressing hMSCs in normoxic culture. Human MSCs transduced with the MIF-overexpressing plasmid (MIFover) and empty vector control (MIFctl) were cultured under normoxia to determine the regulation between MIF activity and AKT activation.
To further investigate the regulatory role of MIF in cellular senescence of hMSCs under hypoxia, we stably transduced hMSCs with a retroviral vector-mediated shRNA to silence the expression of MIF in hMSCs and compared the AKT activity of MIF-knockdown cells with that of the control cells transduced with scrambled shRNA (shScr) plasmid. Our western blot and ELISA results showed that both intra- and extracellular levels of MIF were downregulated in shMIF-transduced hMSCs compared to those in shScr-transduced cells to confirm that MIF activity was successfully knocked down (Fig. 7A, B). With the aforementioned results that overexpression of MIF in hMSCs under normoxia increased the activation of AKT and reduced cellular senescence (Fig. 6), we anticipated that knocking down MIF would decrease the activation of AKT and increase cellular senescence of hMSC in hypoxic culture. Interestingly, the activation of AKT was not decreased in MIF-knockdown hMSCs (Fig. 7A). Moreover, cell morphology of MIF-knockdown hMSCs was similar to that of the control shScr-transduced cells (Fig. 7C). The real-time PCR results showed that the expression levels of p16 and p21 mRNA transcripts were also comparable between MIF-knockdown hMSCs and the control cells (Fig. 7D). The similar trend of no significant difference between shMIF- and shScr-transduced hMSCs was shown in the expression levels of NANOG, OCT3/4, and SOX2 mRNA transcripts (Fig. 7E).
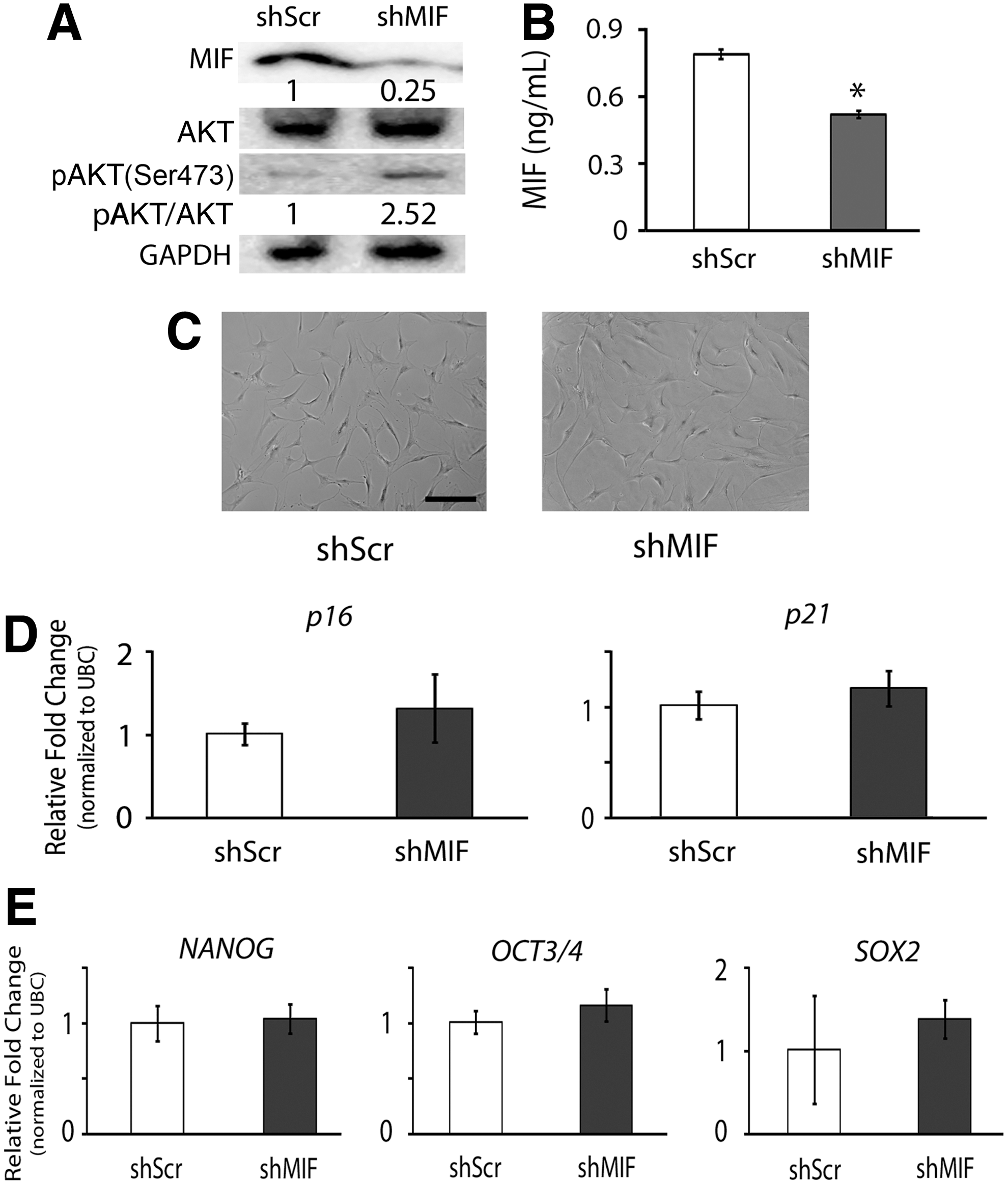
Cell response of MIF-knockdown hMSCs in hypoxic culture. Human MSCs transduced with MIF-targeting shRNA (shMIF) or control scrambled shRNA (shScr) plasmids were cultured under hypoxia to determine the regulation between MIF activity and AKT activation.
Discussion
