
Abstract
Tumorigenicity of human pluripotent stem cells is a major threat limiting their application in cell therapy protocols. It remains unclear, however, whether suppression of tumorigenic potential can be achieved without critically affecting pluripotency. A previous study has identified hyperexpressed genes in cancer stem cells, among which is E2F2, a gene involved in malignant transformation and stem cell self-renewal. Here we tested whether E2F2 knockdown would affect the proliferative capacity and tumorigenicity of human embryonic stem cells (hESC). Transient E2F2 silencing in hESC significantly inhibited expression of the proto-oncogenes BMI1 and HMGA1, in addition to proliferation of hESC, indicated by a higher proportion of cells in G1, fewer cells in G2/M phase, and a reduced capacity to generate hESC colonies in vitro. Nonetheless, E2F2-silenced cells kept expression of typical pluripotency markers and displayed differentiation capacity in vitro. More importantly, E2F2 knockdown in hESC significantly inhibited tumor growth in vivo, which was considerably smaller than tumors generated from control hESC, although displaying typical teratoma traits, a major indicator of pluripotency retention in E2F2-silenced cells. These results suggest that E2F2 knockdown can inhibit hESC proliferation and tumorigenicity without significantly harming stemness, providing a rationale to future protocols aiming at minimizing risks related to therapeutic application of cells and/or products derived from human pluripotent cells.
Introduction
T
In the general rush toward implementing cell therapy for patients with unmet medical needs, acute and long-term safety issues cannot be ignored and should be properly addressed. In that sense, tumor formation persists as a major threat limiting potential therapeutic application of differentiated cells derived from pluripotent ones. Generation of iPS cells itself also involve potential oncogenic hazards [5]. However, since both pluripotency and tumorigenicity are related processes sharing common regulatory pathways [6], lowering the tumorigenic potential of pluripotent cells may not be a straightforward procedure.
Ideally, mechanisms inhibiting tumorigenesis should avoid permanent or uncontrolled modification of the genome and should not significantly affect pluripotency, although the latter issue remains to be clarified. Embryonic stem cells are known to rapidly proliferate [7] and setting proliferation rates of somatic cells to a similar pace, by accelerating cell cycling, improves reprogramming [8,9]. Nonetheless, regulation of ESC proliferation differs from that of somatic cells, involving particular cell cycle proteins [10], which reveals a more complex interplay with pluripotency.
Previous studies have shown that omission of c-MYC expression [11] or additional copies of p53 or Ink4a/ARF [12] do not disturb pluripotency induction of mouse fibroblasts. Reducing the cyclin A1 protein to correct levels has been reported to reduce tumorigenicity of mouse iPS cells, while improving reprogramming to ground state pluripotency [13]. Taken together, these observations suggest that modulation of pathways regulated by classic proto-oncogenes and tumor suppressors, as well as cell cycle regulators, might be a feasible strategy toward reducing tumorigenicity of hESC, with limited impact on stemness.
In this study, we asked whether knockdown of the E2F2 gene encoding a transcription factor with important functions in the control of embryonic development, cell cycle, and apoptosis [14], would inhibit the tumorigenicity of hESC. The canonical E2F control of cell cycle progression, DNA replication, checkpoint control, apoptosis, DNA damage repair, and cell differentiation involves interaction with the tumor suppressor Rb. Such Rb/E2F network is often deregulated in cancer [15]. E2F also induces the transcription of oncogenes typically associated with poor prognosis, such as HMGA1, BMI1, and MELK [16 –18]. The genes, BMI1 and MELK, in particular, are essential for self-renewal of normal and neoplastic stem cells [19 –21]. Function of E2F2, however, is less characterized relative to other members of the E2F family. In astrocytomas, E2F2 directly regulates Bcl-2 gene expression, increasing cell resistance to apoptosis [22], and aberrant E2F2 expression in stem-like cells positively correlates with tumor malignancy [23].
In this study, we found that E2F2 knockdown in hESC caused a significant decrease in HMGA1 and BMI1 expression, followed by a significant reduction in cell proliferation and tumorigenicity, without significantly affecting pluripotency.
