
Abstract
Marrow stromal cells constitute a heterogeneous population of cells, typically isolated after expansion in culture. In vivo, stromal cells often exist in close proximity or in direct contact with monocyte-derived macrophages, yet their interaction with monocytes is largely unexplored. In this report, isolated CD146+ and CD146− stromal cells, as well as immortalized cell lines representative of each (designated HS27a and HS5, respectively), were shown by global DNase I hypersensitive site mapping and principal coordinate analysis to have a lineage association with marrow fibroblasts. Gene expression profiles generated for the CD146+ and CD146− cell lines indicate significant differences in their respective transcriptomes, which translates into differences in secreted factors. Consequently, the conditioned media (CM) from these two populations induce different fates in peripheral blood monocytes. Monocytes incubated in CD146+ CM acquire a tissue macrophage phenotype, whereas monocytes incubated in CM from CD146− cells express markers associated with pre-dendritic cells. Importantly, when CD14+ monocytes are cultured in contact with the CD146+ cells, the combined cell populations, assayed as a unit, show increased levels of transcripts associated with organismal development and hematopoietic regulation. In contrast, the gene expression profile from cocultures of monocytes and CD146− cells does not differ from that obtained when monocytes are cultured with CD146− CM. These in vitro results show that the CD146+ marrow stromal cells together with monocytes increase the expression of genes relevant to hematopoietic regulation. In vivo relevance of these data is suggested by immunohistochemistry of marrow biopsies showing juxtaposed CD146+ cells and CD68+ cells associated with these upregulated proteins.
Introduction
P
A considerable body of work using LTC has identified cells and their products that contribute to ME support of both stem and progenitor cells. To date, there is general agreement regarding the identity of some of the gene products that function within the ME, including CXCL12, angiopoietin, osteopontin, SCF, thrombopoietin, nestin, and Connexin-43 to name a few [4 –7]. However, there is less agreement regarding the identity of the cells that provide these activities [8]. Marrow stromal cells certainly contribute to the ME, but this is an imprecise term that encompasses fibroblasts, osteoblasts, fat cells, reticular cells, and endothelium [9 –15]. Compelling studies have implicated cells lining the endosteum as critical components of the stem cell niche, specifically the osteoblast, as well as an otherwise undefined cell, which also lines the surface of the bone [16,17]. Equally compelling are the data suggesting that sinusoids serve as stem cell niches, with critical functions attributed to perivascular cells [18]. Cells required for periendothelial niche development in vivo are reported to express CD146 (reviewed in Bianco et al. 2013 [19]).
Our efforts to functionally define the critical components of the ME have focused on immortalizing and cloning functionally distinct nonhematopoietic cells present in primary LTC. We have reported extensively on two stromal cell lines, designated HS5 and HS27a, which differ in function: CD146− HS5 secretes growth factors (GM-CSF, G-CSF, IL-6) leading to the proliferation and differentiation of CD34+ cells, whereas CD146+ HS27a cells do not secrete these factors, but do express activities reported to be associated with the stem cell niche [20]. Despite these differences, both cell lines are closely associated with the fibroblast lineage as shown by Principal Coordinates Analysis of DNase I hypersensitive site mapping.
Realizing that marrow stromal cells do not function in isolation, but rather in the context of other cells, we investigated whether monocytes and monocyte-derived macrophages, cells that are clearly present in the marrow, can interact with stromal cells to contribute to the ME milieu. Our in vitro results show that soluble factors secreted by CD146+ HS27a cells, as well as their CD146+ freshly isolated homologues induce CD14+ monocytes to acquire a macrophage phenotype. However, when monocytes are cultured in contact with the CD146+ HS27a cells and the two admixed populations assayed as a unit, the gene expression profile for the unit differs from that obtained by exposing monocytes to HS27a-conditioned media (CM) or by adding the two individual profiles together. Many of the contact-dependent upregulated gene products belong to a functional cluster associated with controlling a multicellular organization such as hematopoietic regulation [e.g., cyclin-dependent kinase 6 (CDK6) and Dickkopf-related protein 3 (DKK3)]. In contrast to this, the gene expression profile from monocytes in contact with CD146− HS5 cells does not differ from that obtained with monocytes in HS5CM or by adding their two profiles together.
Materials and Methods
Marrow and peripheral blood cells from normal donors
Bone marrow aspirates, biopsies, and peripheral blood were obtained from healthy adult donors after informed written consent was obtained using forms approved by the Institutional Review Board (IRB) of the Fred Hutchinson Cancer Research Center (FHCRC) in accordance with the Declaration of Helsinki (FHCRC IRB file numbers 208, 211, 985, 2167). Aspirated marrow was processed either as buffy coat cells or as mononuclear cells isolated by density gradient centrifugation. Long-term marrow cultures were established as modified Dexter cultures using buffy coat cells grown in a medium containing the Iscove's Modified Dulbecco's Medium, 12.5% horse serum, 12.5% fetal calf serum (FCS), hydrocortisone sodium succinate, and sodium pyruvate as previously reported [21]. Thirty immortalized stromal cell clones were isolated from the Dexter-like culture as described [20]. Two of these, designated HS5 and HS27a, have been reported previously and are available through American Tissue Culture Collection (ATCC). Stromal cell lines are maintained in complete media [supplemented RPMI 1640 medium containing 10% FCS (HyClone), L-glutamine, and sodium pyruvate].
