
Abstract
The promise of off-the-shelf cellular therapeutics (CTPs) based on allogeneic induced pluripotent stem cells (iPSCs) may be hindered by alloimmunity, leading many to suggest that such products could be based on a series of human leukocyte antigen (HLA)-typed iPSC lines allowing at least some degree of tissue matching. While based on sound scientific principles, this suggestion presupposes that other immune responses will not be limiting. Technically this approach would present a number of major challenges, the first being the development of a suitably reliable reprogramming method amenable to validation that results in highly consistent iPSC lines. Further, the resulting array of HLA-typed iPSCs would need to be shown to be capable of being manufactured into the same CTP and exhibit comparable quality, safety, and efficacy. When the enormities of these challenges are laid out, it becomes apparent that the manufacturing and product development challenges would be unprecedented. Given the uncertainties and lack of clinical experience with iPSC-based CTPs at this time, the financial costs and commercial risks do not appear to be acceptable.
Introduction
I
GMP, good manufacturing practices; HLA, human leukocyte antigen; iPSC, induced pluripotent stem cell; TSE, transmissible spongiform encephalopathy.
The use of iPSCs for cellular therapeutics (CTPs) is attractive since at first glance they solve some (eg, ethical) of the concerns that have hindered the clinical use of embryonic stem cells [6]. However, the relative inefficiency and complexity of current protocols has led many to conclude that iPSCs will not be cost effective if used to make autologous CTPs [7 –10], suggesting that their use may be limited to allogeneic CTPs.
On the other hand, the concern of immunogenicity may in some instances hinder the development of allogeneic CTPs, although some of the earliest authorized products are based on allogeneic keratinocytes and fibroblasts, such as Dermagraft® and Apiligraf®. Fibroblasts in particular are known to have low immunogenicity and there is also little evidence that these cells persist beyond a few weeks [11 –13]. The body of evidence to support human leukocyte antigen (HLA) matching is overwhelming; however, there are other situations where HLA matching might not be essential; where the cells only need to persist for a short period of time, where the cells are themselves immunomodulatory, such as mesenchymal stem cells, and where the cells will be administered to so-called “immune privileged” sites, such as the eye. The risk of acute rejection and the need or otherwise for HLA matching are likely to depend on the clinical application; further, chronic rejection mechanisms may also prove to be important. In March 2013 there were estimated to be at least six allogeneic CTPs in late-stage clinical trials [14] and their success will be dependent on allogenicity not limiting efficacy. However, it is fair to say that the prospect of immune rejection (or the use of immunosuppression drugs in parallel) is a significant uncertainty in the development of allogeneic CTPs, and the possibility of HLA match could remove this uncertainty in the minds of stakeholders. Further, it is generally assumed that allogeneic CTPs will bring efficiencies of scale and thus theoretically be more cost effective than an equivalent autologous CTP.
A number of groups have attempted to estimate how many individuals would need to be screened to identify potential donors homozygous for the most common HLA types of a particular population. These estimates focus on HLA-A, B, and DR since these are known to be the most important for organ transplantation [15,16]. Estimates and approaches vary; for example, screening 10,000 individuals in the United Kingdom should allow 10 homozygous HLA types to be identified that would provide a potentially beneficial match for up to 67.4% of the U.K. population [8]. Gourraud et al. [9] used a probabilistic model to estimate that screening 26,000 northern Europeans could identify 20 individuals who could provide a match for around 50% of that population. Moreover, the diversity of the HLA system is such that the numbers quickly rise depending on the degree of matching and the desired population coverage, suggesting that the number of lines required for a global product could be significant.
However, other sources of immunogenicity may also prove to be important; for instance, fetal bovine serum has led to hypersensitivity to an immunotherapy CTP in the clinic [17,18]. In vitro cell culture will alter glycosylation of many proteins, an issue well known to the biopharmaceutical industry [19]. If changes in glycosylation patterns can impact the safety and efficacy of single-protein therapeutic, then it stands to reason that it could impact CTP safety and efficacy, and evidence is emerging to support this [20 –22]. So in addition to adaptive immune responses due to HLA or other immunogens, there is a possibility of innate immune responses due to abnormal glycosylation, or indeed a range of other minor histocompatibility antigens and other alterations in the cells due to in vitro culture and differentiation. Despite these uncertainties, Turner et al. [10] recently suggested that the time is right to initiate an international effort to prepare banks of HLA-typed iPSCs suitable for clinical use. Although founded on rational scientific analysis, deeper scrutiny of the scientific and practical implications of undertaking this activity at this time needs to be discussed.
