
Abstract
Cell-based therapies are emerging as an alternative treatment option to promote functional recovery in patients suffering from neurological disorders, which are the major cause of death and permanent disability. The present study aimed to differentiate human dental pulp stem cells (hDPSCs) toward functionally active neuronal cells in vitro. hDPSCs were subjected to a two-step protocol. First, neuronal induction was acquired through the formation of neurospheres, followed by neuronal maturation, based on cAMP and neurotrophin-3 (NT-3) signaling. At the ultrastructural level, it was shown that the intra-spheral microenvironment promoted intercellular communication. hDPSCs grew out of the neurospheres in vitro and established a neurogenic differentiated hDPSC culture (d-hDPSCs) upon cAMP and NT-3 signaling. d-hDPSCs were characterized by the increased expression of neuronal markers such as neuronal nuclei, microtubule-associated protein 2, neural cell adhesion molecule, growth-associated protein 43, synapsin I, and synaptophysin compared with nondifferentiated hDPSCs. Enzyme-linked immunosorbent assay demonstrated that the secretion of brain-derived neurotrophic factor, vascular endothelial growth factor, and nerve growth factor differed between d-hDPSCs and hDPSCs. d-hDPSCs acquired neuronal features, including multiple intercommunicating cytoplasmic extensions and increased vesicular transport, as shown by the electron microscopic observation. Patch clamp analysis demonstrated the functional activity of d-hDPSCs by the presence of tetrodotoxin- and tetraethyl ammonium-sensitive voltage-gated sodium and potassium channels, respectively. A subset of d-hDPSCs was able to fire a single action potential. The results reported in this study demonstrate that hDPSCs are capable of neuronal commitment following neurosphere formation, characterized by distinct morphological and electrophysiological properties of functional neuronal cells.
Introduction
N
Following CNS damage, endogenous repair of the affected tissue by neural stem cells (NSCs) is limited [7,8]. The ideal candidates for stimulating repair in CNS injuries are ex vivo expanded and manipulated NSCs, due to their neurogenic predisposition [9 –12]. Indeed, promising results have been achieved with human NSCs in animal models of neurological disorders, including multiple sclerosis, spinal cord injury, ischemic stroke, Parkinson's and Alzheimer's disease (reviewed in ref. [6]). However, there are arguments that human NSCs might not be suitable for stem cell-based therapies in neurological disorders, contrary to what was originally thought. First, there are ethical considerations regarding the invasive isolation of human NSCs, derived from embryonic and fetal stem cells [13]. Second, researchers experienced difficulties in isolating and culturing NSCs in addition to the low number of cells that can be isolated from the adult human brain [14]. Therefore, there is a need for an easy-accessible alternative stem cell source with a neurogenic differentiation potential that is able to reconstitute the lost neural tissue or with the capacity to stimulate endogenous repair by host NSCs.
Human dental pulp stem cells (hDPSCs) can be cultured under NSC conditions to produce cells with a neurogenic phenotype and to offer a potential alternative source of stem cells, which can be used to produce functional neurons ex vivo. hDPSCs, first described by Gronthos et al. in 2000, can be isolated from extracted third molars and are believed to originate from migrating neural crest cells [15,16]. Furthermore, hDPSCs have been shown to possess mesenchymal stem cell (MSC) characteristics, similar to bone marrow-derived stem cells (BMSCs), and can be isolated with less donor site morbidity [16,17]. hDPSCs, like BMSCs, are able to differentiate in vitro into the classical mesodermal cell lineages, forming bone, cartilage, and fat-producing cells. However, the adipogenic differentiation potential of hDPSCs appears to be less achievable [18,19]. The presence of specific MSC surface markers, CD29, CD44, CD90, CD117, and CD146, can also be used to characterize cultured hDPSCs. In addition, like cultured BMSCs, hDPSCs are negative for CD34 and CD45. However, subsets of CD34+ hDPSCs and MSC were identified by other studies, suggesting that hDPSCs and MSC cultures are a heterogeneous cell population [18,20,21]. Similar to BMSCs, hDPSCs are also thought to possess immunomodulating properties [22], making them good candidates for transplantation studies and/or cell-based therapies.
More recently, researchers gained more interest in the neurogenic properties of hDPSCs due to their neuroectodermal origin. It was shown that hDPSCs are characterized by the basal expression of neurogenic markers [23]. In addition, hDPSCs secrete growth/neurotrophic factors, including brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), and glial cell-derived neurotrophic factor (GDNF) [24,25]. Vascular endothelial growth factor (VEGF) and other proangiogenic growth factors were also found to be present in the hDPSCs secretome [26]. These findings suggest that hDPSCs can provide trophic support to neuronal cells. Not only do hDPSCs show promising results in vitro but also in various in vivo models, encouraging effects have been observed following transplantation of hDPSCs. The proposed mechanisms of action of disease amelioration by the transplanted cells included integration of the transplanted cells in the host brain and/or stimulating the proliferation and differentiation of endogenous NSCs [24,25,27 –31].
In our study, we hypothesized that hDPSCs could be more successfully differentiated to neuronal cells in vitro when neurosphere formation precedes neuronal maturation. Neurosphere formation is considered to be a standard cell culture procedure in which NSCs are propagated and is used to investigate neural precursor characteristics [12]. Furthermore, it is assumed that neurospheres create a suitable microenvironment in which the intra-neurospheral cells differentiate toward neuronal and/or glial precursors [32]. Neurosphere formation is highly dependent on epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) signaling. Moreover, it is necessary to carefully monitor the size of the neurospheres as this influences cell viability and the differentiation capacity of the intra-neurospheral cells [12,33 –35]. After neurosphere formation, the neurospheres are collected and reseeded on an adherent surface allowing attachment of the neurospheres and outgrowth of the cells in serum-free differentiation promoting conditions [36].
Although (h)DPSCs—and other dental tissue-derived stromal cells—were shown to be able to form neurosphere-like structures in vitro, the full neurogenic maturation potential of these sphere-derived cells was not further elucidated [21,37 –40]. Therefore, we established a neurosphere culture by adding trophic support of EGF and bFGF. Subsequent neurogenic maturation was based on cAMP and neurotrophin-3 (NT-3) signaling [21,38,40 –43]. By means of transmission electron microscopy (TEM), the ultrastructural characteristics of intra-neurospheral hDPSCs and their microenvironment were determined. Neurogenic maturated hDPSCs were subjected to immunocytochemical (ICC), polymerase chain reaction (PCR), ultrastructural, and electrophysiological analysis. In addition, an enzyme-linked immunosorbent assay (ELISA) was performed for VEGF, NGF, BDNF, and GDNF to evaluate a differential growth factor secretion profile of hDPSCs before and after neurogenic differentiation.