Marrow mononuclear cells from healthy donors (n=8) were used to establish relatively short-term, primary cultures of marrow stromal cells using a Lineage Cell Depletion Kit and magnetic separation (Miltenyi Biotec) according to the manufacturer's protocol. The cells were cultured in mesenchymal stromal cell (MSC) basal media with MSC stimulatory supplement according to the manufacturer's protocol (ALLCELLS), and then expanded in a 1:1 mixture of the MSC and complete media. The cells were passaged until the desired number of cells was reached (less than eight passages). The absence of contaminating macrophages and other adherent hematopoietic cells was confirmed by flow cytometry using antibodies against CD45, CD14, and CD11c.
Peripheral blood mononuclear cells (PBMC) from healthy donors were isolated by density gradient centrifugation. For large-scale isolations, PBMC apheresis products were obtained from the Large Scale Cell Processing Core of the Core Center of Excellence in Hematology (NIH/NIDDK Grant No. DK056465) after obtaining approval from the FHCRC Institutional Review Board. In both cases, the CD14+ cells were isolated by AutoMACS (Miltenyi Biotec) using a CD14 monoclonal antibody (clone TÜK4; Dako) and anti-mouse immunoglobulin G2a+b-conjugated magnetic microbeads (Miltenyi Biotec). The purity of isolated CD14+ cells was 98% (range, 96%–99%).
Bone marrow biopsies for immunohistochemistry (IHC) were obtained from healthy and arthritic donors during joint replacement surgeries (n=3) and a patient with myelodysplastic syndrome (MDS) after informed written consent was obtained using forms approved by the FHCRC Institutional Review Board (IRB) in accordance with the Declaration of Helsinki. The patient with MDS was previously reported as Patient #20 [22]. Patient biopsies were used for IHC only.
DNase I hypersensitive site mapping
Digital DNase I mapping was performed essentially as described [23]. Briefly, CD146-positive and -negative cell lines (HS27a, HS5) and primary marrow stromal cells (DS20514, DS20518) were grown as described above. We pelleted 1×108 cells and washed them with cold phosphate-buffered saline. We resuspended cell pellets in buffer A (15 mM Tris-Cl (pH 8.0), 15 mM NaCl, 60 mM KCl, 1 mM EDTA (pH 8.0), 0.5 mM EGTA (pH 8.0), 0.5 mM spermidine, and 0.15 mM spermine) to a final concentration of 2×106 cells/mL. Nuclei were obtained by dropwise addition of an equal volume of buffer A containing 0.04% NP-40 to the cells, followed by incubation on ice for 10 min. Nuclei were centrifuged at 1,000 g for 5 min and then resuspended and washed with 25 mL of cold buffer A. Nuclei were resuspended in 2 mL of buffer A at a final concentration of 1×107 nuclei/mL. We performed DNase I (Roche; 10–80 U/mL) digests for 3 min at 37°C in 2 mL volumes of DNase I buffer (13.5 mM Tris-HCl pH 8.0, 87 mM NaCl, 54 mM KCl, 6 mM CaCl2, 0.9 mM EDTA, 0.45 mM EGTA, 0.45 mM spermidine). Reactions were terminated by adding an equal volume (2 mL) of stop buffer [1 M Tris-HCl (pH 8.0), 5 M NaCl, 20% SDS and 0.5 M EDTA (pH 8.0), 10 μg/mL RNase A; Roche] and incubated at 55°C. After 15 min, we added proteinase K (25 μg/mL final concentration) to each digest reaction and incubated for 1 h at 55°C. After DNase I treatments, careful phenol–chloroform extractions were performed. DNase I double-cut fragments and sequencing libraries were constructed as described [24 –26].
Preparation and analysis of CM
HS27a, HS5, and primary stromal cells (3×106) were grown in 10 mL of RPMI 1640-based complete media in T-75 flasks. CM were harvested after 7 days, clarified by centrifugation, and stored at 4°C. The CM were diluted with one volume of the complete media before use. Cytokine levels in the CM were determined using enzyme-linked immunosorbent assays (ELISAs) performed by the Cytokine Laboratory Shared Resource of the FHCRC. Endotoxin and mycoplasma levels of cells, media, cytokines, and all other reagents were undetectable using a Limulus Amebocyte Lysate test kit (Charles River Laboratories) and a 4′6-diamidino-2-phenylindole (DAPI)-based cytochemical detection method, respectively. Both assays were performed by the Biologics Production Facility at the FHCRC.
Flow-based analysis of labeled cells
Primary marrow stromal cells, HS27a, and HS5 cells were detached from culture plates by incubation at 37°C for 10 to 30 min in the Cell Dissociation Buffer (Gibco), and washed with the Hank's Balanced Salt Solution (HBSS; Gibco). Nonspecific binding to FcR was blocked by incubating in a 10% FcR-blocking solution (Miltenyi Biotec) for >20 min at 4°C. Cells were then stained with PE- or FITC-conjugated antibodies. Control staining was performed simultaneously using isotype-matched, irrelevant antibodies also directly conjugated to PE or FITC. Cells were washed twice, and propidium iodide was added as a marker to exclude dead cells. The fluorescence intensity of the cells was analyzed by flow cytometry (FACSCalibur; Becton Dickinson). The antibodies used are PE-conjugated anti-CD146 (BD Biosciences) and FITC-conjugated antibodies against CD45 and CD14 (BD Biosciences).