In this study, we show that hypoxic conditions increase the production of intra- and extracellular MIF, and thus, the upregulated levels of MIF increase the activation of AKT in hMSCs to delay cellular senescence. Our findings demonstrate that both MIF and AKT play a crucial role in the regulation of hMSC senescence under hypoxia. Regulation of AKT activity by MIF in the cell types other than MSCs has been reported by several groups [39,51 –53]. For example, studies have shown that the introduction of exogenous MIF in culture increases the activation of AKT in MEFs, Jurkat T cells, and human dermal microvascular endothelial cells [39,51 –53] and that knocking down MIF using siRNA leads to a drastic decrease of AKT phosphorylation in pancreatic cancer cells [39]. To the best of our knowledge, our new findings that upregulate the expression of MIF by hypoxia increases the activation of AKT in hMSCs have not been reported before.
Hypoxic culture as a tool has been shown to delay cellular senescence of various cell types [14,17]. Previous studies have reported several potential mechanisms, such as reduction of oxidative stress [54], alteration of energy metabolism [15], and activation of hypoxia-inducible factor [14], that are involved in the regulation of cellular senescence mediated by hypoxia. In this study, we show that the activation of AKT by hypoxia is able to delay cellular senescence of hMSCs, suggesting a crucial role of activated AKT in the downregulation of cellular senescence under hypoxic conditions. AKT signaling has been demonstrated to be critical to cell survival and growth [55]. In this study, we demonstrate that the activation of AKT is the key to reduction of cellular senescence in hMSCs. It is worthwhile to mention that the effect of AKT signaling activation on modulation of cellular senescence may be cell-type dependent [18,56]. Previously, a study has shown that the activation of AKT signaling decreases cellular senescence induced by the DNA alkylating agent temozolomide in human U87MG glioblastoma cells [57], and another study has reported that blocking the PI3K/AKT signaling pathway not only increases senescence of human skin-derived precursors but also inhibits self-renewal of the cells [19]. Similarly, we have shown that the downregulation of AKT activity increases the progression of cellular senescence of hMSCs in hypoxic culture.