Materials and Methods
Cell culture media and chemicals
The alpha modification of the minimum essential medium (α-MEM), 1:1 ratio of the Dulbecco's modified Eagle's medium and F12 medium (DMEM/F12), the Neurobasal medium, penicillin, streptomycin,
Isolation and cell culture of hDPSCs
Human dental pulps were obtained from both male and female patients aged from 15 to 25 years (n=9; average age=18 years and 5 months) undergoing routine extraction of third molars for orthodontic reasons at the Department of Maxillofacial Surgery, Ziekenhuis Oost-Limburg (Genk, Belgium) with informed consent of the patient or through their legal guardians in the case of underaged patients (<18 years). This study was approved by the Medical Ethical Committee of the Hasselt University (13/0104u). Obtaining the pulp tissue and subsequent isolation of the hDPSCs were performed using the explant method, as previously described [19]. Briefly, after mechanical fractioning of the tooth, the dental pulp tissue was carefully isolated using forceps and the tissue was rinsed in α-MEM supplemented with 2 mM
Neurosphere generation and neurogenic differentiation of hDPSCs
Neurospheres were generated by seeding hDPSCs (n=9 different cell donors) at a density of 7.5×103 cells/cm2 in Hydrocell® 6 cm Ø Petri dishes (Thermo Fisher Scientific, Inc.) in DMEM/F12 supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 2% B27 supplement, 20 ng/mL EGF, and 20 ng/mL bFGF. The medium was changed every 3–4 days. Floating neurospheres were kept in the culture for 6–8 days while carefully monitoring that the diameter of the spheres did not exceed 250 μm as this is crucial for neurosphere viability [35]. Free-floating neurospheres were collected and fixed in 2% glutaraldehyde for TEM analysis or rinsed with phosphate-buffered saline (PBS) and resuspended in the neurogenic maturation medium. Subsequently, the collected neurospheres were seeded on glass or plastic (Thermanox®; Electron Microscopy Sciences, Hatfield, PA) coverslips, Petri dishes, or in culture plates, which were previously coated with 15 μg/mL poly-
Neurogenic maturation was induced through the addition of the Neurobasal medium supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM
Control samples
Control samples (n=9) were obtained by seeding hDPSCs in the standard culture medium at the following densities: 1×104 cells/cm2 on glass and Thermanox coverslips for ICC and TEM analysis, 5×103 cells/cm2 in Petri dishes for electrophysiological recordings, and 2×104 cells/cm2 for ELISA and RNA isolation. All cell culture materials were precoated with 15 μg/mL PLO and 2 μg/mL Laminine. After 24 h, the culture medium was changed to the Neurobasal medium supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM
Transmission electron microscopy
Free-floating neurospheres and samples cultured on plastic Thermanox coverslips were prepared for ultrastructural analysis, as previously described [19]. Briefly, cells fixed with 2% glutaraldehyde were postfixated with 2% osmiumtetroxide in 0.05 M sodium cacodylate buffer (pH=7.3) for 1 h at 4°C. Dehydration of the samples was performed by ascending concentrations of aceton. Dehydrated samples were impregnated overnight in a 1:1 mixture of aceton and araldite epoxy resin at room temperature. After impregnation, samples were embedded in araldite epoxy resin at 60°C using the pop-off method [48]. Embedded samples were cut into slices of 40–60 nm with the Leica EM UC6 microtome (Leica, Wetzal, Germany) and transferred to 0.7% formvar-coated copper grids (Aurion, Wageningen, The Netherlands). The samples were contrasted with 0.5% uranyl acetate and a stabilized solution of lead citrate using the Leica EM AC20 (Leica). TEM analysis was performed with the Philips EM208 S electron microscope (Philips, Eindhoven, The Netherlands) equipped with the Morada Soft Imaging System camera with corresponding iTEM-FEI software (Olympus SIS, Münster, Germany).
Immunocytochemistry
Cells seeded on PLO/laminine-coated glass coverslips were fixed in 4% PFA, and immunostainings were performed according to a standardized protocol. In the case of an intracellular epitope, cells were permeabilized with 0.05% Triton X-100 in PBS for 30 min at 4°C. Afterward, 10% donkey serum was used to block nonspecific binding sites. Cells were incubated for 1 h at room temperature or overnight at 4°C with the primary antibodies. Afterward, cells were incubated for 30 min at room temperature with the appropriate secondary antibody. The primary and secondary antibodies that were used in this study are listed in Table 1. Nuclei were counterstained with 4,6-diamidino-2-phenylindole and coverslips were mounted with the antifade mounting medium (Dako, Glostrup, Denmark) on glass slides. Negative controls were included in each staining in which the staining procedure was performed in parallel with the other samples but with the omission of the primary antibody. Micrographs were taken with the Nikon Eclipse 80i Fluorescence Microscope equipped with the 2MBWc digital sight camera and NIS-elements software. The mean fluorescence intensity (MFI) was quantified by measuring the total fluorescent area divided by the total number of cells on the micrographs. Threshold levels for the fluorescent signal were kept constant throughout all quantifications. For each marker, representative images from five different patient samples were used and at least 300 cells were counted per sample.
MM, mouse monoclonal antibody; RM, rabbit monoclonal antibody; RP, rabbit polyclonal antibody; GAP.43, growth-associated protein 43; MAP-2, microtubule-associated protein 2; NCAM, neural cell adhesion molecule; GFAP, glial fibrillary acid protein; IgG, immunoglobulin G.
Enzyme-linked immunosorbent assay
ELISAs were performed for BDNF, NGF, VEGF, and GDNF (Raybiotech®; Norcross, GA). The conditioned medium of d-hDPSCs and hDPSCs was collected, as described previously, and was centrifuged at 300 g to exclude the uptake of cellular material before aliquoting. Subsequently, samples were stored at −80°C until needed. ELISAs were performed according to the guidelines of the manufacturer (available online at
Reverse transcriptase–PCR
Whole RNA was isolated with the Arcturus PicoPure RNA Isolation Kit (Thermo Fisher Scientific, Inc.) according to the instructions of the manufacturer (available online at
After optimization of the PCR reaction with the gradient PCR method, the cDNA samples were amplified with the primers and annealing temperatures (Tm) listed in Table 2. Primers for neural cell adhesion molecule (NCAM) and microtubule-associated protein 2 (MAP-2) were adopted from Nourbakhsh et al. [51]. PCR reactions were performed with the MyCycler thermal cycler (Bio-Rad Laboratories). The PCR products were analyzed by gel electrophoresis on a 1.2% agarose gel and stained with ethidiumbromide (2.5 μM; Merck, Darmstadt, Germany). A UV transilluminator (Bio-Rad Laboratories) was used to visualize and photograph the amplicons.
Patch-clamp recordings and electrophysiological measurements
Voltage-clamp recordings were performed using the whole-cell configuration of the patch-clamp technique [52]. For electrophysiological recordings, the growth medium was replaced with a bath solution containing 145 mM NaCl, 1.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, and 10 mM glucose, which was adjusted to pH 7.4 with NaOH (∼307 mOsm/kg). When appropriate, this extracellular solution was supplemented with tetrodotoxin (1 μM, TTX; Alomone Labs, Jerusalem, Israel) or tetraethyl ammonium (35 mM, TEA). These chemicals were applied using a fast perfusion system (SF-77B; Warner Instruments, Holliston, MA) by rapidly moving the solution interface across the cell surface. Micropipettes (2–5 MΩ resistance) were fabricated from 1.5-mm (o.d.) borosilicate glass capillary tubes (Hilgenberg, Malsfeld, Germany). The pipette solution contained 125 mM KCl, 1 mM CaCl2, 2 mM MgCl2, 2 mM Mg-ATP, 2 mM Na2ATP, 10 mM HEPES, 10 mM EGTA and was adjusted to pH 7.2 with KOH (∼283 mOsm/kg). All recordings were carried out at room temperature using a computer-controlled patch-clamp amplifier (EPC-10; HEKA Electronics, Lambrecht, Germany) and Patchmaster software (HEKA Electronics). Residual capacitances and leak currents were eliminated by means of a P/6 protocol. Currents were filtered at 2.9 kHz, sampled at 10 kHz, and stored on a computer hard disk for later analysis. Cells were clamped at a holding potential of −70 mV during 100 ms. Current patterns were obtained by depolarizing the cell membrane from the holding potential to voltages between −80 and +60 mV at intervals of 10 mV. Pulse duration was 50 ms. Na+ and K+ current amplitudes were measured, respectively, at the peak inward and outward value. The changes in current amplitudes were expressed as changes in current densities to correct for cell size (pA/pF). Action potentials were recorded under current clamp conditions in which the cells were stimulated with 50–300 pA current injection during 2,000 ms. Data were analyzed off-line with Fitmaster v2x69 software (HEKA Electronics).