Microarray hybridization and data analysis
CD14+ cells were cultured in the presence of CM for 2 days, and total RNA was isolated from the CD14+ cells as described above. In some experiments, CD14+ cells were cultured together with HS27a cells, and both were harvested together. Total RNA was then isolated from the combined populations. Double-stranded cDNA and cRNA were synthesized from 2–5 μg of total RNA and hybridized to Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays (Affymetrix) using the manufacturer's standard protocols. The Genomics Shared Resource at FHCRC performed microarray hybridization.
To explore the effects of coculturing CD14+ and HS27a cells, data sets from CD14+ cells, HS27a cells, CD14+ cells cultured in HS27a CM, and CD14+ cells cultured with HS27a cells were RMA normalized using the Bioconductor package affy [27], followed by filtering using Affymetrix MAS5 absent/present calls, where only those probes that were called present in all samples for at least one condition were retained. A variance filter was subsequently applied using the shorth function in the Bioconductor package genefilter. Pairwise significance testing for the CD14+ and HS27a coculture vs. HS27a cells was performed using the Bioconductor package limma [28], and P values were corrected for multiple testing using the false discovery rate (FDR) method of Benjamini and Hochberg [29]. Using a |log2(ratio)| ≥1 with the FDR set to 0.1%, we identified 631 probes as differentially expressed. Using this set of differentially expressed probes, we median centered each probe's normalized signal across all conditions and performed k-means clustering (Euclidean distance similarity matrix). Clustering and heat map presentation were performed using the TM4 suite module MultiExperimental Viewer (MeV) open source software [30]. The same analysis was also done using data sets from HS5 cells, CD14+ cells, CD14+ cells cultured in HS5 CM, and CD14+ cells cultured with HS5 cells. Affymetrix CEL files are available in the Gene Expression Omnibus microarray expression database (
Over-representation of GO biological process terms in Clusters 3 and 8 were determined using a standard hypergeometric test in the Bioconductor package GOstats [31]. In the hypergeometric tests, the gene lists used as input comprised probeset identifiers with Entrez Gene IDs from each cluster, using a universe, which comprised those genes that passed previous signal and variance filtering steps, described above. A GO term was considered to be significantly overrepresented when P value ≤10−4.
Immunohistochemistry of CD146+ cells and macrophages in bone marrow
The Experimental Histopathology Laboratory at the FHCRC performed dual IHC to detect CD146+ cells, CD68+or CD163+ macrophages, and CDK6+ cells in marrow biopsies. Single IHC was also performed to detect DKK3. Summary of the reagents and protocols for the IHC are shown in Supplementary Tables S1 and S2 (Supplementary Data are available online at
Results
Global epigenome analysis of marrow stromal cells using DNase I hypersensitivity site mapping shows CD146+ marrow stromal cells are associated with the fibroblast lineage