MIF is a proinflammatory cytokine capable of regulating various biological activities. In addition to its well-established role in immune response [27,34,35], several studies have shown that MIF maintains neural stem cell properties [58] and suppresses cellular senescence [40,41]. Particularly, Welford et al. have reported that the upregulation of MIF under hypoxic conditions suppresses senescence of MEFs [41]. Similarly, our study has shown that MIF in hypoxic culture plays an important role as a key factor in the regulation of cellular senescence and maintenance of stem cell potency in hMSCs. Moreover, we have demonstrated a potential mechanism by which hypoxia induces hMSCs to modulate cellular senescence through increased intra- and extracellular levels of MIF. In addition to the identified mechanism that extracellular MIF binds to its receptor CD74 to induce AKT activation [51,52], it is likely that MIF can activate intracellular mechanisms to modulate cellular senescence of hMSCs in hypoxic culture. A previous study has shown that intracellular MIF binds to p53 transcription factor to suppress cellular senescence [40]. It is not clear if the extracellular MIF/CD74/AKT signaling converges with the intracellular MIF/p53 signaling to regulate cellular senescence. Further investigation is needed to clarify the relationship between these two signaling pathways on hypoxia-induced regulation of hMSC senescence.
Our MIF-misexpression results show that overexpressing MIF in normoxia-cultured hMSCs increases AKT activation, reduces cellular senescence, and enhances stem cell potency, whereas knocking down MIF expression in hypoxia-cultured hMSCs does not lead to the results of reduced AKT activation, increased cellular senescence, and decreased stem cell potency. It is possible that under hypoxic conditions, MIF-knockdown cells are able to modulate production and/or secretion of senescence-associated secretory factors to interfere activation of signaling pathways, including AKT signaling and regulation of cellular senescence. It is known that cellular senescence induces secretion of soluble cytokines, and the changes in cytokine secretion are termed senescence-associated secretory phenotype (SASP) [59] or senescence-messaging secretome [60]. SASP factors include IL-1α, IL-1β, IL-6, IL-8, CXC chemokine ligand-1, CC chemokine ligand-2, CXC chemokine receptor-2, and VEGF [61]. It has been reported that SASP factors are involved in the regulation of AKT signaling [62]. For example, in senescent cells, increased IL-6 is capable of interacting with AKT signaling [63]. Similarly, MIF has also been shown capable of regulating the activity of SASP factors. For example, MIF can increase the expression of IL-6 [64] and other cytokines, including IL-8 [65], IL-2 [35], and interferon gamma [35]. Collectively, these previous findings suggest that SASP factors are involved in the regulation of MIF/AKT-mediated cellular senescence, which may be able to explain why in our results MIF-knockdown cells in hypoxic culture showed increased AKT activation and no difference in cellular senescence and stem cell potency. Moreover, it is also possible that the cells are able to activate signaling pathways other than AKT signaling to compensate for the effect of MIF-knockdown on cellular senescence. Several previous publications have reported that such a compensation response to restore critical cellular functions during cell apoptosis and senescence occurs in response to knockdown of target molecules [66 –68]. With that being said, our findings from the MIF overexpression study clearly demonstrate that cellular senescence of hMSCs under hypoxic conditions is regulated through the mechanism that soluble MIF released from hMSCs induces AKT activation.
Cellular senescence occurring during in vitro culture has been considered one of the major challenges encountered in hMSC-based regenerative medicine. Our findings shed light on one of the potential mechanisms underlying hypoxia-regulated cellular senescence in hMSCs and likely contribute to better understanding of how hypoxic conditions delay the progression of hMSC senescence. The findings also suggest that it may be beneficial to use exogenous MIF in hMSC culture to delay cellular senescence and maintain stem cell potency for translational applications of cell therapies and regenerative medicine [36,39,51]. However, it is important to note that a potential limitation with the use of MIF in hMSC culture is its unknown side effects. Several studies have reported that MIF is upregulated in various types of cancer cells [69 –71] and may play a role in linking chronic inflammation and cancer development [72]. Therefore, it is important to fully identify the role of MIF in tumor biology before using it for hMSC culture.
Footnotes
Acknowledgments
We thank Dr. Matthew Squire at the Department of Orthopedics and Rehabilitation for providing human femoral heads for MSC isolation. We also thank Dr. Justin Palumbo for proofreading the article.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.