
Abstract
Notch has a broad range of regulatory functions in many developmental processes, including hematopoiesis, neurogenesis, and angiogenesis. Notch has several key functional regions such as the RBP-Jκ/CBF1 association module (RAM) domain, nuclear localization signals (NLS), and ankyrin (ANK) repeats. However, previous reports assessing the level of importance of these domains in the Notch signaling pathway are controversial. In this study, we have assessed the level of contribution of each Notch domain to the regulation of mammalian neural stem cells in vivo as well as in vitro. Reporter assays and real-time polymerase chain reactions show that the ANK repeats and RAM domain are indispensable to the transactivation of Notch target genes, whereas a nuclear export signal (NES)-fused Notch intracellular domain (NICD) mutant defective in nuclear localization exerts a level of activity comparable to unmodified NICD. Transactivational ability appears to be tightly coupled to Notch functions during brain development. Unlike ANK repeats and RAM domain deletion mutants, NES-NICD recapitulates NICD features such as promotion of astrogenesis at the expense of neurogenesis in vitro and enhancement of neural stem cell character in vivo. Our data support the previous observation that intranuclear localization is not essential to the oncogenesis of Notch1 in certain types of cells and imply the importance of the noncanonical Notch signaling pathway in the regulation of mammalian neural stem cells.
Introduction
T
NICD, the active form of Notch, contains several functional domains, including a recombinant signal binding protein for the RBP-Jκ/CBF1 association module (RAM) domain that mediates association with CBF1, nuclear localization signals (NLS), ankyrin (ANK) repeats, a transactivational domain, and conserved proline/glutamic acid/serine/threonine-rich motifs (PEST domain) leading to the degradation of NICD [5,6]. Many studies have shown that ANK repeats are required for Notch functions such as transcriptional activation and induction of oncogenesis [7 –9]. However, there are conflicting reports on the importance of the NLS and RAM domains. The NLS is important in the inhibition of myogenic differentiation of 3T3 cells [10], and transformation of HC11 mouse mammary epithelial cells [7] and E1A-immortalized baby rat kidney cells (RKE) [11]; whereas it is dispensable for the induction of T cell leukemia/lymphoma when Notch is expressed in the bone marrow [12]. The RAM domain shows a wide spectrum of importance in Notch-dependent cellular events. It is essential for the survival of neuronal precursor cells [13] and is partially responsible for the inhibitory effects of Notch on osteoblastogenesis [9]. The RAM domain is not required for Hes1 promoter transactivation in ST-2 stromal cells [9], inhibition of myogenesis [14], T-cell leukemogenesis [12], or transformation of RKE [11,15].
In this study, we investigate the contribution of the RAM domain, ANK repeats, and NLS to Notch function in the mammalian nervous system. Our results show that the RAM domain and ANK repeats are crucial for Notch function during brain development. In contrast, a Notch mutant that resides exclusively in the cytoplasm maintained a comparable level of activity with unmodified NICD in all aspects examined in this study. These results indicate that the cytosolic form of Notch can recapitulate the effects of nuclear NICD in the mammalian nervous system and shed new light on the noncanonical Notch signaling pathway during brain development.
Materials and Methods
Plasmid construction
The NICD sequence was amplified using the primers NICD-F (5′-GGATCCGCCACCATGGCTCCATGGCCAGCTCTG-3′) and NICD-R (5′-GGATCCCTAAGCGTAATCTGGAGAACATCGTATGGGTAGCTCGAGCTGTCCAACAGG-3′) with GNIA [2] as a template. A nuclear export signal (NES) was fused to the 3′ end of NICD by PCR using the primers NICD-F and NES-NICD-R (5′-GGATCCCTAAGCGTAATCTGGAGAACATCGTATGGGTACAGGTCCAGGCCGGCCAGCTTCAGGGCCAGGCTCGAGCTGTCCAACAGG-3′) producing NES-NICD. ΔRAM-F (5′-GGATCCGCCACCATGGCTCCAAAGAAGAAGCGTAAGGTAGAGACCAAGAAGTTCCGGTT-3′) and NICD-R primers were used to generate the ΔRAM mutant. The ΔRAM-F primer contains the SV40 NLS sequence.
The deletion mutant lacking the ANK repeat, ΔANK, was generated by site-directed mutagenesis using the following primers: 5′-TGCTGACCTGCGCATGTCTGCCATGGTGCGGCTTTTGGATGAGTACAACC-3′ and the complementary reverse primer. All amplified NICD and NICD mutant sequences were initially cloned into pGem-T Easy vector (Promega, Madison, WI) and then introduced into the BamHI site of MSIG [16].