Although marrow stromal cells share some similarities with pericytes and endothelial cells, their lineage association has not been established [32]. To address this question and to gain a genome-wide perspective, we used digital DNase I analysis of HS27a, HS5, primary CD146+ marrow stromal cells, as well as cells from defined cell populations with endothelial, hematopoietic, epithelial, and fibroblastoid origins to map chromatin accessibility (Supplementary Table S3). Digital DNase I profiling is highly reproducible and enables quantitative delineation of chromatin accessibility, including both classical DNase I hypersensitive sites as well as regions of general chromatin accessibility marked by DNase I sensitivity. This enables the construction of DNase I hypersensitivity site (DHS) landscapes [23,33] for each cell/tissue type. Figure 1 shows a principal coordinate analysis of landscapes for HS27a (CD146+), HS5 (CD146−), and freshly isolated marrow stromal cells from healthy donors, which show clear separation from the clusters of endothelial landscapes (P<0.005) and hematopoietic landscapes (P<0.005), but is not distinguishable from the cluster of fibroblast landscapes (P=0.63) [significance test by a two-sided K-S test (Kolmogorov–Smirnov test) using R]. The DHS files are available in the Gene Expression Omnibus series accession GSE29692 (
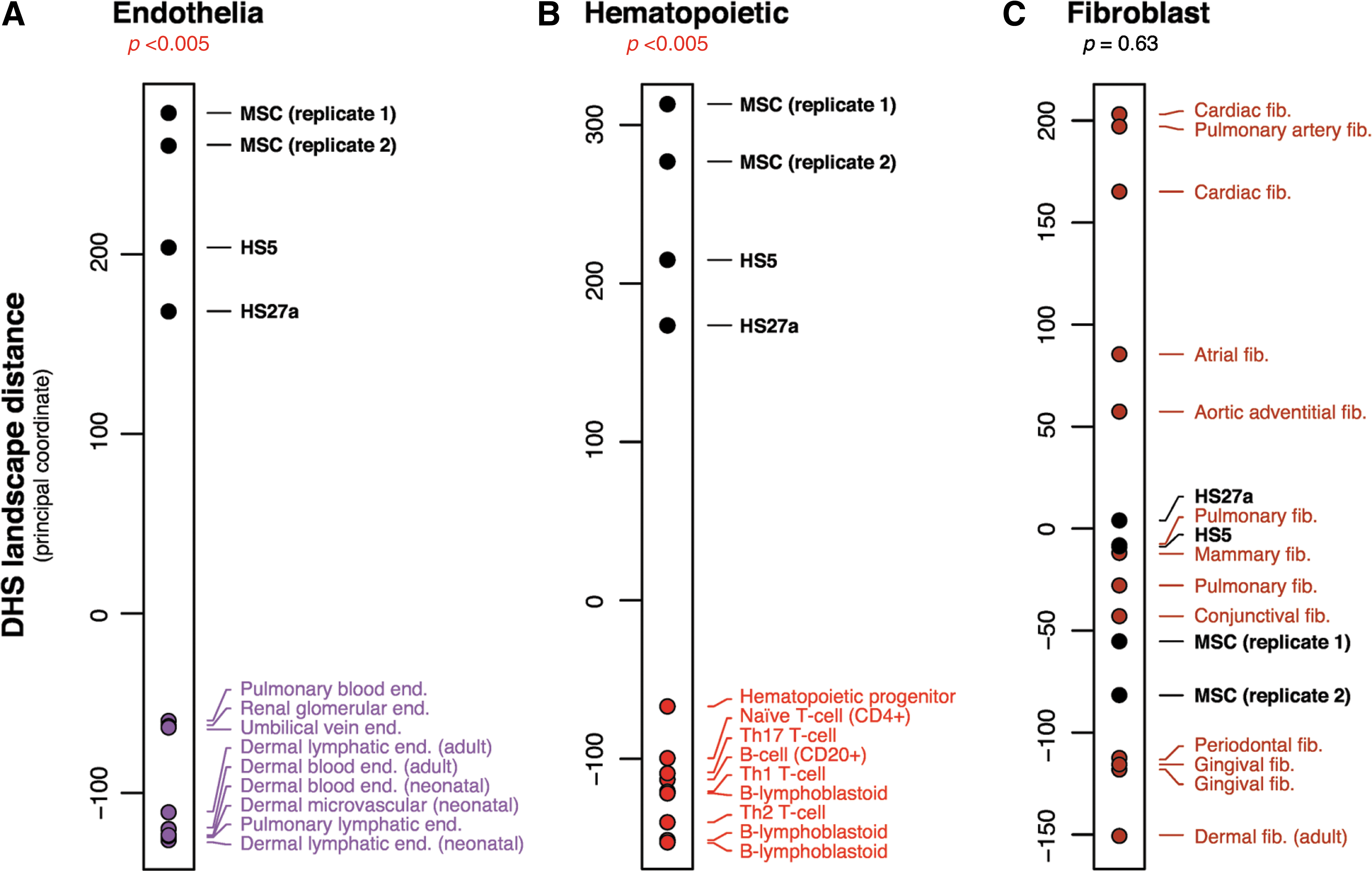
Three 1-dimensional principal coordinate analyses using the DHS landscape (linear patterning of DHSs) for each lineage class combined with data of primary marrow stromal cells from healthy donors and marrow stromal cell lines (HS5, HS27a). The plot shows the relative positioning of the primary stromal cells and the cell lines with clusters of endothelial in panel
CD146+ cells represent a subset of stromal cells
Figure 2A shows a flow cytometric analysis of CD146 expressed by primary marrow stromal cells and the two cloned stromal cell lines. Primary marrow stromal cells show heterogeneous expression of CD146, whereas HS27a cells express a more homogenous level of CD146 as determined by fluorescence intensity. In contrast, HS5 cells are negative for CD146. Differential expression of CD146 was transcriptionally regulated as confirmed by quantitative PCR (Fig. 2B). These data indicate that CD146 can be used to distinguish these two functionally distinct marrow stromal cell populations. Functional differences have been reported previously for HS27a and HS5, and complete array analyses of genes expressed by HS27a and HS5 can be accessed at GSE9390 and GSE10595 at

Expression of CD146 in stromal cells.
Flow analysis of stromal cells in primary cultures indicates that the proportion of CD146+ cells can vary from 10%–54% among cultures. To eliminate this variation while retaining adequate numbers of cells, most studies were designed to use HS27a cells, which closely resemble the primary CD146+ cells and serve as a consistent and unlimited source of CD146+ cells.
CM from HS27a and HS5 cells induce different monocyte fates
The first studies investigated the effects of CM from HS27a and HS5 on the behavior of normal monocytes in culture. Inverted phase-contrast imaging of monocytes after 2 days in culture showed they acquired a macrophage phenotype in the presence of CM from either HS27a or primary stromal cells (Fig. 3A, C). Cells incubated in HS5 CM remained unattached and retained their original morphology (Fig. 3B). This morphology is similar to the monocytes differentiating to dendritic cells (DC) in vitro. By adding recombinant cytokines IL-4 and TNFα to the HS5 CM, a distinct population of CD14−/MHC-DR++/CD80+/CD86+cells were generated, similar to the standard DC phenotype (Fig. 3D, E).