Materials and Methods
Cell culture
The hESC line H9 was kindly provided by LANCE (Laboratório Nacional de Células-tronco Embrionárias–Universidade Federal do Rio de Janeiro, Brazil). Cells were cultured under a feeder-free system using Matrigel-coated plates (BD Bioscience) with conditioned cell growth media (mTeSR–StemCell Technology), at 37°C, 5% CO2, and 90%–95% humidity, retaining their undifferentiated state. Growth media were changed daily and culture passage was performed when cells reached 80% confluence, by enzymatic dissociation with Accutase (StemCell Technology).
E2F2 knockdown
Plasmids carrying shRNA against E2F2 (shE2F2) (SureSilencing™ shRNA Plasmids; SABiosciences) were transfected into hESC using Lipofectamine™ RNAiMAX (Life Technologies) according to the manufacturer's instructions. Briefly, hESC were seeded in a six-well plate (1×105 cells/well), suspended in 0.5 μg shE2F2/lipofectamine solution, and incubated at room temperature for 20 min. Afterward, a conditioned medium was added to the suspension and cells were transferred into a Matrigel-coated plate for overnight cultivation. In the second day of transfection, the medium was replaced with a fresh conditioned medium. Another round of transfection was performed (48 h after the first transfection), following the same procedure described. Cells transfected with nonspecific DNA were used as negative control (CT−). Positive control cells were treated with Lipofectamine RNAiMAX, as the other experimental groups, but received no plasmids. After 24 h of the second transfection, cells were selected by cultivation with 250 μg/mL neomycin (G418–Life Technologies) for 3 days, with daily medium changing. Transfected hESC colonies were maintained in the conditioned medium until their use in downstream experiments.
Quantification of gene expression by real-time PCR
Total RNA was extracted from hESCs using the RNeasy® Mini Kit (Qiagen, Inc.), according to the manufacturer's instructions. The RNA concentration was estimated by absorbance at 260 with NANO DROP (Qiagen, Inc.). The A260/A280 ratios in the range of 1.8–2.0 were considered satisfactory for purity standards. Denaturing agarose gel electrophoresis was used to assess the RNA quality. cDNA was synthesized from 1 μg of total RNA using the oligo-dT primer and 200 U of Superscript II Reverse Transcriptase (Life Technologies). Quantitative PCR was performed in a 7500 Real-Time PCR System Thermal Cycler (Applied Biosystems) by the SYBR-Green approach (Platinum SYBR Green qPCR SuperMix-UDG; Life Technologies). Amplification specificity was assessed by dissociation curve analysis. Expression of the housekeeping gene Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as endogenous control. Quantification was based on linear regression analysis from standard curves determined for each gene analyzed, with amplification efficiencies ranging from 90% to 100%. The cDNA samples were subjected to PCR amplification using specific primers designed in different exons and annealing temperature varying from 58°C to 68°C (Supplementary Table S1; Supplementary Data are available online at
Cell cycle and colony assays
The hESC cycling activity was analyzed using the Guava Cell Cycle Reagent Kit (Millipore—Guava Technologies) according to the manufacturer's procedure. Briefly, 1×105 cells per sample were harvested and dissociated until single cell suspension for staining. Cells were then fixed in cold 70% ethanol at 4°C for 24 h, washed in 1×PBS, and incubated in Guava Cell Cycle Reagent at room temperature for 30 min, shielded from light. Samples were analyzed in the Guava EasyCyte 5HT™ Flow Cytometer (Millipore—Guava Technologies) using GuavaSoft 2.1 software. Proliferation of hESC was also analyzed by the size and number of cell colonies grown in vitro as previously described [24]. Briefly, cells were cultivated under low attachment conditions and alkaline phosphatase-positive colonies were examined in an Axio Vert.A1 Microscope (Carl Zeiss) by the AxioVision 4.8.2 software. The number of colonies was determined according to their sizes 100–200, 200–500, and 500–1,500 μm. Samples were analyzed in duplicate.
Analysis of pluripotency markers
Pluripotency of transfected cells were examined by the FlowCellect™ Human ESC Surface Marker (HESCA-1, SSEA1, and SSEA4) Characterization Kit (Millipore), following the manufacturer's instructions. Intracellular levels of the pluripotent factors OCT4 and SOX2 were also determined in permeabilized hESC. Samples were analyzed in a Guava EasyCyte 5HT Flow Cytometer (Millipore—Guava Technologies).