Statistical analysis
Statistical analysis was performed using Graphpad Prism 5 software (Graphpad, San Diego, CA). Experimental groups were compared by means of the nonparametric Mann–Whitney U test. Patch-clamp data were assumed to be normally distributed and were analyzed with a two-tailed unpaired t-test or repeated measures analysis of variance followed by the Bonferroni post-test. Differences were considered statistically significant at P-values ≤0.05. Data were expressed as mean±standard error of the mean except for the electrophysiological recordings, which were expressed as mean±standard error.
Results
hDPSCs acquire a neuronal morphology during the differentiation process
Throughout the two-step differentiation process, hDPSCs underwent multiple morphological adaptations ranging from the formation of neurospheres to establishing a culture of neuron-like cells with a large perikaryon and intercommunicating cytoplasmic extensions (Fig. 1). In the first step, explant-derived hDPSCs grown as a monolayer were transformed to free-floating neurospheres in low-attachment Petri dishes (Fig. 1A, B). hDPSCs formed neurospheres after 24–48 h in culture and after 6–8 days, the obtained neurospheres were transferred to a pre-coated surface, allowing outgrowth of neurosphere cells (Fig. 1C). After 4 weeks in maturation-promoting conditions, hDPSCs acquired morphological characteristics of neuronal cells, characterized by large round perikarya with a peripheral halo (Fig. 1D, encircled) and multiple cytoplasmic extensions that formed an intercellular network (Fig. 1D, arrowheads).
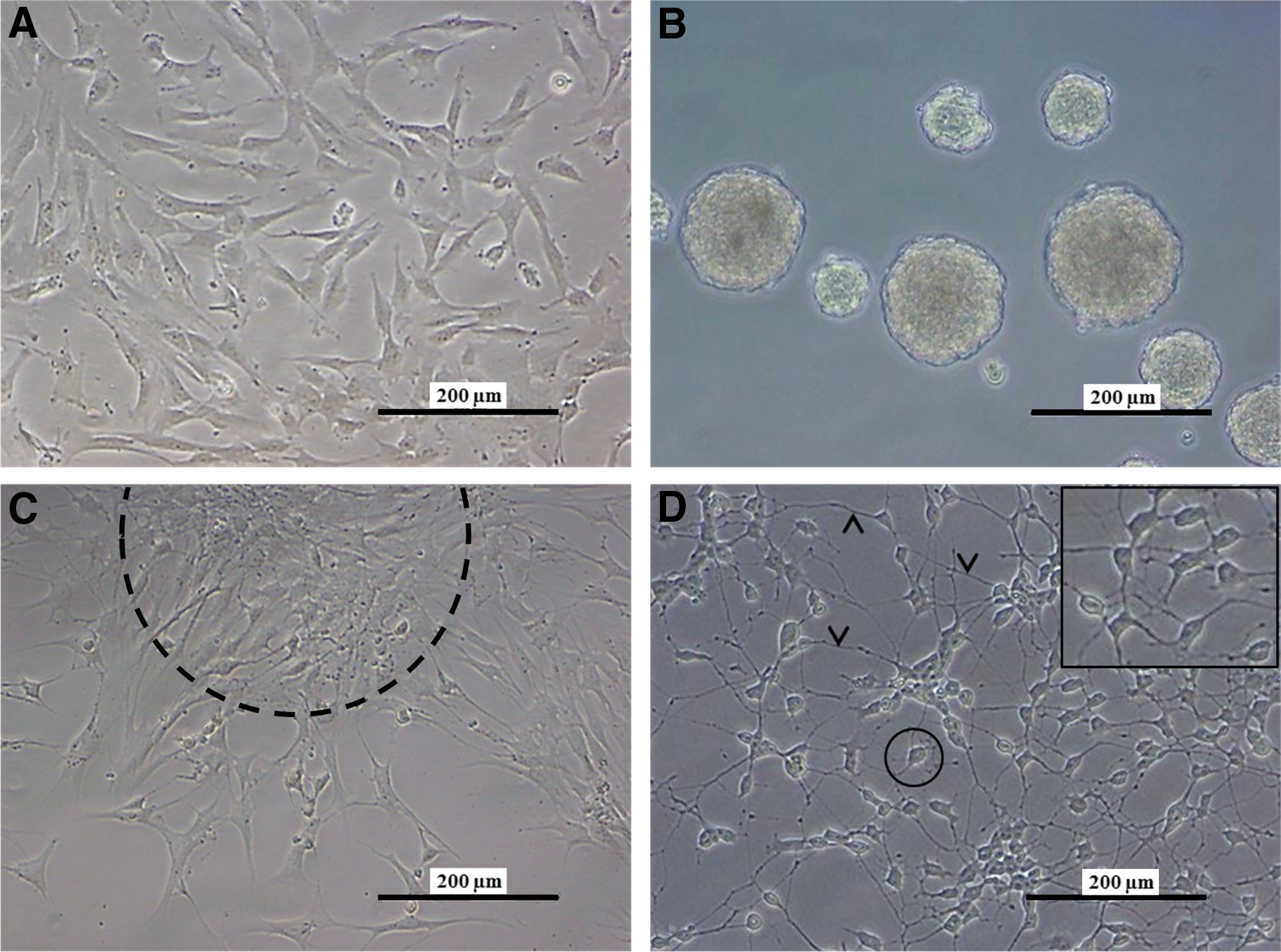
Human dental pulp stem cells (hDPSCs) acquired a neuronal morphology during the differentiation process. Explant-derived hDPSCs were cultured as a monolayer
Ultrastructural characteristics of intra-neurospheral hDPSCs
To gain insight into the intercellular interactions that take place within the neurospheres, these spheres were subjected to TEM analysis (Fig. 2). Intra-neurospheral hDPSCs appeared as elongated cells with prominent nucleoli (Fig. 2A, asterisk), a prominent Golgi apparatus and a dilated rough endoplasmatic reticulum (RER) (Fig. 2B, black arrow and asterisk, respectively), suggesting an increased metabolic activity within neurospheres in vitro. These features were more prominent in the cells at the center of the neurosphere. However, areas of intracellular vacuolization and loss of cell integrity were observed in a subset of intra-neurospheral hDPSCs (Fig. 2A, arrowheads). This loss of cell integrity was reflected in a decrease in cell viability of ∼45.74% (standard deviation=7.56%; n=6 individual neurosphere generation experiments) as quantified by neurosphere dissociation with Accutase and subsequent cell number determination with trypan blue exclusion (data not shown).