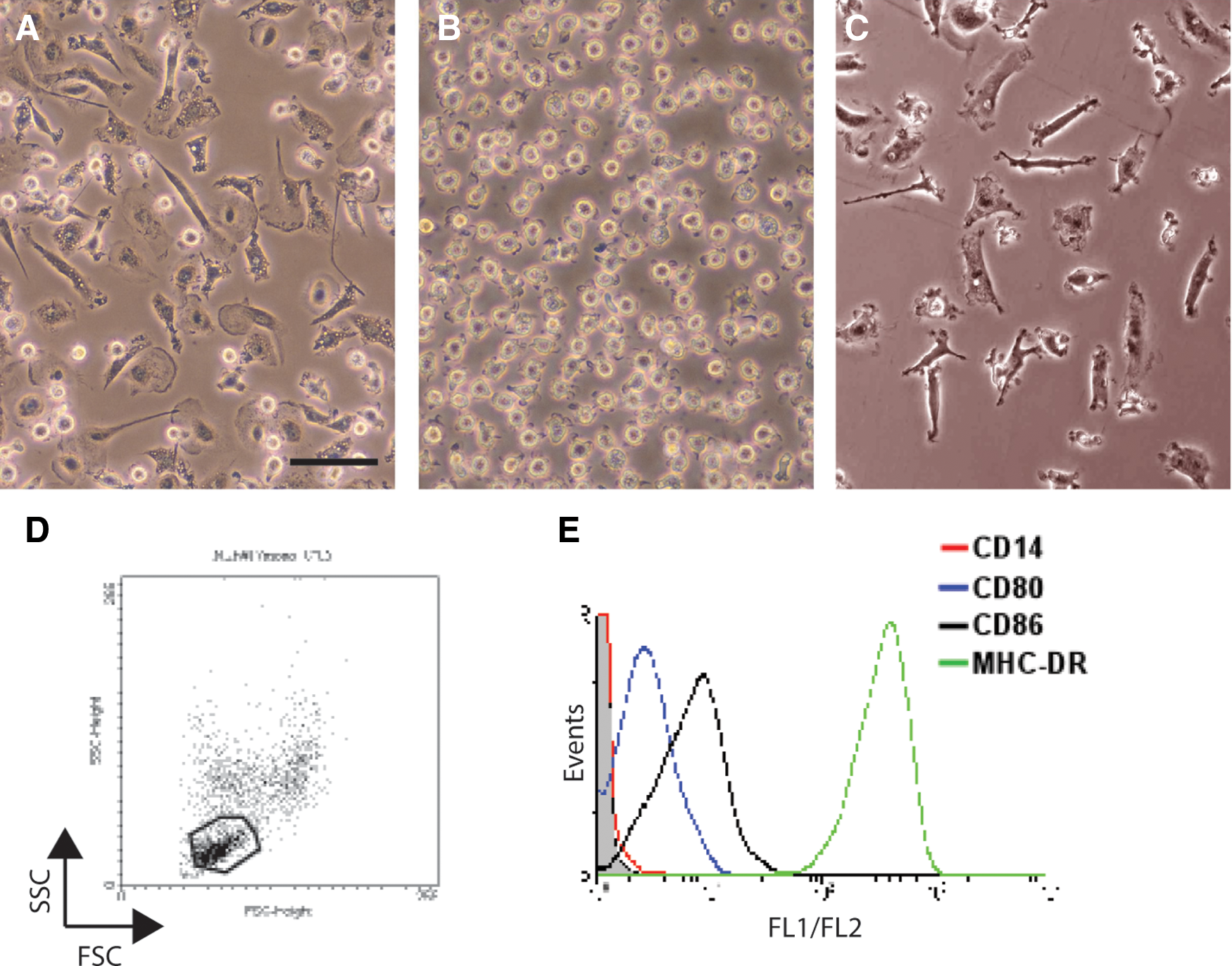
Morphological and phenotypic changes of CD14+ monocytes after culture in conditioned media (CM) from primary and immortalized stromal cells.
Global gene expression profiling using microarray technology showed HS27a CM-dependent changes in gene expression in monocytes after 2 days of culture (Cluster 8 in Fig. 4). The gene list was used to test associations using GOstats (Table 1). Changes included expression of macrophage markers such as secreted proteins (MMP-9 and SEPP1), macrophage membrane proteins (CSF1R/MCSFR, MRC1, ITGB2, FCGR3A/B (CD16), and FCGRT), and transcription factor (MAF), all in agreement with the macrophage lineage.