Western blot
NIH3T3 cells were lysed using RIPA buffer with protease and phosphatase inhibitor cocktail. Equal amounts of protein were resolved by 10% (w/v) sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Pierce, Rockford, IL). The membranes were blocked with TBST (150 mM NaCl, 10 mM Tris/HCl, 0.1% (v/v) Tween 20, pH 8.0) containing 1% (w/v) BSA (Invitrogen, Carlsbad, CA), and incubated with anti-HA or anti-β-actin (both from Sigma, St. Louis, MO) primary antibodies diluted in blocking solution at 4°C overnight. The membranes were then incubated with horseradish peroxidase-conjugated goat anti-mouse or rabbit IgG secondary antibody (Pierce) at room temperature. The protein bands were visualized with the enhanced chemiluminescence system (Millipore, Temecula, CA) and X-Omat film (Kodak, Rochester, NY).
Luciferase assay
Three different luciferase reporter constructs, JH23 (4×CBF1-binding sites and the basal SV40 promoter-luc) [17], Hes1 promoter-luc [18], and Hey2 promoter-luc [19], were used to measure the transactivation activity of Notch. 5×105 of NIH3T3 cells were plated in a six-well dish and transiently cotransfected with each luciferase reporter construct and NICD expression vector. The luciferase assay was performed according to the manufacturer's protocol (Promega). Briefly, transfected cells were harvested and lysed in 200 μL of 1×Reporter Lysis Buffer for Dual Luciferase Assay. Twenty microliters of lysate was mixed with the firefly Luciferase Assay Reagent II, and the luminescent signal was measured using a luminometer. Stop and Glo Reagent (100 μL) was added to the same tube. The Renilla luciferase vector ptk-Rluc was cotransfected to normalize for transfection efficiency.
Reverse transcription and quantitative real-time PCR
Total RNA was prepared from cultured cells using Trizol reagent (Invitrogen), and cDNAs were synthesized from 1 μg of each RNA sample by using an oligo(dT) primer and AMV-RT enzyme (Roche, Indianapolis, IN). Real-time quantitative PCR was performed according to the Smart Cycler System's (Takara, Shiga, Japan) protocol using the following primers: Hes1 forward, 5′-TACCCCAGCCAGTGTCAACA-3′, Hes1 reverse, 5′-TCTTGCCCTTCGCCTCTTC-3′, Hes5 forward, 5′-CAAGGAGAAAAACCGACTGC-3′, Hes5 reverse, 5′-GCTGGAAGTGGTAAAGCAGC-3′, Hey1 forward, 5′-TGAGCTGAGAAGGCTGGTAC-3′, Hey1 reverse, 5′-ACCCCAAACTCCGATAGTCC-3′, Hey2 forward, 5′-CCCATGTCGCCTATCCACAT-3′, Hey2 reverse, 5′-TGGCATCCGAAGAGCAGAAT-3′, GAPDH forward, 5′-AGCCTCGTCCCGTAGACAA-3′, and GAPDH reverse, 5′-AATCTCCACTTTGCCACTGC-3′.
Retroviral vector production and transduction
The method of retroviral vector production has been previously described [20]. Briefly, the retroviral construct was transfected into 293T cells with gag-pol (pCA-gag-pol) and env-expressing vector (VSV-G) using Lipofectamine-PLUS reagent (Invitrogen). Supernatant was collected at 48 h after transfection, filtered through a 0.45 μm filter, and frozen at −80°C until use. Concentrated viral stocks were prepared by ultracentrifugation at 25,000 rpm for 90 min at 4°C in an SW28 rotor (Beckman-Coulter, Fullerton, CA). Pellets were resuspended in 50 μL of phosphate-buffered saline (PBS) at 4°C for about 12 h, and virus aliquots were stored at −80°C. For transduction, NIH3T3 cells were seeded at 1×105 in six-well plates on the previous day. Viral supernatants were added in the presence of polybrene (final concentration 8 μg/mL) (Sigma). The viral titer was determined by measuring the percentage of green fluorescent protein (GFP) or HA-positive NIH3T3 cells transduced with different dilutions of virus stock.