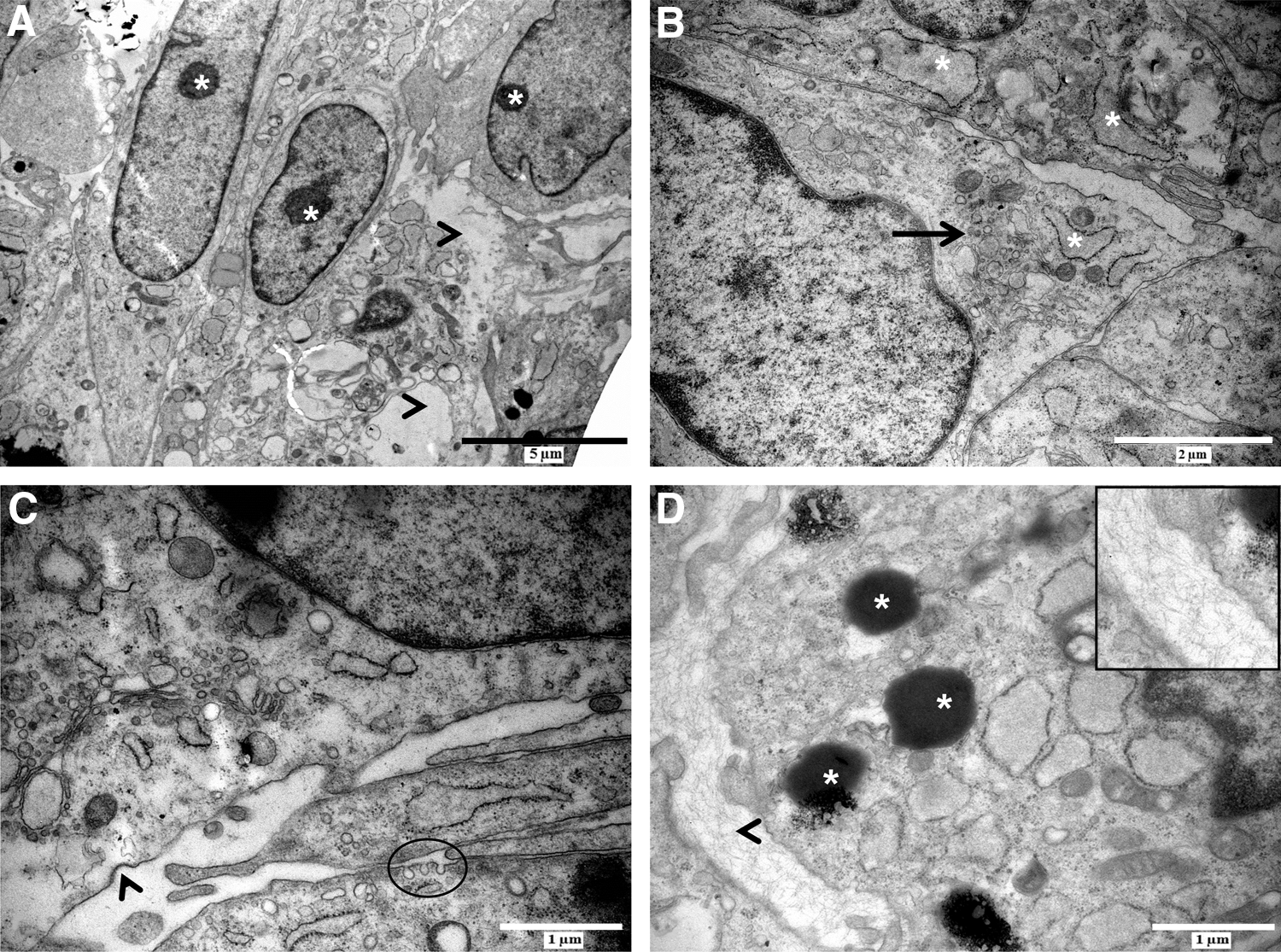
Intra-neurospheral hDPSCs are characterized by an increased metabolic activity and intercellular communication. Intra-neurospheral hDPSCs were characterized by a prominent nucleolus [
In addition to the increased metabolic activity of intra-neurospheral hDPSCs, intercellular communication also seemed to be stimulated. Vesicular transport was observed at cell–cell contact zones as demonstrated by the presence of coated pits and vesicles suggesting signs of endo- or exocytosis (Fig. 2C, arrowhead and circle). Intra-neurospheral hDPSCs produced electron-dense vesicles containing an unknown substance, and extracellular matrix (ECM) deposits were observed between adjacent cells (Fig. 2D, asterisk and arrowhead, respectively).
ICC, PCR, and secretome analysis of d-hDPSCs
After the 4-week neuronal maturation period, the neurogenic d-hDPSCs showed immune reactivity for neuron-related markers (Fig. 3A–F). A quantitative analysis for the level of immune reactivity was performed for both the number of immune reactive cells and the MFI per cell. The data demonstrated an increase in neuronal nuclei (NeuN; Fig. 3A, B), MAP-2 (Fig. 3C, D), NCAM (Fig. 3E, F), growth-associated protein 43 (GAP.43; Fig. 3G, H), synaptophysin (Fig. 3I, J), synapsin I (Fig. 3K, L), and glial fibrillary acid protein (GFAP, Fig. 3M, N) immune reactivity in d-hDPSCs compared with hDPSCs. GFAP and NCAM showed a perinuclear staining pattern and were not present in the cytoplasmic extensions of d-hDPSCs. More detailed images of synapsin 1 and synaptophysin expression in d-hDPSCs are presented in Supplementary Fig. S1. A2B5 (Fig. 3O, P) expression was observed in <2% of d-hDPSCs. Nestin expression was decreased in d-hDPSCs compared with controls (Fig. 3Q, R). No fluorescent signal was detected in the negative controls. The differential expression of neuron-related markers in d-hDPSCs and hDPSCs was also demonstrated by measuring the MFI per cell (Fig. 3T). A significant difference (P≤0.05) between NeuN, MAP-2, NCAM, synaptophysin, synapsin I, GFAP, and nestin expression was observed between hDPSCs and d-hDPSCS (n=5). Similarly, the percentage of cells that were immune reactive for these markers also increased significantly in d-hDPSCs compared with hDPSCs (Fig. 3S). In addition to evaluating the expression of neuronal markers in d-hDPSCs compared with controls, the percentage and MFI per cell of the stem cell markers CD29, CD44, and Stro-1 were also evaluated. No significant difference in the percentage of immune-positive cells and MFI per cell was detected between d-hDPSCs and hDPSCs (Supplementary Fig. S2). A detailed statistical analysis of the percentage of immune reactive cells and the MFI per cell can be found in Supplementary Table S1. The differential expression of MAP-2 and NCAM was confirmed by RT-PCR (Fig. 3U). However, the expression of the neuron-specific genes, Mash-1 and SCG10, could not be observed in both hDPSCs and d-hDPSCs (n=4).