In vitro cell differentiation assays
Spontaneous differentiation in vitro was evaluated through embryoid body (EB) formation. Cells were dissociated until single cells, plated into ultralow adhesion plates (Corning), and cultured in suspension in an EB medium consisting of DMEM/F12+GlutaMax, 15% knockout serum replacement (KSR), 1% MEM-nonessential amino acids (NEAA), and 0.1 mM β–mercaptoethanol (β-ME) (all from Gibco Invitrogen) for 7 days. The medium was changed every other day until complete EB formation, as described by Fernandes et al. [25]. EBs higher than 200 μm were counted for statistical analysis. hESCs were also subjected to neural differentiation according to Baharvand et al. [26]. Cells were cultured for 4–5 days in the absence of feeder layer (Matrigel) with the conditioned medium (mTeSR). The generation of neural rosettes was induced with 4 μM retinoic acid (RA; Sigma-Aldrich) in the differentiation medium (DMEM/F12+GlutaMax, 1% NEAA, and 0.1 mM β-ME) with 5% KSR and without bFGF, for 2 weeks. Neural tube-like structures, obtained in the third week of differentiation by adding 25 ng/mL bFGF, were selected manually and plated on 5 μg/mL laminin and 15 μg/mL poly L-ornithine (both Sigma-Aldrich) precoated culture dishes (Corning) and cultured in the neurobasal medium supplemented with 1% N2 and 2% B27 (all from Gibco Invitrogen) for 30 days.
Teratoma assay
Teratoma formation was assessed by subcutaneously injecting 5×106 hESC, suspended in 300 μL of Matrigel, into the right flank of 4- to 8-week-old Balb/C nude mice (n=9) [27]. Tumors were measured every other day with a digital caliper. Animals were sacrificed when tumors reached a volume of approximately 1,500 mm3. Teratomas were weighed and analyzed by hematoxylin and eosin (HE) staining. The tumor volume was calculated by the following formula: volume (mm3)=width diameter2×length diameter×0.5. The generation of intracranial tumors was analyzed by injecting 5×106 hESC, suspended in 5 μL DMEM/F12 without KSR, with a high precision microsyringe (model 701RN; Hamilton Co.) into the right striatum at a 0.5 μL/min rate, using a stereotaxic equipment (Insight, BR). Brain tissue was evaluated by HE staining 30 days postintracranial implantation of cells. Animals were kept at 22°C–24°C, under a controlled 12-h light–12-h dark cycle, with free access to food and water. All efforts were made to minimize animal suffering, as proposed by the International Ethical Guideline for Biomedical Research (CIOMS/OMS, 1985) and the study was approved by the Internal Review Board of the Federal University of São Paulo (CEP No. 2096/08).
Statistical analysis
All experiments used triplicate samples and three independent experiments were carried out. Data were analyzed by ANOVA with the Bonferroni post hoc test. Significance was established at the P≤0.05 level. Results are expressed as mean±SD.
Results
E2F2 knockdown affects expression of selected proto-oncogenes in hESC
Double transfection of hESC with shRNA plasmids specifically inhibited E2F2 expression, reaching a silencing level of about 60% after 7 days of initial transfection (P<0.001) (Fig. 1A). A significant reduction in the expression levels of the E2F targeted proto-oncogenes BMI1 and HMGA was also found in hESC with silenced E2F2, compared with control cells. Equivalent silencing levels (40% for BMI1 and 41% for HMGA) were verified for these two proto-oncogenes after 7 days of initial transfection of hESC with shE2F2 plasmids (Fig. 1B, C). However, under the same experimental conditions, expression of other classical proto-oncogenes in hESC was not affected by the E2F2 knockdown. As indicated in Fig. 1D–F, expression of c-MYC, MELK, and b-MYB, the last two genes also known to be involved in stem cell self-renewal, was not significantly changed after 7 days of initial E2F2 silencing.