Animals and in vivo injection into the embryonic brain
All animal protocols were approved by the Institutional Review Board and conducted in the Laboratory Animal Research Center of Sungkyunkwan University. Timed pregnant CD1 mice (Orient Bio, Osan, Korea) were used for viral injections, and embryos were considered 0.5 days old (embryonic day 0.5, E0.5) when a vaginal plug was detected in the morning. Virus delivery into the telencephalic ventricle was performed at E9.5, using the ultrasound backscatter microscopy (UBM) system as previously described [2,21]. Briefly, pregnant mice were anesthetized with Zoletil 50 (Virbac, Carros, France) and Rompun (Bayer Korea, Korea). The uterus was exteriorized, and the fetuses were scanned, using an ultrasound biomicroscopic imaging system (Vevo660; VisualSonics, Toronto, ON, Canada). Ultracentrifuge-concentrated viruses were injected into the telencephalic ventricle of E9.5 or E13.5 embryos.
Immunofluorescence of brain sections and cell quantification
Standard immunofluorescence procedures were used for visualization of GFP and lineage-specific marker gene expression in retroviral vector-injected animals using the following primary antibodies: anti-GFP (Invitrogen), anti-βIII-tubulin (TuJ1)(Covance, Princeton, NJ), anti-Sox2 (Chemicon, Temecula, CA), and anti-HA antibody (Sigma). Briefly, embryonic brains at E14.5 or E17.5 were fixed in 4% paraformaldehyde and cryosectioned. Sections were washed in PBS, then blocked for 1 h with PBS containing 1% fetal bovine serum and 0.2% Triton X-100. Samples were incubated with primary antibodies diluted in blocking buffer overnight at 4°C. Sections were washed thrice in PBST (PBS/0.1% Tween-20) and incubated for 1 h at room temperature with Alexa-488 and -555 conjugated secondary antibodies (Invitrogen) diluted in blocking solution. Immunostained samples were further counterstained with Hoechst 33342. To count the number of GFP+, or GFP/lineage marker double-positive cells, images of the dorsal telencephalic region were taken from each embryo, for a total of three or four embryos per each NICD mutant, with a Zeiss LSM 510 confocal microscope with an Axiovert 100 M system (Zeiss, Oberkochen, Germany). Yellow-colored cells were counted as double positives for GFP and each lineage-specific marker. Ventricular zone (VZ)/subventricular zone (SVZ) and extraventricular regions (including intermediate zone, cortical plate/marginal zone) of telencephalon were determined by cell density and laminar architecture visualized by Hoechst staining.
Neural progenitor cell preparation, in vitro differentiation, and double immunostaining
Primary neural progenitor cells were prepared from the lateral and medial ganglionic eminences of E14.5 embryos. Dissected brain tissue was minced, washed thrice with PBS, and incubated in 0.25% trypsin (Invitrogen) at 37°C for 5 min. DNase and ovomucoid trypsin inhibitor (both from Worthington, Freehold, NJ) were added, and samples were triturated using a fire-polished Pasteur pipette. Cells were washed twice with DMEM/F12 media, resuspended in PBS, and run through a 40 μm cell strainer (Falcon, Franklin Lakes, NJ). Before in vitro differentiation, primary neural progenitors were transduced with the concentrated retroviral vectors expressing NICDs. After 48 h, cells were seeded on poly-L-ornithine and laminin (both from Sigma)-coated plates and incubated for another 2 or 3 days in DMEM/F12 containing 2% FBS. For double immunostaining, differentiated cells were fixed and processed for immunostaining as described earlier [2]. As primary antibodies, we used antibodies to GFP (Invitrogen), βIII-tubulin (TuJ1), GFAP (Invitrogen), and S100β (Abcam, Cambridge, MA). Alexa-488 and -555 conjugated secondary antibodies (Invitrogen) were used for double fluorescence. Fluorescent images were obtained with an upright microscope (Eclipse 80i; Nikon, Tokyo, Japan) equipped with a GFP filter set.
Results
Characterization of mutant forms of NICD used in this study
To map the regions that are important for Notch function, modifications were introduced into the intracellular domain of Notch1 lacking its C-terminal region, including the PEST domain (Fig. 1A). This PEST-deleted form of NICD shows constitutive signaling activity; it effectively promotes radial glial identity and increases the frequency of neurosphere formation [2,20]. The modifications included deletion of the ANK repeats (ΔANK) or RAM domain (ΔRAM), and insertion of the nucleus export signal (NES) derived from heat-stable protein kinase inhibitor (PKI) [22] to the C-terminal end of NICD (NES-NICD) [11]. Since the first NLS sequence overlaps with the RAM domain, the NLS of simian virus 40 (SV40) large T antigen was added to the ΔRAM mutant to rescue the possible loss of nuclear localization associated with the RAM domain deletion.