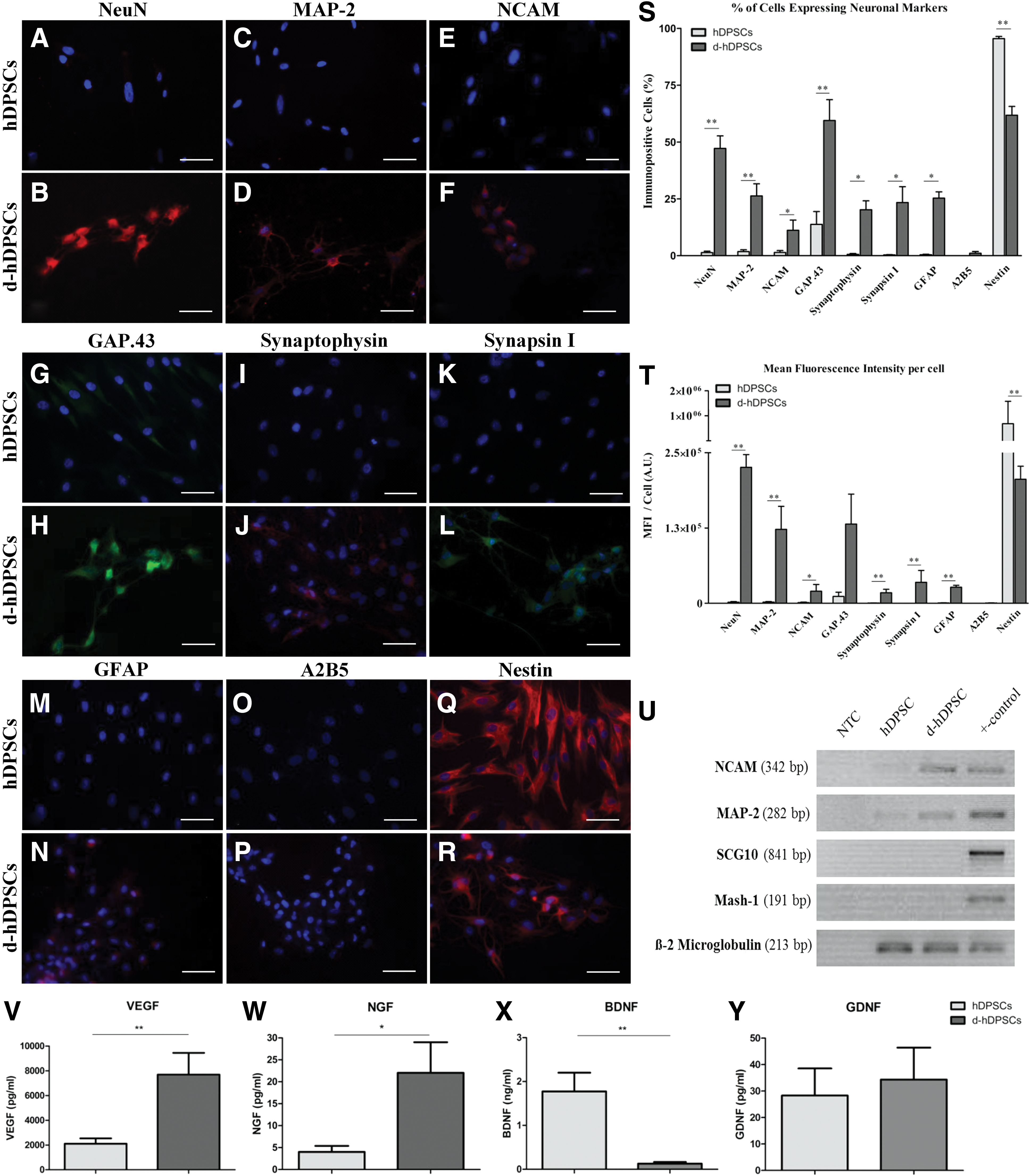
Differentiated hDPSCs (d-hDPSCs) acquired immune reactivity for neuron-related markers and showed an increased expression of neuronal genes in addition to an altered secretome. Compared with controls, neurogenic maturation induced the expression of neuronal nuclei [NeuN;
The secretome of hDPSCs was compared with that of d-hDPSCs to investigate if neurogenic differentiation of hDPSCs altered the secretome of these cells (Fig. 3J–M). ELISA of the conditioned medium of hDPSCs and d-hDPSCs demonstrated a significant increase in VEGF (n=6; P=0.0022) and NGF (n=5; P=0.0159) secretion by d-hDPSCs compared with hDPSCs (Fig. 3V, W). BDNF (n=6; P=0.0022) secretion was significantly decreased after neurogenic maturation (Fig. 3X). Differential secretion of GDNF (n=6; P=1.00) by hDPSCs and d-hDPSCs could not be observed (Fig. 3Y).
Ultrastructural characteristics of d-hDPSCs
d-hDPSCs were subjected to an ultrastructural analysis using TEM (Fig. 4). The perikaryon of d-hDPSCs was characterized by a large central nucleus with a prominent nucleolus (Fig. 4A, asterisk). Abundant organelles associated with the metabolic activity and protein synthesis were present in the cytoplasm of d-hDPSCs (Fig. 4B). These organelles included an extended Golgi apparatus and RER (Fig. 4B, bracket and white arrows), indicating increased packing of proteins in membrane-bound vesicles. In addition, d-hDPSCs developed multiple cytoplasmic extensions containing longitudinally aligned cytoskeletal elements and varicosities along their length (Fig. 4C, between arrowheads, Fig. 4D, arrowheads). Interestingly, intercellular contact sites containing mitochondria were observed at these varicosities (Fig. 4D, circle). A detailed analysis of intercellular contact zones showed the presence of electron-dense vesicles (Fig. 4E, asterisk) at the distal fraction of the cytoplasmic extension. Moreover, multiple small vesicles (Fig. 4E, bracket) and granular material were present in the intercellular cleft, and the internalization of membrane-bound vesicles by receptor-mediated mechanisms was observed (Fig. 4E, arrowheads). Multivesicular bodies (Fig. 4F, white arrows, MVB) were found in the vicinity of the coated pits (Fig. 4F, arrowheads) at the distal parts of the cytoplasmic extensions.
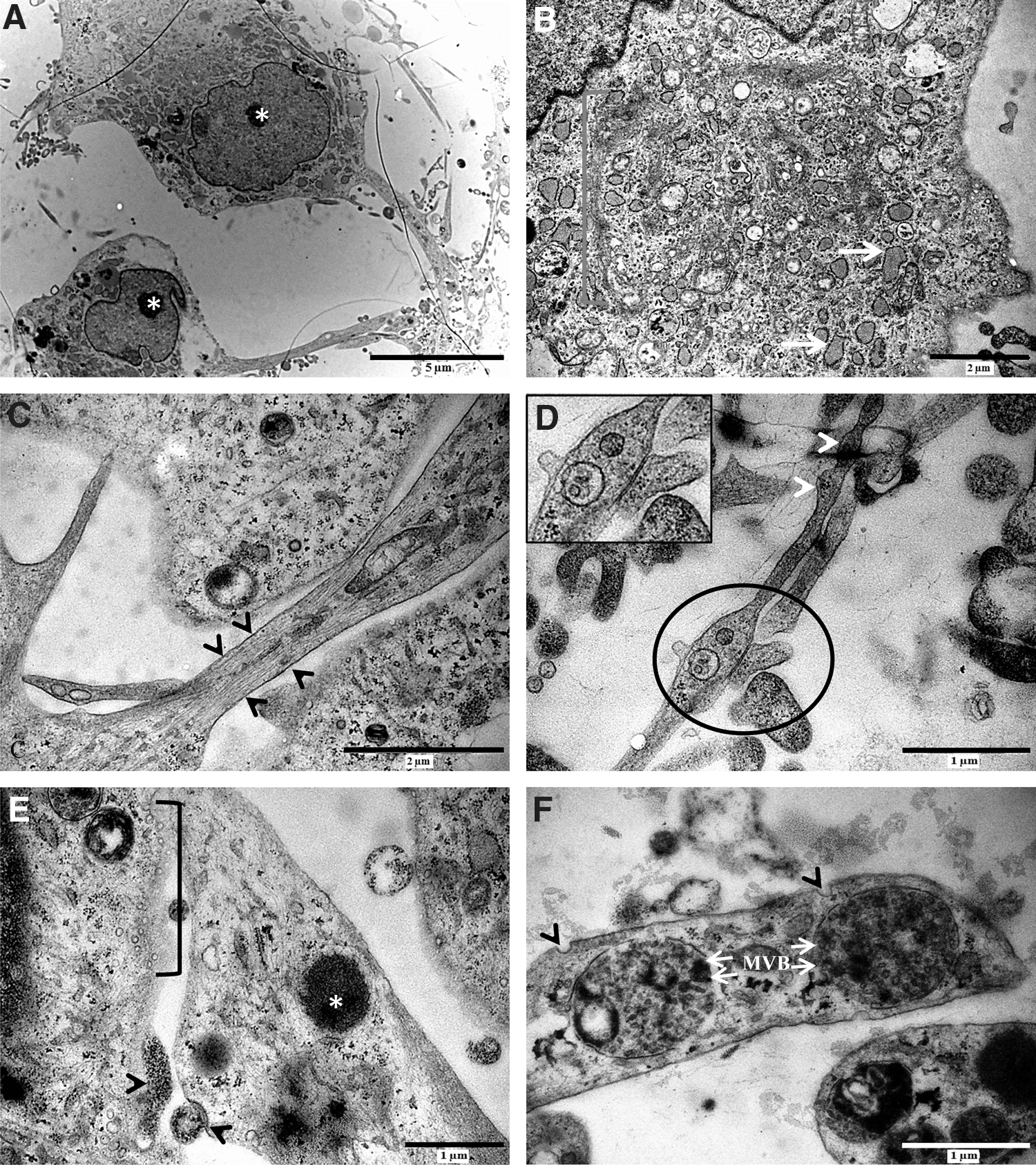
d-hDPSCs developed ultrastructural features of neuronal cells. The perikaryon of d-hDPSCs was characterized by a large central nucleus with a prominent nucleolus [
Electrophysiological properties of d-hDPSC