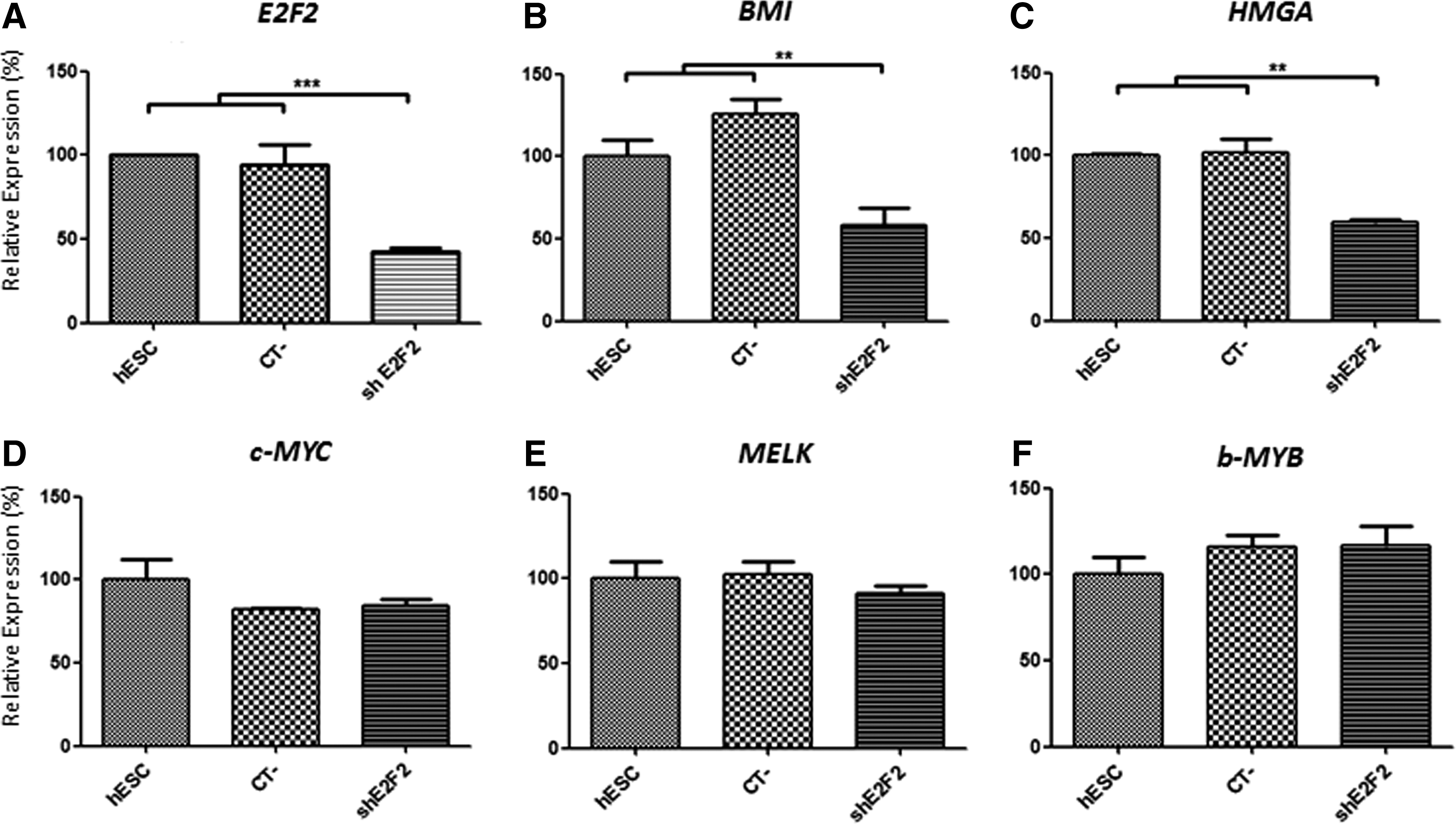
Expression of proto-oncogenes after E2F2 knockdown in human embryonic stem cells (hESC). In addition to E2F2
E2F2 knockdown significantly inhibits hESC proliferation
Cell cycle analysis of hESC subjected to E2F2 silencing indicated a lower percentage of cells at both the S (P<0.01) and G2/M phases (P<0.001), as well as a higher amount of cells at G1 (P<0.001), compared with control cells (Fig. 2A). To confirm the effects of E2F2 knockdown on cell proliferation, the amount of hESC colonies developed in vitro under E2F2 silencing was determined. In agreement with the previously verified cell cycle inhibition, the capacity of hESC to generate colonies was significantly affected by the E2F2 knockdown. A reduced amount of hESC colonies of large (500–1,500 μm) diameter were detected after 4 and 7 days of growth under E2F2 silencing (Fig. 2B). Earlier culture periods were not suitable for such evaluation since most hESC colonies were still underdeveloped (below 200 μm). In some samples, hESC treated with scrambled shRNA (CT−) also yielded lower amounts of colonies compared with positive control cells, mostly after 7 days of growth, although not achieving overall significantly reduced levels.