Expression and subcellular localization of mutant NICDs.
To facilitate detection of the expression of these NICD derivatives, an HA tag was fused to the C-terminal ends. The NICD mutants and GFP reporter gene were expressed from the retroviral vector MSIG [16]. MSIG contains the murine stem cell virus long terminal repeat that drives gene expression and an internal ribosome entry sequence which enables bicistronic expression of a gene of interest and GFP. The retroviral constructs were transfected into 293T cells using a three-plasmid transfection method [20], and viral supernatants were taken to transduce NIH3T3 cells. Western blot analysis of cellular lysates from the transduced NIH3T3 cells using an anti-HA antibody showed that all NICD constructs expressed comparable amounts of protein at the predicted size (Fig. 1B). Immunofluorescence revealed that all NICD mutants localized exclusively to the nucleus except for NES-NICD (Fig. 1C). Fusion of the NES to NICD resulted in complete exportation of the protein from the nucleus to the cytoplasm despite the existence of two NLS sequences on NICD. Since a significant level of NLS-deleted NICD proteins still remained in the nucleus in previous studies [10,12], our result suggests that insertion of the NES sequence derived from PKI can eliminate NICD from the nucleus more efficiently than NLS deletion.
Measurement of the transactivational ability of NICD mutants
To examine the impact of the NICD modifications on the transactivation of target genes, a luciferase reporter assay was performed. A reporter construct driven by a CBF1-responsive element (CBFRE, four CBF1-binding sites, and the basal SV40 promoter) [1] was introduced into NIH3T3 cells along with the NICD expression vectors. NICD increased expression of the reporter gene and this activation decreased dramatically on deletion of the ANK repeats or RAM domain (Fig. 2A). Surprisingly, NES-NICD still possessed approximately 60% transactivation ability compared with wild-type NICD. Thus, we focused our efforts on comparing NICD and NES-NICD with other Notch reporter plasmids, and found that NES-NICD increased Hes1 promoter activity at a 70% level of NICD (Fig. 2B). Interestingly, the difference in Hey2 promoter-activating ability between NICD and NES-NICD was not statistically significant (Fig. 2C). Overall, similar tendencies were also observed in C17.2, an immortalized mouse neural stem cell line derived from neonatal cerebellum cells [23] (Fig. 2D–F).

Comparison of transactivational ability of NICD mutants. NIH3T3 cells and C17.2 cells were transfected with NICD mutant vectors along with CBFRE-luc
These results were confirmed further by quantitative real-time PCR analysis of endogenous Notch target genes. Mouse primary neural stem cells prepared from E14.5 embryonic brain were transduced with retroviral vectors expressing NICD or NES-NICD, and target mRNA expression was measured. The amounts of Hes1, Hey1, and Hey2 mRNAs were induced by NES-NICD to a level statistically comparable with that increased by NICD (Fig. 2G, I, J), and NES-NICD still induced Hes5 mRNA expression at a 60% level of NICD (Fig. 2J). However, the increase of Hes1 mRNA was less prominent (within 1.5-fold) in both NICD and NES-NICD-infected cells than that of other target genes. This result might be explained by the fact that the Hes1 protein negatively regulates its own promoter [24]. Taken together, our data, generated from diverse experimental settings using different techniques (reporter assay and quantitative-RT PCR) and different experimental cell models (NIH3T3, C17.2, and primary neural stem cells), indicate that NES-NICD retains a significant level of the transcriptional activation function of the canonical Notch pathway, despite its exclusive presence in the cytoplasm.
Effect of NICD mutants on neural stem cell character in vivo
The observation that NES-NICD quite efficiently activated transcription of the Notch target genes raised the possibility that the cytosolic form of NICD could enhance neural stem cell character, one of the very well-known functions of Notch in development of the mammalian central nervous system. In vivo Notch activity in the embryonic brain has been examined by a method using UBM image-guided gene delivery (UIGD) [21,2]. In these studies, a retroviral vector bicistronically expressing NICD and a reporter gene was injected into the telencephalic ventricle of embryonic age 9.5 (E9.5) embryos and the pattern of transgene expression in the brain was analyzed at E14.5. While cells transduced with control vectors were dispersed throughout the brain, ones transduced with NICD-expressing vectors were found mostly in the VZ where neural stem cells reside.