To evaluate the functional maturation of d-hDPSCs, whole-cell patch-clamp recordings were performed on three parallel d-hDPSCs and control hDPSCs cultures (Fig. 5). The I–V relationship of voltage-dependent potassium and sodium currents was recorded on both d-hDPSCs (n=10) and hDPSCs (n=12) (Fig. 5A, B). Voltage-dependent potassium currents were present in d-hDPSCs, but not in hDPSCs (Fig. 5A). These currents were activated at membrane potential more positive than −10 mV and showed a typical delayed rectifier I–V profile. A statistically significant increase in outward currents was reached at +20 mV (P=0.0408) and more positive membrane potentials. A peak value of 35.65±15.6 pA/pF was measured at +60 mV. Furthermore, these outward potassium currents could be completely and reversibly blocked by 35 mM TEA (Fig. 5C) (n=6). Similarly, the I–V relationship of voltage-dependent sodium currents recorded on both experimental conditions showed the presence of these currents in d-hDPSCs, but not in hDPSCs (Fig. 5B). These inward sodium currents were activated at a threshold between −40 and −30 mV and statistically significant currents were observed from −20 to +60 mV (P-value at −20 mV=0.0136). A peak (negative) value of −25.9±11.4 pA/pF was reached at a membrane potential of 0 mV. These voltage-dependent sodium currents could be reversibly blocked by 1 μM TTX (Fig. 5D) (n=6). A significant decrease in voltage-dependent sodium currents by applying TTX was observed at a membrane potential of −10 mV (P=0.0289) and more positive membrane potentials. Consequently, after washing out TTX, the voltage-dependent sodium currents were restored. No significant difference in the amplitude of voltage-dependent sodium currents was observed between control traces and after washout of TTX. Representative traces of sodium (Fig. 5E) and potassium currents (Fig. 5F) recorded in d-hDPSCs under nonblocking (black trace), blocking (red trace), and washout (green trace) conditions confirmed the reversible blocking of voltage-gated sodium channels by TTX and voltage-gated potassium channels by TEA. Finally, upon current stimulation (50–300 pA), a single action potential was observed in a subset of d-hDPSCs (n=3) (Fig. 5G). This action potential was characterized by an initial depolarization of the membrane potential in d-hDPSCs followed by incomplete repolarization of the membrane potential. Repetitive action potential firing was not observed.
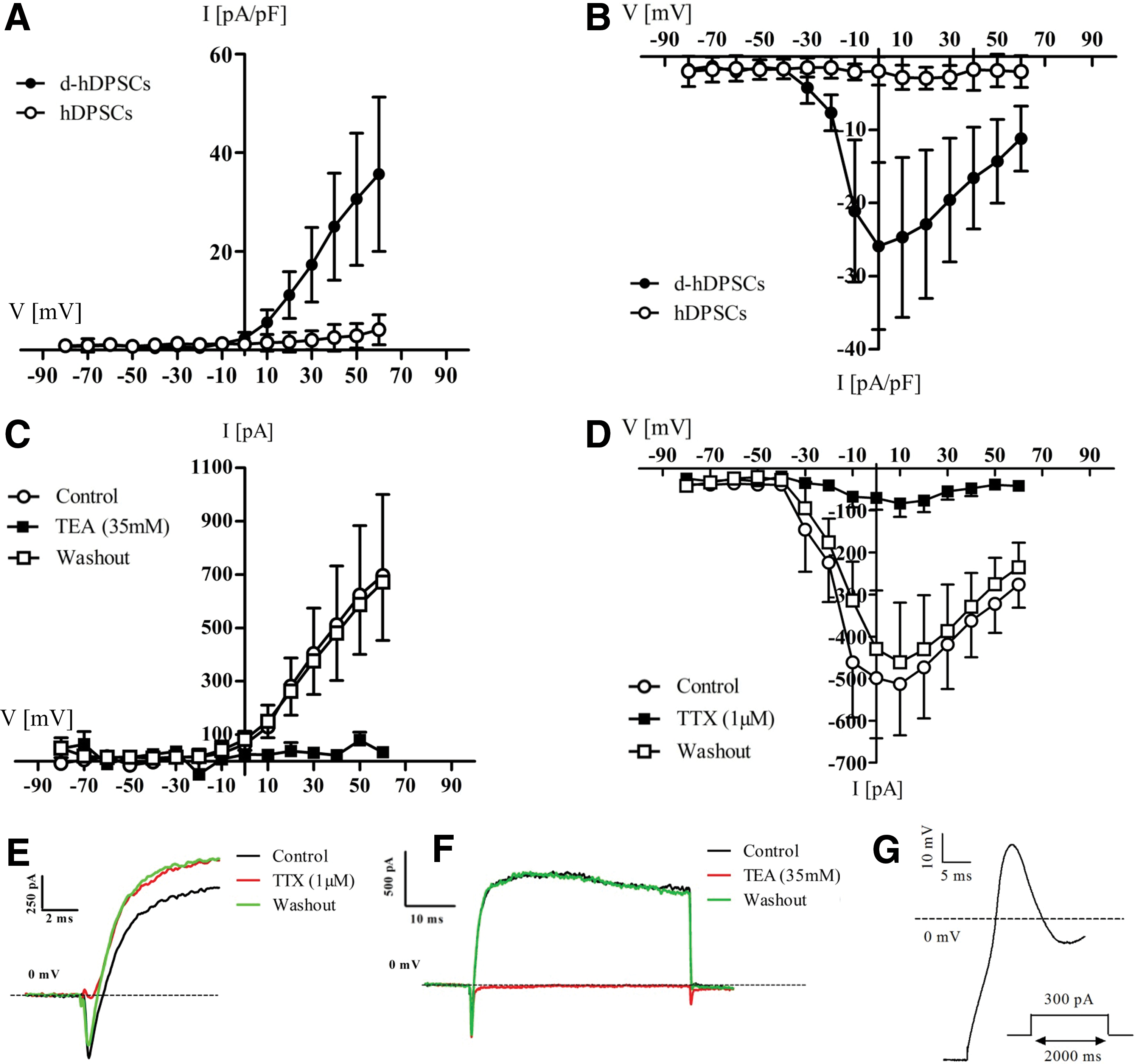
Voltage-dependent sodium and potassium channels in d-hDPSCs could be selectively blocked by tetrodotoxin (TTX) and tetraethyl ammonium (TEA), respectively. The I–V relationship of voltage-dependent potassium and sodium currents was recorded on both d-hDPSCs (n=10) and hDPSCs (n=12) [
Discussion
In the present study, we described a novel approach to differentiate hDPSCs toward cells with a neuronal phenotype. Apart from acquiring morphological features of neuronal cells, a subset of differentiated cells also showed a neurogenic associated electrophysiological profile. This was demonstrated by the presence of voltage-sensitive sodium and potassium currents that could be reversibly blocked by TTX and TEA, respectively, in addition to the firing of a single action potential upon current stimulation.