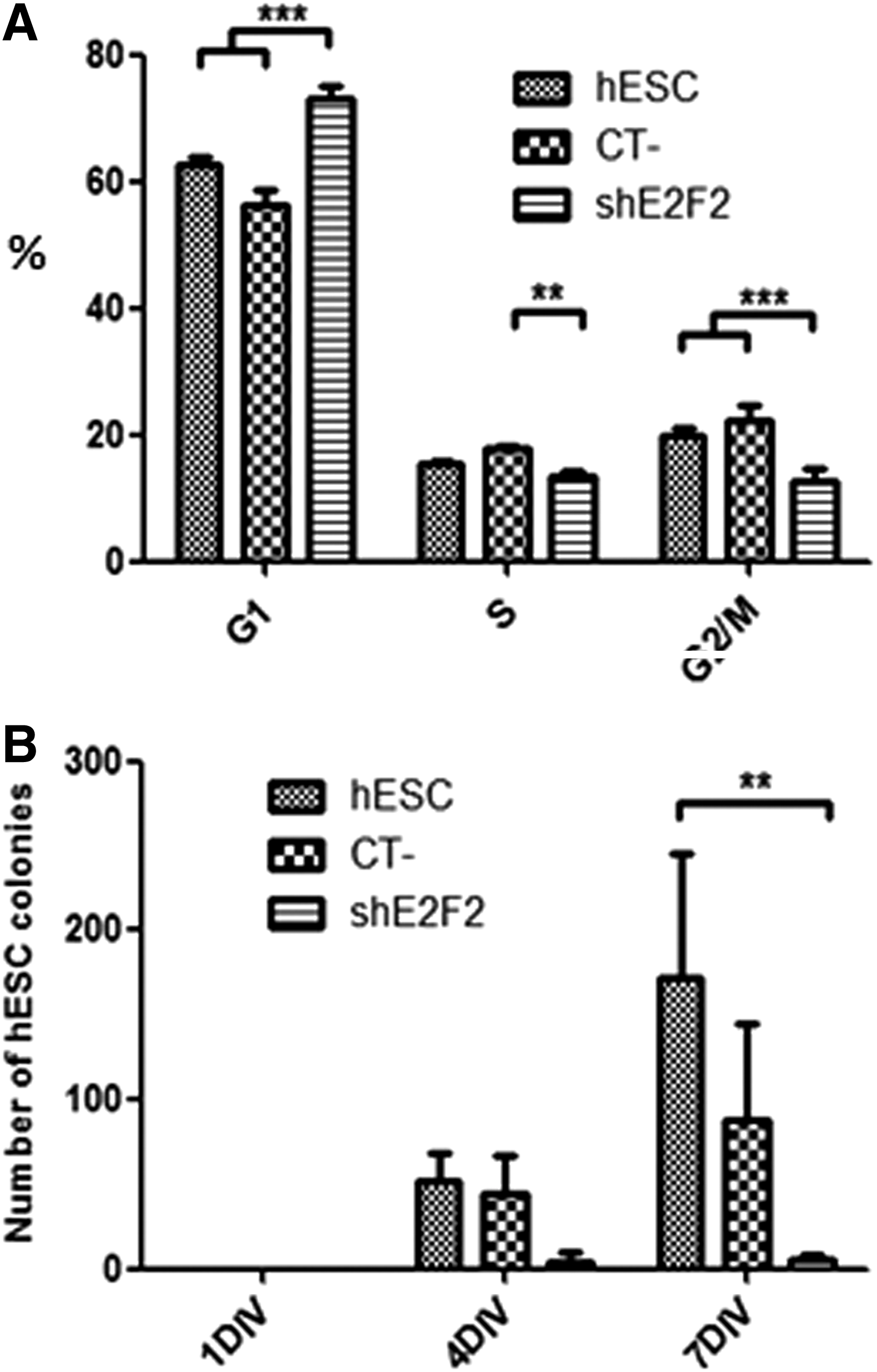
E2F2 knockdown significantly inhibits hESC proliferation.
hESC retain stemness despite E2F2 knockdown
In spite of the reduced proliferative activity of hESC verified upon E2F2 knockdown, under such conditions, no evident signs of pluripotency loss were found. The proportion of pluripotent cells based on the expression of typical membrane surface markers (HESCA+/SSEA4+/SSEA1− immunophenotype) was not significantly altered in the E2F2–silenced group, compared with controls (Fig. 3A). These results were further confirmed when determining the intracellular levels of pluripotency factors. Seven days post-E2F2 knockdown, most hESC were still positive for OCT4 and SOX2 expression (98% and 96% of the total cell population analyzed, respectively), displaying frequency and signal levels similar to positive control cells (Fig. 3B).