CTPs Versus Transplantation
The success of allogeneic transplantation has obvious parallels for allogeneic CTPs where autologous donation is unsuitable (eg, due to underlying disease) or undesirable (eg, cost). However, to understand whether the approach of HLA matching, which would require a number of HLA-typed cell banks, is adaptable to CTPs, we must first consider how they differ from transplantation.
A transplant involves “like-for-like” replacement of diseased or damaged cells, tissues, or organs from the recipient (patient) with healthy cells, tissues, or organs from a donor (different individual, except where autologous), without modification (Fig. 1A) [23]. To do this successfully certain simple (minimal) manipulations of the donor material may be required (eg, purification and cryopreservation) but importantly these do not alter the essential function(s) of the graft. As long as any manipulation does not intentionally alter the function of the graft, and accepting some transient or permanent loss of graft function that may occur during the whole procedure, transplantation does not require extensive quality control. In contrast, CTPs (Fig. 1B, C) are intentionally not transplants; they are alternative attempts to treat disease and/or loss of tissue or organ function by other means. The cells used are usually substantially manipulated, typically expanded to achieve adequate numbers, and/or manipulated in other ways to overcome normal physiological limitations (eg, differentiated, activated, and administered in nonphysiological concentrations). These ex vivo manipulations pose a range of potential risks to the patient; adventitious agents may be unintentionally introduced during manufacturing, and culture expansion and other manipulations may alter their immunogenicity. Further, it is necessary to confirm that the intended function(s) of the cells is present, and confirm that any unintentional effects are balanced with the intentional effects. Hence, such products are now regulated in most jurisdictions in the same or similar way to medicines rather than transplants or practice of medicine [24,25]; these differences also impact how you approach cell banking.
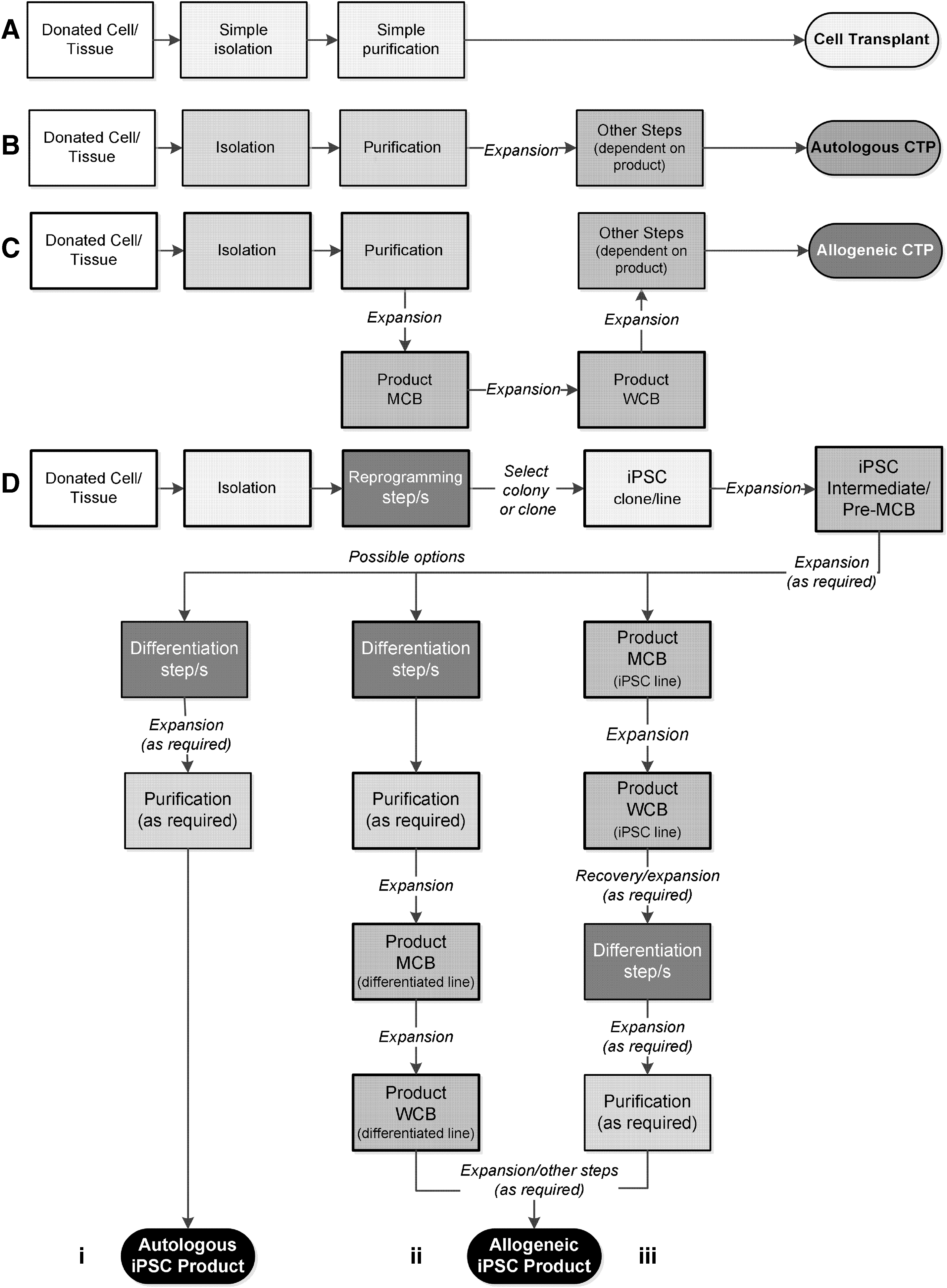
Schematics of typical approaches to cell therapy manufacturing. The likelihood of a manufacturing step alters cell characteristics (degree of manipulation) is graded from white background (unaltered), minimal changes (pale gray) through increasing degrees of substantial changes (to black). The resulting product is shaded with respect to likely cumulative changes with respect to the starting material (darker shading greater likelihood of changes). Steps leading to master cell bank/working cell bank (MCB/WCB) system (where used) are identified by thick border lines. Frozen intermediates may also be included in any of these approaches for manufacturing flexibility.