Similar to previous studies, NICD-expressing cells were retained in the VZ of E14.5 brains, whereas control GFP-positive cells were dispersed throughout both the VZ and postmitotic zones (TuJ1-stained region) (Fig. 3A). NICD mutants lacking ANK or RAM domains appeared to have lost the ability to enhance neural stem cell character, as the infected cells were found throughout the whole brain region. On the other hand, NES-NICD still exerted its influence on the maintenance of neural stem cell character, as most of the NES-NICD-expressing cells were detected in the VZ. Consistent with the E14.5 results, E17.5 brains also showed higher fractions of NICD- and NES-NICD-expressing cells in the neocortical VZ and SVZ than ΔANK- or ΔRAM-expressing cells (Fig. 3B). Furthermore, the majority of NICD or NES-NICD-infected cells in the VZ were colabeled with Sox2, indicating that these cells are neural stem cells (Fig. 3D).
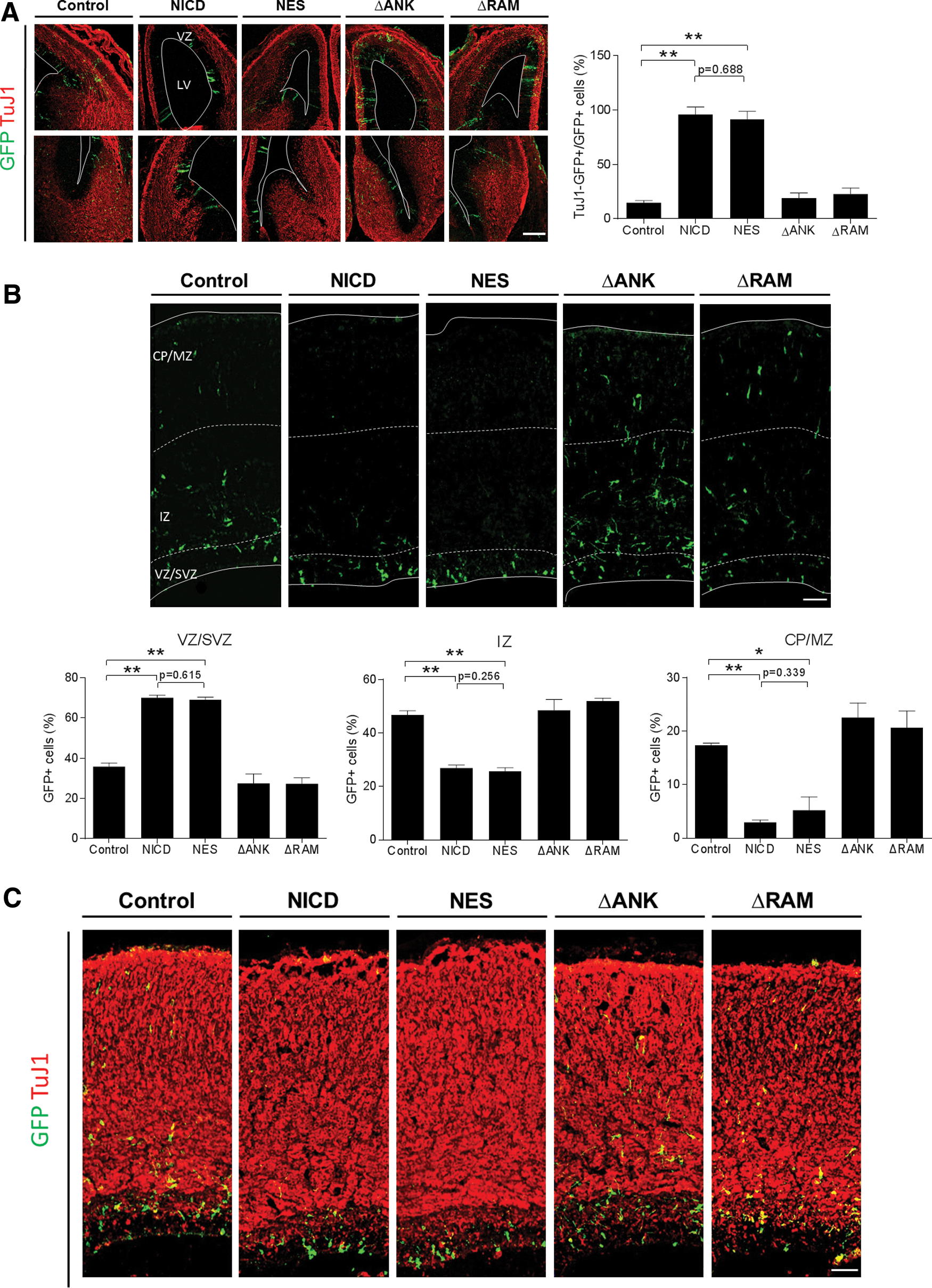
Effects of NICD mutants on neural stem cell character in vivo. Mouse embryos were injected intraventricularly at E9.5 or E13.5 with retroviral vectors expressing NICD mutants and GFP and examined at E14.5
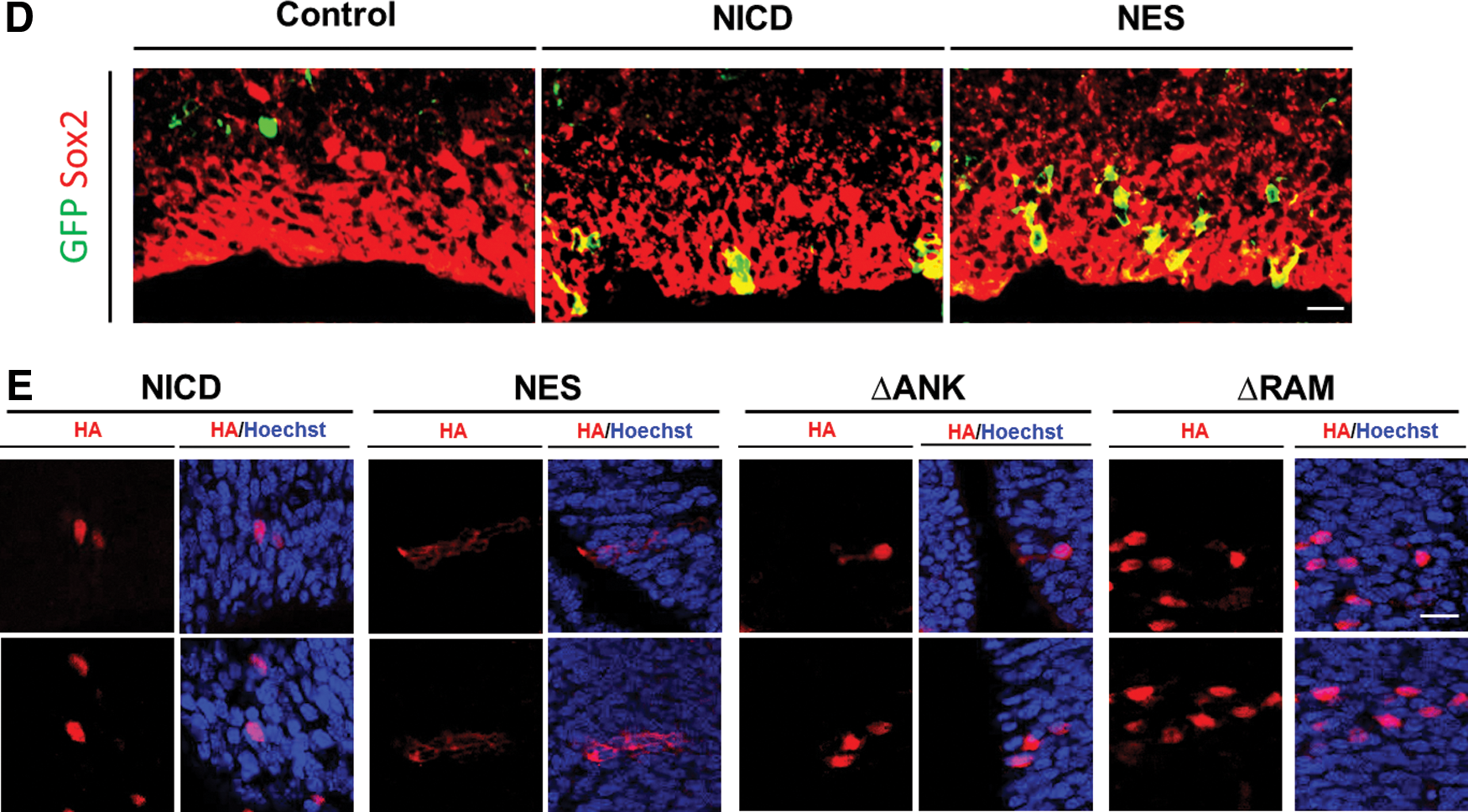
To be certain that NES-NICD was not expressed in the nucleus, confocal immunofluorescence microscopy was used to show that this protein localized to the cytoplasm of transduced cells in mouse brains (Fig. 3E). These results indicate that, consistent with the transcriptional activation results described earlier, the ANK repeats and RAM domain are indispensable for the promotion of neural stem cell character in vivo, whereas nuclear localization is not an essential requirement for this function.