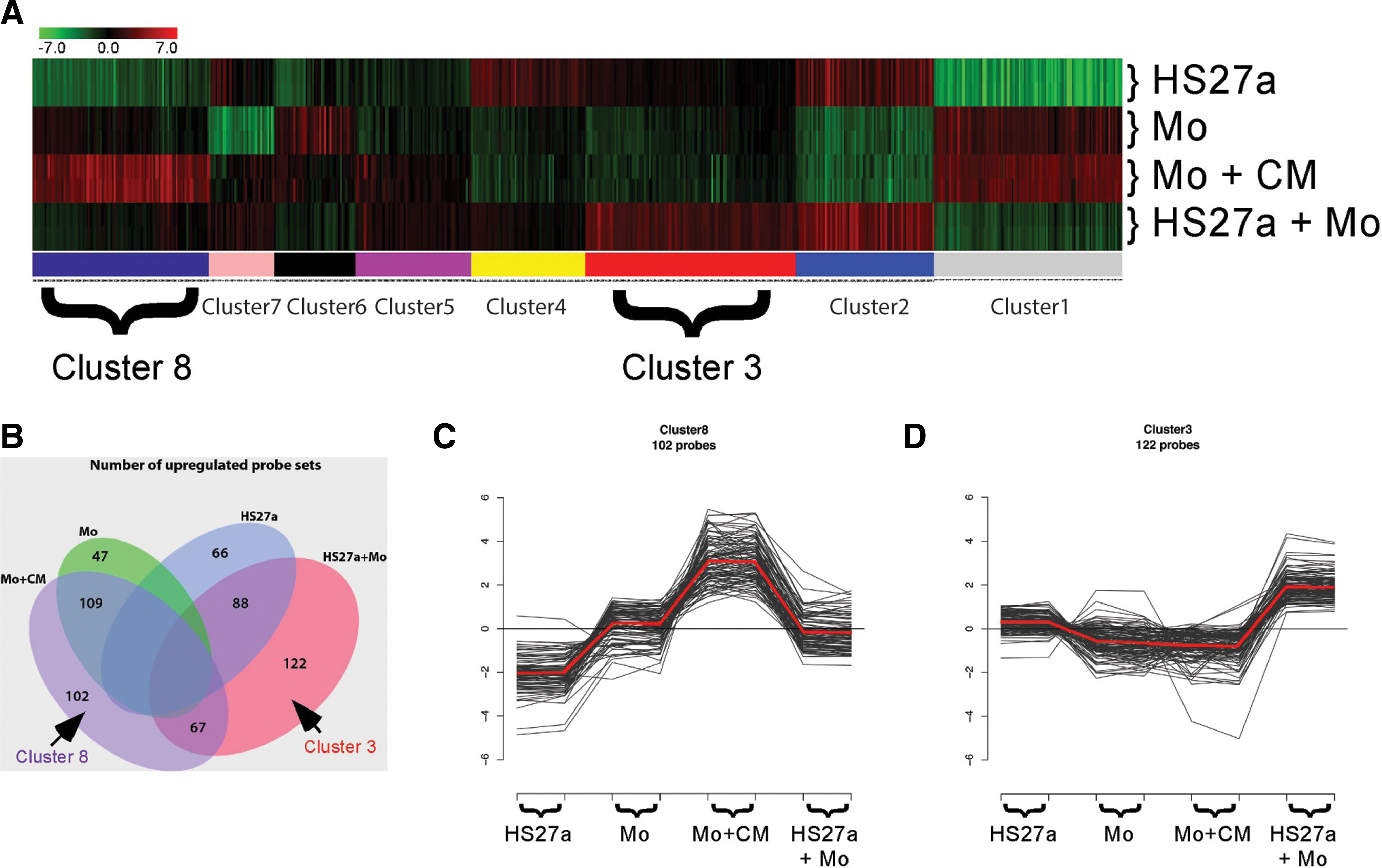
K-means clustering analysis of microarray data obtained from HS27a cells, freshly isolated peripheral blood CD14+ monocytes (Mo), CD14+ cells cultured in HS27a conditioned media (Mo+CM), and cocultures of HS27a and CD14+ cells (HS27a+Mo). Normalized log2 intensities were probewise median centered across all conditions (see Materials and Methods section for details).
A different pattern of gene expression was detected when monocytes were exposed to HS5 CM (Table 2). Some genes (MMP9, CSF1R, and MAF) from Cluster 8 in Fig. 4 were also upregulated in monocytes cultured in HS5 CM. Monocyte genes uniquely upregulated by HS5 CM included secreted proteins (PPBP and CXCL5/ENA-78) and membrane proteins (CD209/DS-SIGN and SLAMF8). Other differentially expressed genes, SEPP1, FCGR3A/B (CD16), and FCGRT are absent in HS5 CM stimulated monocytes, but present in those exposed to HS27 CM. These data support the conclusion that the secreted factors from the two cloned stromal cell lines induce different monocyte fates.
Changes in the combined transcriptome of HS27a and monocyte-derived macrophages assayed together as a microenvironmental unit
In vivo, marrow stromal cells do not function in isolation, but rather in the context of other cells. Therefore, HS27a cells were cultured in contact with monocytes from healthy donors, and the admixed population of cells was harvested and analyzed as a unit. This was necessary to avoid significant cell lysis that resulted from efforts to separate these two cell types. Increases in gene expression that could not be attributed to adding the individual profiles together or to the effects of soluble factors defined in Table 1, are considered the result of contact-dependent interactions between monocytes and HS27a. Global gene expression profiling analysis captured more than 100 gene probes uniquely upregulated in the cocultures of HS27a and monocytes (Cluster 3 in Fig. 4). Table 3 provides a GO term association using GOstats in the ME unit compared with either cell type alone as well as monocytes cultured with CM. Changes detected included those associated with cell cycle (CDK6 and CDKN2B), lipoprotein metabolism (PCSK9 and LDLR), adhesion (VCAM1 and ITGA11), extracellular matrix (DKK3 and BMP1), cytokines (VEGFA and PDGFA), and Notch signaling (NOTCH3 and HES1).