Different Cell Banks for Different Purposes
Cells are banked for diverse reasons, which can be sufficiently different to preclude their use beyond the original intent. As mentioned earlier, cells for transplantation must be banked without significant manipulation, while cells banked for CTPs are manipulated (eg, expanded) to provide a supply of sufficiently consistent starting material for manufacturing multiple batches of CTPs with acceptably uniform quality. When this is contrasted with the objectives of research cell banks (Table 1) it is clear that their purpose is very different on many levels.
A key objective of CTP manufacturing process development is to make a consistent product since without a consistent product you cannot hope to see a consistent clinical effect. Consequently, minimizing sources of manufacturing variability is a high priority. Defining the quality of biological materials is relatively difficult compared to chemical materials [26]; the more complex the biological material the more difficult it is to define its quality and control its variability. Two broad approaches are taken to control this source of variability in starting and raw materials, depending on the nature of the material (Fig. 2). For cells the first of these strategies is generally inappropriate, except perhaps to pool multiple donations from the same donor. The second strategy is typically applied to allogeneic CTPs; except that due to the limited expansion capacity of most cells, the risk of transformation, and the likely need for cellular heterogeneity, clonal selection is not applied. For iPSCs it is possible that not just a single colony but single-cell selection and expansion (at least once) will be beneficial and improve the consistency of the final product, but it is yet to be determined whether this will be feasible or desirable. These details aside, banking of cells for CTPs has the intent to provide a large pool of cells of a uniform quality such that many batches of CTPs can be manufactured of sufficiently similar quality. However, unlike biotech cell substrates, cell banks for CTPs will not always be sufficiently large or stable to supply for the whole-product lifecycle, necessitating that new master cell bank (MCB) is introduced from time to time. In theory at least, iPSC banks could be sufficiently large to supply the entire product lifecycle, which when the difficulties in established comparability for CTPs are considered could bring significant advantages.