Pluripotency of hESC is preserved after E2F2 knockdown.
hESC with silenced E2F2 were also capable of forming EBs, indicating a preserved capacity to spontaneously differentiate and form three-dimensional multicellular aggregates in vitro. However, as shown in Fig. 3C, a somewhat reduced amount of EBs (≥200 μm in diameter) was developed from hESC kept for 7 days under E2F2 silencing, compared with controls. This result, nonetheless, is in agreement with the initial cell proliferation inhibition observed after E2F2 knockdown. When subjected to directed neural differentiation conditions, control and E2F2-silenced cells responded in a similar fashion to such stimulus. As shown in Fig. 4, typical neural rosettes comprising columnar progenitor cells were found in the initial differentiation stage (Fig. 4A–C). A later cell organization into neural tube-like structures was also observed after bFGF addition to the medium (Fig. 4D–F). At a later differentiation stage, numerous processes and fibers grew out from the neural tube-like structures and some migrating cells could be visualized in association with these structures (Fig. 4G–I). The generation of neural rosettes also occurred when control cells and E2F2-silenced hESC where directly injected into mouse brains (Fig. 4J–L), an indication that the capacity to respond to in vivo neural differentiation cues was also maintained.
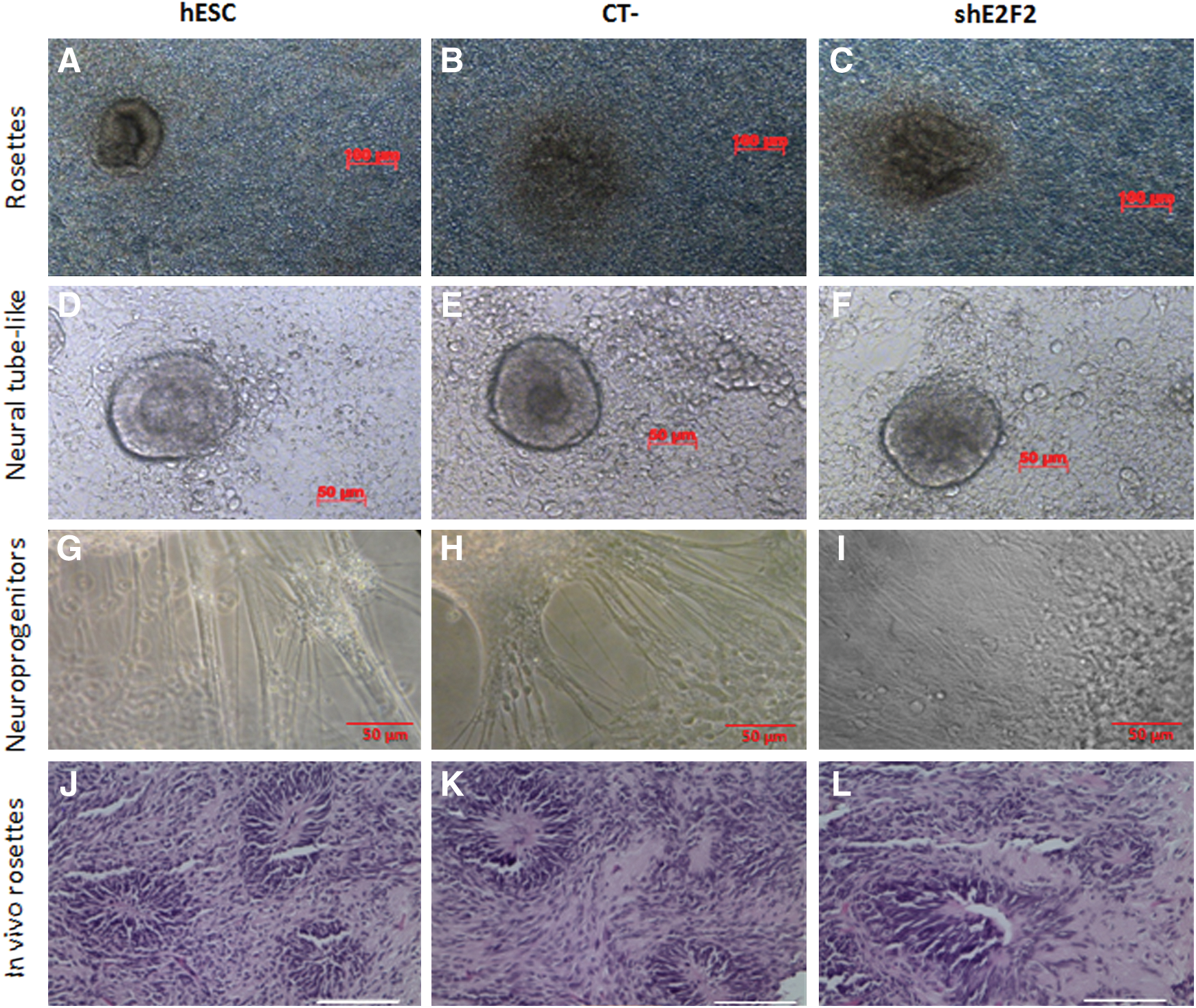
E2F2-silenced hESC respond to directed differentiation cues. Cultures of hESC subjected to neural differentiation stimuli in vitro displayed rosette
E2F2 knockdown suppresses tumorigenicity of hESC
When injected subcutaneously in nude mice, E2F2-silenced hESC were capable of generating teratomas, a further indication of stemness retention. Nontheless, tumors developed from E2F2-silenced hESC grew at a lower pace compared with tumor growth kinetics from control cells, reaching significantly smaller volumes 60 days postinjection (Fig. 5A, B). Mice bearing E2F2-silenced hESC teratomas