In contrast to the results obtained with HS27a cells, results obtained from cocultures of HS5 and monocytes did not differ significantly from monocytes cultured with HS5 CM (Table 3). These data further emphasize the functional differences that exist between these two marrow stromal cell lines.
CD146+ cells are found in contact with macrophages in marrow biopsies
Dual labeling of marrow biopsies using IHC for CD146 and CD68 or CD163 was used to detect stromal cells and macrophages, respectively. As seen in Fig. 5A–D, areas with increased CD146 staining also contain CD163/CD68+ cells, presumably macrophages, some of which are conspicuous for their juxtaposition with bone and other nonvascular areas (Fig. 5E–F).
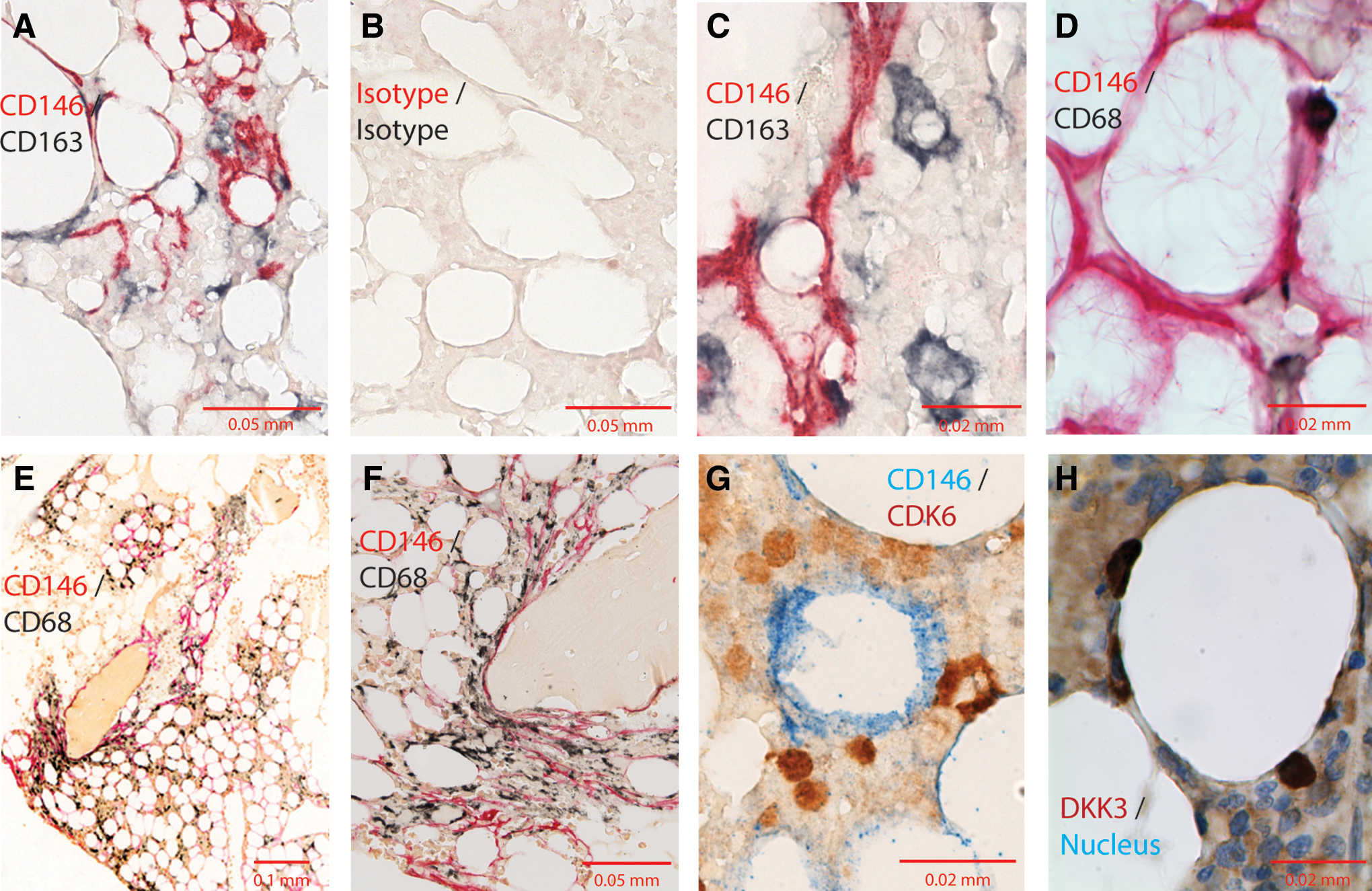
Immunohistochemical detection of CD146+ cells and CD163+ or CD68+ macrophages in marrow biopsies. Marrow biopsies were subjected to dual immunohistochemistry.
CD146+ cells were also found in contact with cells expressing DKK3 and CDK6. Figure 5G–H show IHC of bone marrow biopsies for CD146 and two of the proteins encoded by the contact-dependent upregulated genes (CDK6 and DKK3) chosen from Cluster 3 in Fig. 4 and Table 2. The CDK6- or DKK3-positive cells had monocytoid morphology, and could be found in close contact with CD146+ stromal cells as shown in Fig. 5G–H.