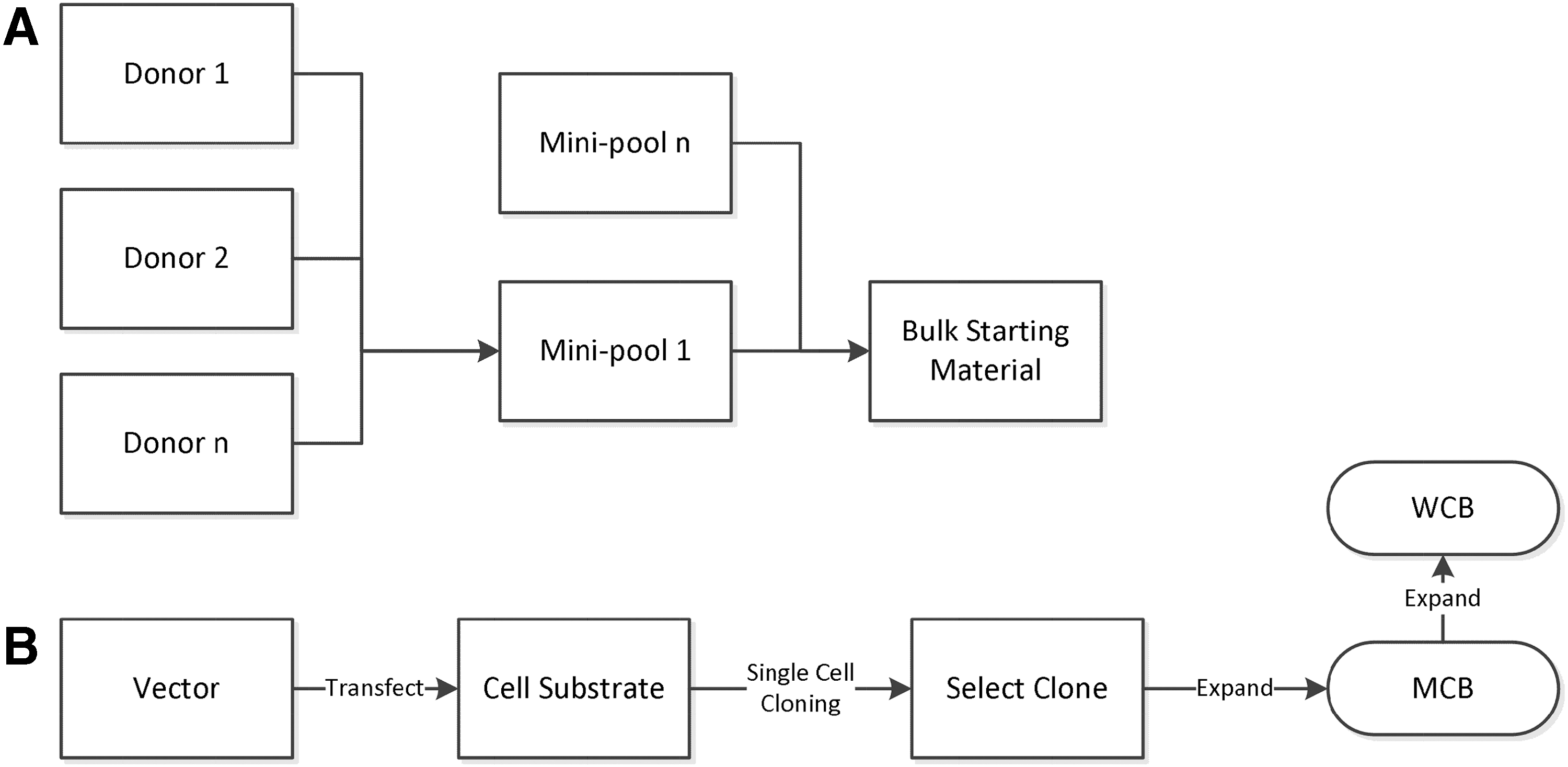
Methods to control the variability of biological starting and raw materials.