To neuronally differentiate hDPSCs, we combined protocols that have been applied in other studies into a two-step protocol. Previous protocols were able to differentiate hDPSCs to functional neuronal cells, as determined by the presence of voltage-dependent sodium and potassium currents. However, these studies did not observe action potential firing by the differentiated cells, addressing the incomplete differentiation of hDPSCs to neuronal cells [41,42]. Therefore, this two-step protocol was implemented to improve the differentiation outcome. First, we adopted the neuronal induction step based on EGF and FGF-2 signaling [42] and combined these essential signaling molecules with the formation of 3-dimensional neurosphere structures. Neurosphere formation is a common culturing technique for NSC as it is considered to create a favorable microenvironment for intercellular interactions [12,21,38 –40]. Furthermore, it has been shown that a close physical contact between neural progenitor cells is essential for neuronal commitment, which can also be achieved by neurosphere generation [53,54]. Second, we used a neuronal maturation protocol based on increased cAMP levels and NT-3 signaling that was previously implied by Kiraly et al. [41]. Elevated intracellular cAMP is thought to play an essential role in sustaining neurogenic differentiation of early neuronal committed cells, whereas NT-3 signaling is essential for neurogenic maturation [43,55,56]. In the study by Kiraly et al., neuronal induction was achieved by epigenetic reprogramming, which was replaced in this study by the formation of neurospheres [41].
To our knowledge, this article presents the first ultrastructural data of the microenvironment within neurospheres derived from dental pulp stem cells. When comparing hDPSC-derived neurospheres with the scarce ultrastructural data on NSC-derived neurospheres, some differential and corresponding features were observed [57,58]. In these studies, multiple apoptotic bodies were found inside the cytoplasm of intra-neurospheral cells. These were not observed in our neurosphere culture, although we observed loss of cell integrity with a subsequent loss of cells that could be used for neurogenic maturation. Moreover, in the present study, ECM deposits were found that presumably contributed to the microenvironment and structural integrity of the 3-dimensional neurospheres. A similar result of the present study, compared with the studies by Bez et al. [57] and Zhao et al. [58], was the increase in intercellular communication inside the neurospheres, which reflects the augmented metabolic activity and demand that is observed within the spheres. However, in the studies by Bez et al. [57] and Zhao et al. [58], intercellular communication was achieved by means of gap junctions, whereas in our culture, paracrine signaling via vesicular transport appeared to be the main form of cell communication. Nonetheless, it can be concluded that by forming neurospheres, a microenvironment is created that influences the metabolic activity of the intra-neurospheral cells and that allows the development of an improved intercellular communication network.
In determining the expression level of neuron-related markers, markers were chosen that were not already expressed in a large fraction of hDPSCs before differentiation [23]. Therefore, NeuN, MAP-2, NCAM, and GAP.43 were selected as neuron-related markers to discriminate between hDPSCs and d-hDPSCs. In addition, synapsin I and synaptophysin were used to evaluate the production of synaptic vesicles. Furthermore, A2B5 was used to identify oligodendroglial cells, whereas GFAP was used to evaluate differentiation of hDPSCs toward astrocytes, although GFAP is also found to be expressed by neuronal precursors [59]. Using these markers, a significant difference in marker expression between hDPSCs and d-hDPSCs was observed, both in the percentage of immune-positive cells and in the correlating MFI. The expression of neuronal and synaptic markers was significantly increased in d-hDPSCs, except for the MFI per cell for GAP.43, which showed a high interpatient variability. Nestin expression was significantly reduced following differentiation, suggesting that a subset of d-hDPSCs shifted toward a more mature neuronal phenotype. A2B5 expression was only detected in <2% of d-hDPSCs, indicating that d-hDPSCs did not differentiate toward oligodendroglial cells. Immune reactivity for GFAP suggested that a subset of d-hDPSCs differentiated toward astrocytes, although the perinuclear staining pattern suggests incomplete differentiation. In addition to neural markers, the percentage of immune-positive cells and the MFI per cell was also assessed for the stem cell markers CD29, CD44, and Stro-1. Both the percentages of immune-positive cells and MFI per cell were not significantly reduced following neuronal differentiation, although the MFI per cell shows a declining trend. Although markers such as CD29 and CD44 are often used to characterize MSC populations, it has been shown that neural stem/precursor cells also express these markers [60,61].
Taken together, these results suggest that d-hDPSCs acquire a neuronal phenotype, based on an increase in the MFI per cell and the percentage of immune-positive cells for neuron-associated and synaptic markers. However, the percentage of NCAM-positive cells was lower than that in a previous report of Arthur et al. for the basal expression in both hDPSCs and d-hDPSCs [42]. Nourbakhsh et al. also reported a higher percentage of NCAM-positive cells before and after neuronal differentiation conditions, although in lower percentages than Arthur et al. and in stem cells derived from the pulp of human deciduous teeth [51]. This discrepancy between the present study and earlier reports can be attributed to the fact that we used a lower and upper fluorescent threshold when determining the percentage of immune-positive cells. Therefore, a very strict standardized quantification method was used compared with nonstandardized quantification methods, such as the visual observation of the amount of immune-positive cells per field of view. In addition, other factors that might influence the outcome of the NCAM reactivity are noted. First, to obtain their stem cell culture, Arthur et al. [42] and Nourbakhsh et al. [51] used an isolation method based on enzymatic digestion of the pulp tissue, whereas we opted for the explant method. Previously, it was shown that the isolation methodology does not have an effect on the stem cell properties and multilineage differentiation potential of hDPSCS. However, the influence on neurogenic differentiation potential and basal neuronal marker expression has not been evaluated [18]. Second, the culture medium in which the control cells were grown differed between the present study and the study of Arthur et al. [42] and Nourbakhsh et al. [51]. We opted to culture the control cells in the neurogenic maturation medium minus cAMP and NT-3 48 h before harvesting the material or fixing the cells. Other studies cultured the control samples in the standard culture medium by which they are unable to assess the influence of the basal medium without growth factors on which their differentiation promoting conditions are built. Nonetheless, the increased expression of both MAP-2 and NCAM was confirmed with PCR indicating active transcription of these proteins at the RNA level. Both hDPSCs and d-hDPSCs failed to express the early neuronal gene Mash-1 and the late neuronal gene SCG10. The failure of hDPSCs to express Mash-1 suggests that although hDPSCs are of neuroectodermal origin and that they express neuronal markers at the protein level, they should not be labeled as neural progenitor cells that are known to express Mash-1 [62,63]. The absence of Mash-1 expression in d-hDPSCs can be explained by the advanced maturation stage of d-hDPSCs compared with hDPSCs. Although Mash-1 is frequently used as a genetic marker for cells with neuronal differentiation potential, Mash-1 is also expressed in tumor cells, such as the SH-SY5Y cells that were used as a positive control [64]. This explains why neuronally differentiated SH-SY5Y expressed both Mash-1 and SCG10. The lack of SCG10 expression in d-hDPSCs can be attributed to incomplete neuronal maturation as this gene is expressed in mature neurons [65,66].
In addition to evaluating the neurogenic differentiation of hDPSCs based on protein and gene expression, the secretome of hDPSCs and d-hDPSCs was examined in more detail. Interestingly, an altered secretion pattern could be observed between hDPSCs and d-hDPSCs for the growth factors/neurotrophins BDNF, NGF, and VEGF while the secretome of both cell populations contained a similar concentration of GDNF. Depending on the CNS pathology, these growth factors/neurotrophins are known to have diverse effects on the host tissue, including neuroprotection and the stimulation of host NSC activation to improve endogenous repair [29,31,67 –69].
The concentration of BDNF was significantly higher in hDPSCs compared with d-hDPSCs. It can be suggested that this decrease in BDNF secretion following neurogenic differentiation can be attributed to the characteristic BDNF secretion mechanism in neurons, assuming d-hDPSCs acquired neuronal characteristics. In neurons, it was shown that endogenous release of BDNF is limited in unstimulated neurons [70,71]. Following depolarization, BDNF secretion is triggered through calcium influx-dependent mechanisms [71 –73]. In the present d-hDPSC culture, it can be assumed that intercellular stimulation is insufficient to generate adequate calcium influx to activate BDNF secretion. Nonetheless, BDNF secretion by grafted cells can have potential applications in Alzheimer's disease in which BDNF secretion is known to be decreased in the hippocampus [74,75] and in ischaemic stroke where BDNF signaling is thought to ameliorate the disease outcome [76,77]. In addition to its trophic and protective effects, it has also been shown that BDNF can promote seizures when injected into the hippocampus due to increased excitatory signaling. Therefore, caution needs to be taken into account when using a BDNF-based therapeutic strategy [78].