Effects of NICD mutants on the differentiation of neural stem cells in vitro
Next, the roles of the Notch domains in the cell fate choice of neural stem cells were investigated. Under differentiation conditions, Notch activation prevents neural stem cells from giving rise to neurons [3,20]. Primary neural stem cells prepared from E14.5 embryos were transduced with the same titer of retroviral vectors expressing NICD mutants, followed by differentiation with DMEM/F12 containing 2% fetal bovine serum. Four days later, the samples were subject to immunostaining. As shown in Fig. 4A–C, NICD dramatically inhibited neuronal differentiation (TuJ1+) while promoting astrocytic fate (GFAP+ or S100β+). Consistent with other results in this study, NES-NICD showed a similar level of inhibition of neurogenesis and promotion of astrogenesis to that of unmodified NICD, whereas ΔANK and ΔRAM mutants were severely compromised. These results indicate that the consequence of forced expression of NICD in the cytoplasm during cell fate choice was indistinguishable from nuclear NICD.
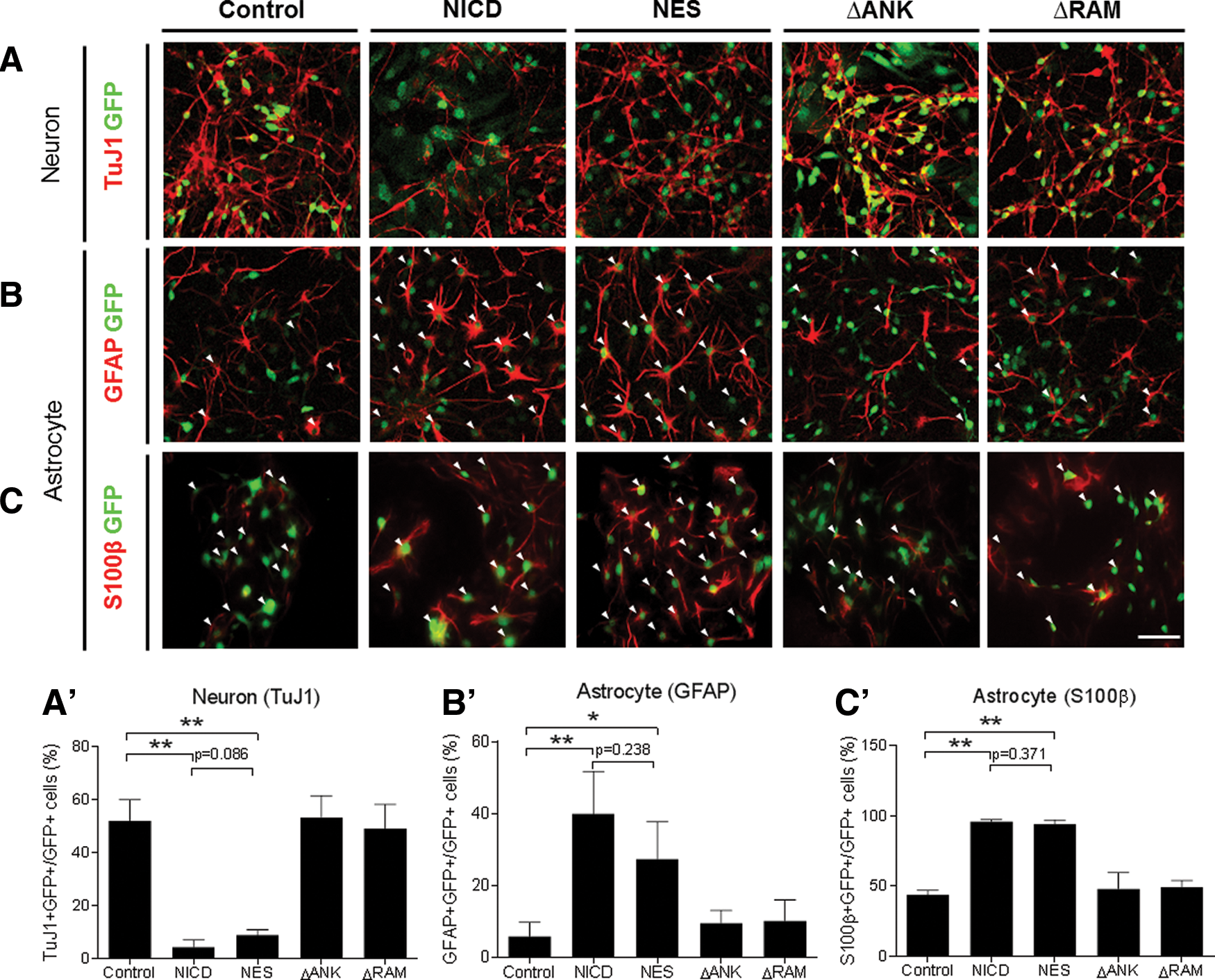
Effects of NICD mutants on neural stem cell differentiation in vitro.