were further monitored untill 80 days postinjection, when tumor volumes were still below 250 mm3 and exponential tumor growth was absent. Conversely, teratomas developed from control hESC reached similar volumes (250 mm3) within 45 days and were already over 1,500 mm3 in volume at day 60, postinjection.

E2F2 knockdown inhibits tumorigenicity of hESC.
When excised, teratomas had a mixed consistency displaying solid areas combined with fluid-filled cysts. A careful histological analysis of teratomas generated from control and E2F2-silenced cells revealed tissue structures representing all three germ layers. As shown in Fig. 5C, various structures could be detected, including gut-like epithelial tissues (endoderm), muscle, cartilage, and adipose tissues (mesoderm), as well as neural tissue (ectoderm).
Discussion
The function of the E2F2 transcription factor in tumorigenesis is poorly characterized, relative to other members of the E2F family. Studies with mouse models have reported both tumor suppression and pro-oncogenic activities. Whereas mice lacking E2F2 (either E2F2 +/− or E2F2 −/−) provide evidence of a haploinsufficient tumor suppressor function for E2F2 in murine models of lymphomagenesis [28] and MYC-driven mammary tumors [29], mice overexpressing E2F2 were reported to develop thymomas [30]. Mouse fibroblasts stably overexpressing E2F2 were also found highly prone to oncogenic transformation [31].
In human cancers, however, most studies support a pro-oncogenic role for E2F2. E2F2 has been shown to positively regulate MYCN transcription in aggressive neuroblastomas [32] and to induce cell proliferation in breast cancer cells, under control of the AP-1 transcription factor [33]. High E2F2 expression has also been associated with poor prognosis. In breast cancer, increased E2F2 expression was reported to be associated with poor patient survival [34]. In glioblastoma, E2F2 was found hyperexpressed in CD133+ tumor cells, and its expression correlated with the tumor malignancy grade [23].