Furthermore, NGF and VEGF secretion was enhanced following neurogenic differentiation compared with nondifferentiated hDPSCs. Similar to BDNF, NGF is also thought to have potential applications in both Alzheimer's disease and ischaemic stroke. In ischaemic stroke, NGF is thought to have a beneficial effects [67,79], whereas in Alzheimer's disease, the use of NGF as a therapeutic agent is a topic of debate [80,81]. VEGF has been shown to have various effects in CNS pathologies, which improved disease symptoms and outcome. The potential mechanisms of action of VEGF were not only limited to improving blood supply to brain lesions by inducing angiogenesis but also due to increased neurogenesis and neuroprotection following VEGF treatment in animal models of neurological disorders [82 –86]. Finally, GDNF is known for its ability to enhance survival of dopaminergic neurons, opening possibilities for treating Parkinson's disease [87 –90]. Taken together, hDPSCs already secrete multiple growth factors/neurotrophins, of which the secretion can be either promoted or downregulated by subjecting hDPSCs to neuronal differentiation. The differential secretome of these cells broadens potential applications should hDPSCs, d-hDPSCs, or a combination of both be used as a growth factor delivery vehicle in cell-based therapies.
In the present study, the assessment of neuronal maturation of hDPSCs was investigated more in detail with TEM to gain insight into the acquired neuron-specific characteristics of d-hDPSCs. d-hDPSCs were characterized by a large central nucleus with a prominent nucleolus and various cytoplasmic extensions that formed an intercellular network. In addition, these cytoplasmic extensions were aligned in parallel along with the cytoskeletal components within the extensions, suggesting the possibility of antero- and retrograde transport. Both vesicular and electrical communications are potential communication mechanisms between adjacent d-hDPSCs. Electrical communication is suggested by gap junctions that were observed at cell–cell contact sites along cytoplasmic extensions. Furthermore, these contact sites appeared clustered at varicosities where mitochondria were found in close proximity to the intercellular contacts. Vesicular transport was demonstrated by the presence of both electron-dense and electron-lucent vesicles at the distal part of the cytoplasmic extensions. In addition, the active secretion and absorption of communicating vesicles was observed. Interestingly, MVB were found in the vicinity of coated pits. MVB are intracellular organelles that are comprised of multiple vesicles enclosed by an outer membrane that are thought to play an important role in endocytosis and protein trafficking in neurons and other nonneural cells [91,92]. In neuronal cells, it is thought that MVB play an essential role in the retrograde transport of neurotrophic factors, such as BDNF and GDNF [93]. The presence of these organelles confirmed the increased inter- and intracellular communication in d-hDPSCs. Together, these distinct neuronal features suggest a successful neuronal maturation of d-hDPSCs [91].
Although d-hDPSCs acquired morphological characteristics of neurons, this does not confirm a successful differentiation toward functional neurons. Therefore, a patch-clamp analysis was performed. The electrophysiological recordings of d-hDPSCs showed that voltage-gated sodium and potassium currents were present after differentiation. The selective blocking of these currents by, respectively, TTX and TEA confirmed their ion specificity. Compared to previous studies by Arthur et al. and Kiraly et al., some corresponding observations can be made [41,42]: both studies were able to find TTX-sensitive sodium channels. However, Arthur et al. did not examine the presence of voltage-gated potassium channels. The voltage-gated potassium channels reported by Kiraly et al. [41] could only be partially blocked by TEA suggesting that the concentration of TEA used in that study (5 mM compared with 35 mM TEA in our study) was insufficient to block all potassium channels. Moreover, in the study by Kiraly et al. [41], it was not shown whether the observed ion channels could be reversibly blocked by TTX and TEA. Furthermore, the differentiated cell population was also split up based on their electrophysiological profile showing the presence of d-hDPSCs displaying sodium currents with premature and mature characteristics. In the present study, a subset of differentiated cells is seen that displayed larger inward or outward currents (peak values of −60.025±18.0 pA/pF and +87.45±18.21 pA/pF; n=4) together with cells displaying more premature characteristics. This mixture of cells in different developmental stages explains the large standard error on electrophysiological data and is also reflected in the PCR data for Mash-1 and SCG10. We decided to show the electrophysiological profile of the general d-hDPSCs population, providing a more realistic overview of the obtained cells being aware that it is a heterogenic population. Nonetheless, d-hDPSCs acquired voltage-sensitive sodium and potassium currents that could be reversibly blocked by TTX and TEA, respectively. In addition to voltage-gated sodium and potassium currents, we were able to observe the firing of a single action potential by a subset of d-hDPSCs. However, a train of repeated action potential firing after stimulation was not observed, which would be the ultimate proof of functional neurons. The failure to fire repeated action potentials might be attributed to the gating kinetics of the delayed rectifier potassium channels, that is, the high activation potential (−10 mV), resulting in an incomplete repolarization. The incomplete repolarization failed to deactivate sodium channels, which would be necessary for repetitive firing. A similar observation was made in a study by Wislet-Gendebien et al. where MSC were cocultured with cerebellar granule neurons [94]. In this study, the firing of an action potential by differentiated MSC was observed, but these cells also failed to fire repeated action potentials. Although differentiating cells were kept on maturation-promoting conditions for 4 weeks, this incubation period might be insufficient for hDPSCs to reach full neuronal maturation. It has been shown that there is a progressive increase in current amplitude during neuronal maturation and ultimately, action potential firing [95]. Moreover, recent studies reported that differentiating human embryonic stem cells and induced pluripotent stem cells (iPSCs) to neurons take 30–50 days to acquire a functional neuronal culture [96 –99]. In addition, human embryonic stem cells and iPSCs are considered to be pluripotent and thus more capable of differentiating toward other cell types compared with hDPSCs, which are considered multipotent, although they express pluripotency markers [100,101]. Therefore, it can be postulated that a 4-week maturation period is insufficient for hDPSCs to acquire a fully functional neuronal phenotype.
Conclusion
The present study aimed to provide a new successful protocol to differentiate hDPSCs toward functionally active neurons. Although promising results were achieved, establishing a completely functional d-hDPSC culture remains a challenge. The results in this study demonstrated that hDPSCs are capable of neuronal commitment with distinct features of neuronal cells as demonstrated by morphological and electrophysiological characteristics. In addition, exposing hDPSCs to the neurogenic differentiation protocol altered the secretion of selected growth factors/neurotrophins. Future research is needed to identify to what extent and by which mechanism hDPSCs and/or d-hDPSCs could be used to ameliorate the disease outcome in neurological disorders.
Footnotes
Acknowledgments
A word of gratitude goes out to Mr. Marc Jans and Mrs. Jeanine Santermans for their indispensable help in preparing TEM specimens and practical expertise with ICC. In addition, we thank Mrs. Petra Bex for performing patch-clamp recordings. This research was supported by grants to Wendy Martens and Jessica Ratajczak from the Research Foundation-Flanders (“Fonds Wetenschappelijk onderzoek Vlaanderen-FWO”, grant no. G029112N and G089213N). Petra Hilkens benefits from a PhD scholarship of the FWO and Annelies Bronckaers is a postdoctoral fellow of the FWO.
Author Disclosure Statement
The authors have nothing to disclose and no competing interest exists.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.