Discussion
The initial goal of this study was to assess the significance of the RAM domain and NLS of NICD because their role is under dispute, whereas ANK repeats have been known to be critical to the Notch signaling pathway with almost no exceptions. The NLS and RAM domains have been considered essential factors, because the basic assumption of the canonical Notch pathway is the binding of NICD to CBF1 through the RAM domain after translocation to the nucleus. However, deletion of the RAM domain and NLS produced controversial results, showing no effect or a modest to severe defect, depending on the experimental settings.
Before our assessment of Notch regions in this study, no in vivo evidence existed that clarified whether Notch requires each domain in physiological conditions. To address this question, the UIGD technique was used to determine the importance of these major Notch domains in the developing mammalian brain. Our data indicate that both the RAM domain and ANK repeats appear to be essential for Notch function in the mammalian nervous system in vitro and in vivo. Unexpectedly, addition of NES to NICD did not affect the function of Notch significantly in all our experimental settings, although it effectively blocked nuclear localization of NICD. These results are consistent with the previous observation that intranuclear localization is not essential to the oncogenesis of Notch1 [12].
The NLS deletion done in most previous studies to impair nuclear localization may cause some problems. Two NLSs are located in different positions on the NICD. Therefore, these two should be deleted simultaneously to efficiently inhibit the movement of NICD to the nucleus. In addition, it is possible that an unknown important region exists near the NLS and if so, deletion of this region would produce misleading data in determining the importance of nuclear localization. Indeed, a third CBF1 interaction domain overlaps with the NLS sequence in Drosophila Notch [25]. These drawbacks were successfully overcome by an NES fusion that did not delete any amino acids from NICD [11]. However, we could not completely exclude the possibility that NES-NICD may initially enter the nucleus to activate the Notch signaling pathway and then be exported to the cytoplasm by the NES. Currently, there is no effective way to prevent translocation of Notch into the nucleus without deletion of an NLS or the addition of an NES. If a chemical or a tool that inhibits the nuclear import of only Notch but not other nuclear proteins is developed, it will help elucidate our understanding of the importance of nuclear localization of NICD and the noncanonical Notch signaling pathway.
Our results may appear puzzling at first sight, as an interaction with CBF1 in the nucleus has been believed to be a prerequisite for Notch functions. However, we are not the first group to pay close attention to the importance of the cytoplasmic role of NICD. As explained in the Introduction, accumulating evidence implicates the noncanonical Notch signaling pathway acting in the cytoplasm in various cellular functions. To explain these apparently illogical observations, cross-talk between Notch and other signaling pathways in the cytoplasm has been suggested. Pioneering evidence was provided in Androutsellis-Theotokis et al. that Notch receptor activation induces temporally controlled Akt-mTor-Stat3 activation in the cytoplasm followed by induction of Hes3 and Shh in the nucleus, resulting in enhanced survival of neural stem cells [26]. It was also shown that an interaction with the mTor-Rictor complex that induces Akt activation is responsible for non-nuclear NICD-mediated inhibition of apoptosis [27]. To rule out the nuclear role of Notch, membrane-tethered NICD was used. In addition to the Akt pathway, Notch/RTK in cell fate specification, Notch/Wnt in postnatal hair growth, and Notch/TGF in osteoblastogenesis are examples of newly identified cross-talk between Notch and other regulatory molecules present in the cytoplasm [28]. Our study expands the understanding of the noncanonical and cytosolic functions of Notch in neural stem cell maintenance and regulation of neurogenesis/astrogenesis. Further studies are required to determine which regulatory pathways interact with NICD in the cytoplasm to fulfill a crucial role during mammalian brain development.
Footnotes
Acknowledgments
This work was supported by the Basic Science Research Programs through the National Research Foundation of Korea (#2012R1A1A2003185 and #2012R1A1A2008018) funded by the Ministry of Education.
Author Disclosure Statement
No competing financial interests exist.