One evidence linking E2F2 and pluripotency was provided by a recent study using an ectopic xenograft model of prostate cancer, where E2F2 silencing by let-7a, a microRNA known to regulate expression of the pluripotent factor OCT4, caused suppression of tumorigenesis in nude mice [35]. Another recent study integrating whole trancriptome and binding site analyzes suggested E2F as a central regulator of self-renewal of both hESC and hIPS cells [36], through modulation of WNT and FGF pathways. In line with such evidence, the present study demonstrated for the first time that E2F2 knockdown significantly inhibits self-renewal and in vivo tumorigenicity without affecting the stemness of hESC. In mouse ESC, loss-of-function mutations in the Gpc4 gene encoding a member of the heparin sulfate proteoglycan superfamily were also reported to reduce tumorigenicity. Interestingly, Gpc4 also modulates WNT and FGF pathways, but the inhibition of tumorigenicity may be related to premature cell differentiation since Gpc4 loss-of-function significantly accelerated ESC differentiation [37]. Similar results were reported for mouse iPS cells, in which cyclin A1 knockdown-mediated inhibition of tumorigenicity was partly attributed to an enhanced cell differentiation capability [13]. In the present study, however, no significant changes in hESC differentiation were noted, suggesting differences in the mechanism of tumor inhibition by E2F2 knockdown.
Since pluripotency and tumorigenicity are linked processes controlled by common pathways [6], the mechanism by which E2F2 inhibits hESC tumorigenicity may involve regulation of typical proto-oncogenes that do not affect ESC differentiation pathways. Indeed, the inhibition of BMI and HMGA1 genes observed after E2F2 knockdown are in agreement with this notion. However, other target proto-oncogenes c-MYC, MELK, and b-MYB, were not affected by the E2F2 knockdown, indicating that such mechanism is all but ordinary.
Interestingly, in the ESC context, c-MYC, MELK, and b-MYB proteins are important for cell functionality. High levels of c-MYC have been implicated in the maintenance of ESC fitness at the onset of differentiation [38]. Similarly, MELK (maternal embryonic leucine zipper kinase), a member of the Snf1/AMPK family of serine/threonine kinases, is involved in the early and late stages of embryonic development, where it regulates the germ cell line [39] as well as neural differentiation, the latter action also involving the participation of b-MYB [40]. Low levels of b-MYB, on the other hand, have been reported to induce aneuploidy and differentiation-associated cell death in murine ESC [41]. EB and teratoma formation as well as full ectodermic, mesodermic, and endodermic differentiation capacity were detected in E2F2-silenced hESC, indicating that they remained functional despite the interference in gene expression. The preservation of c-MYC, MELK, and b-MYB expression after E2F2 knockdown in hESC might explain, at least in part, the absence of significant inhibition of pluripotency and differentiation capability found in this study.
Lower levels of E2F factors may also increase the amount of free Rb, favoring its interaction with other protein partners such as Rb binding proteins (RBBPs). Interestingly, RBBPs bound to Rb have been detected in hESC and implicated in the maintenance of their pluripotency [42]. In fact, proper cellular levels of certain cell cycle proteins, including cyclin A1 and Rem2 GTPase, have been reported relevant for achieving and sustaining pluripotency [8,13].
In addition to cell proliferation, E2F2 may also regulate genes affecting other hallmarks of cancer in ESC, such as cell survival and energy/metabolism. Indeed, in vivo experiments with E2F1-3 knockout mice showed that E2F1-3 function as transcriptional activators necessary for ESC survival. In their transit-amplifying cell progeny, E2F1-3 optimally activate expression of E2F responsive targets, inducing ectopic cell divisions [43]. In silico analyses have also indicated that E2F directly targets metabolic network/energy generation pathways driving self-renewal of human pluripotent stem cells [36]. All these mechanisms may contribute to the E2F2-mediated effects on hESC tumorigenicity and further studies shall help elucidate this question.
In sum, our findings suggest a role for E2F2 in regulating hESC self-renewal and tumorigenicity. The reported effects upon E2F2 knockdown provide a rationale for inhibition of hESC tumorigenicity, with limited effects on pluripotency and differentiation capacity. Envisioned therapeutic applications of specialized cells generated from pluripotent cells will likely require some degree of in vitro cell manipulation following regulatory guidelines, before administration into patients. In such procedures, transient and specific silencing of E2F2 gene or use of small molecule inhibitors of E2F2 could restrain proliferation of partially differentiated cells and the undesirable occurrence of tumors. Further developments of this strategy may help circumvent safety issues concerning future therapeutic application of cells and/or products derived from human pluripotent cells.
Footnotes
Acknowledgments
This work was supported by grants from FAPESP-CEPID, CNPq, and INCT-CETGEN. Daniela Emi Suzuki and Adriana Miti Nakahata were recipients of CNPq and CAPES-PNPD fellowships, respectively.
Author Disclosure Statement
The authors disclose no potential conflicts of interest in connection with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.