Discussion
The ordered production of blood cells requires a hierarchy of extrinsic signals provided by cells in the marrow ME working together to control stem cell fate and lineage-specific maturation [10,12,15]. Studies designed to identify the cellular components of the various ME units have resulted in an appreciation that several types of nonhematopoietic cells, including endothelium and osteoblasts, may contribute to the ME function [10,35,36], with particular emphasis on a CD146+ cell reported to be required for establishing the stem cell niche [37]. CD146 expression has been associated with endothelial cells, and the requirement for a CD146+ cell to establish a niche has been interpreted to support a preferred perivascular site for the stem cell niche. Our results reported here corroborate that CD146 is expressed on the majority of stromal cells. However, our results from DNase I hypersensitivity site mapping indicate that these CD146+ stromal cells are more closely associated with fibroblasts than the endothelium. In addition, the IHC shown in Fig. 5 reveals that the CD146+ cells are not restricted to perivascular areas, but can be detected throughout the marrow and juxtaposed to bone. A more recent report suggests that bone marrow macrophages are also required to maintain HSC niches [38]. The suggestion that both macrophages and CD146+ cells are required for niche generation/maintenance supports the data presented here that a CD146+ marrow stromal cell secretes activities that induce monocytes to a macrophage phenotype, and then the two in close proximity contribute gene products relevant to hematopoietic niches.
It is generally accepted that stromal cells are stable elements in the ME with low cycling activity, and are not replaced by standard hematopoietic stem cell transplantation [39]. Nevertheless, they play a direct role in the stem cell niche by influencing cell fate decisions through the expression of proteins, like SDF and Jagged, which bind progenitor surface receptors CXCR4 and Notch, respectively. Binding of CXCR4 to SDF retains progenitors in the marrow, whereas Jagged-Notch signaling prevents progenitor responses to cytokines like G-CSF. Regardless of the relatively static nature of stroma, the impact of these stromal signals on hematopoiesis can be modulated. For example, soluble factors secreted by stromal cells stimulate monocytes to secrete osteopontin which, in turn, downregulates Notch expression on progenitors [40]. The loss of Notch on progenitors indirectly reduces the impact of Jagged expressed by stroma. Modulation could also be a direct effect of stromal–monocyte interactions exemplified by the upregulation of DKK3 expression, presumably in the macrophage of the ME unit as suggested by the IHC results. Given that DKK3 can antagonize Wnt signaling, which in turn has significant effects on the repopulating potential of early hematopoietic stem cells, the increased local production of DKK3 could alter the function of the local ME unit [41,42]. Since monocytes circulate and can be recruited from the blood, their number within an ME unit, and possibly their function, could change rapidly.
In vivo, blood monocytes transmigrate the endothelium and come under the influence of the tissue-specific ME they enter [43,44]. They can differentiate in vitro as well as in vivo into DC, osteoclasts, or macrophages, which perform functions relevant to the status of the surrounding tissue [45 –48]. Monocyte–stroma interactions may be of particular importance in hematopoietic diseases like MDS, in which we have shown that monocytes derived from the MDS clone fail to respond appropriately to stromal signals [22]. In contrast to stromal cells, macrophages are derived from hematopoietic stem cells, and are replaced following stem cell transplantation. The fact that a hematopoietic-derived cell could play a critical role in the ME may help clarify the confusion of why some stromal defects can be cured by transplantation, when stromal cells clearly remain host in origin following stem cell transplantation [39].
In summary, our results make three points. First, CD146, known to be expressed by endothelial cells, is also confirmed to be expressed by a subset of marrow stromal cells, which are most closely associated with fibroblasts. Therefore, the presence of CD146 cells in the stem cell niche does not necessarily support the conclusion that the niche is perivascular. Second, circulating monocytes, which can enter any tissue, have several functional phenotypes with the potential to significantly alter the molecular composition of their immediate milieu. Third, isolating and characterizing defined cell populations from bone marrow is not sufficient for defining their role in the hematopoietic ME. These various cell types do not function in isolation, but in the context of other cells and should therefore be evaluated in conjunction with other cells.
Footnotes
Acknowledgments
The authors are grateful for research funding from the National Institutes of Health, Bethesda, MD grants P30 DK056465 and U01 HL099993 to BTS, HG004592 to JAS, and HL094374. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health nor its subsidiary Institutes and Centers.
The authors thank Dr. Shelly Heimfeld for providing leukapheresis products, Dr. Lynn Graf for preparing cells and RNA for microarrays, Paul St. Laurent, Ludmila Golubev, Gretchen Johnson, and Alla Nikitine for maintaining cell cultures, flow cytometry, and preparing CM, Dr. Julie Randolph-Habecker and Tracy Goodpaster for IHC, Drs. Jack Lionberger, Brent Wood, and H. Joachim Deeg for providing bone marrow specimens, Dr. George Sale for histopathological consultation, Ryan Basom for assisting with microarry analysis, and Helen Crawford and Bonnie Larson for help in preparing the manuscript. Additionally, we would like to thank Rajinder Kaul, Theresa K Canfield, Erika Giste, and the UW HTGU for managing sample transfer, cell culture, DNase I library creation, and Illumina sequencing, Alex Reynolds for graphic design work, and Sam John and Andrew Stergachis for their help with the DNase I methods and analysis.
Author Disclosure Statement
The authors have no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.