A question that needs to be addressed is whether the case exists for banking iPSCs for future, and yet to be determined, clinical use? Designing a process and appropriate characterization for the preparation of a cell bank where its eventual uses are unknown would be scientifically unsound. However, in the case of iPSCs, it could be argued that a pre-MCB (also called master seed stock) could be prepared providing the resulting bank retained its pluripotency and met defined criteria such as those identified in Table 2A. Where there is the intention that eventual CTPs will utilize multiple iPSCs, there would still be the problem of determining how to demonstrate that they are suitably comparable (discussed in the next section) without prior knowledge of what they will be used for. Consequently, there would remain a risk that some aspects of the cell banking process altered the iPSC characteristics and rendered it unsuitable for some uses, but since they have the potential to be used for so many future therapies this risk may be acceptable.
Intended cell functions: dependent on intended use, that is, capacity to be differentiated into cell type of interest and ability to exert intended biological effects associated with assumed MoA in selected indication(s).
For cell lines to be comparable they should have equivalent safety and efficacy for the intended use, but this assessment must also allow for normal donor variability. The subjective nature of this assessment also brings considerable uncertainty.
CTPs, cellular therapeutics; MoA, mechanism/s of action.
Any strategy to reprogram a cell is by its very nature a substantial manipulation of the original characteristics of the cell. Aside from the obvious concerns as to whether any one approach will result in undesirable characteristics, the first issue to address is which method is sufficiently reproducible to allow a series of cell lines suitable for clinical use. Key considerations for this are provided in Table 1. Where only a single iPSC line will be used for a CTP, characterization would focus on understanding what changes have occurred, and other studies would specifically address how these changes might impact safety and desired function. In this scenario the objective would be to identify the best iPSC line to take forward.
Existing repositories of iPSC lines will vary considerably not least because they have been prepared by a variety of different approaches using different starting and raw materials in a variety of facilities that may or may not have validated the process used. For an allogeneic CTP prepared from a single iPSC line these differences are immaterial, so long as the process utilized was devised with clinical use in mind (eg, applied good manufacturing practices [GMP], controlled adventitious agent risks, etc.). As with a biotech cell substrate, the provenance of the cell would need to be established, and suitable data provided to regulatory authorities to justify the suitability of the iPSC line for clinical use. Consequently, selection of a single allogeneic iPSC line from existing repositories will be possible, so long as they have been stored with clinical use in mind.
However, the use of existing iPSC lines will almost certainly be restricted to allogeneic CTPs based on a single iPSC line unless they have all been prepared by the same validated process. Further, the same principles also apply to privately banked autologous iPSCs; the reasons for this are discussed in more detail in the next section.
Establishing Comparability Will Be Difficult
These problems are compounded when you consider what would be required to utilize a number of iPSC lines to make a single licensed CTP for a particular indication. It would first be necessary to demonstrate that each cell line was essentially comparable within the realms of inherent donor variability; likely key issues to be addressed are detailed in Table 2B. Since a myriad of small subtle differences will be present between two facilities even following the same manufacturing and testing protocols, if these are prepared in more than one facility the facilities would need to be cross-validated (process and methods) and shown to be comparable. This is not a trivial task, and procedures would also need to be in place to reconfirm equivalence of the two sites on a regular basis to ensure that the processes and test methods do not drift apart from each other. Adding a third, fourth, and so on would progressively complicate validation and demonstration of comparability between all sites and quickly become unmanageable; so a single site would be the safest way to ensure consistency.
Comparability of starting cell populations and resulting CTP is a very difficult concept, unlike, for instance, a recombinant protein, the exact chemical structure cannot be determined. Consequently direct comparison of structure cannot contribute to any assessment of comparability; this brings significant uncertainty. A detailed discussion of comparability is beyond the current discussion; however, if we accept that the more a process could lead to cell changes (the greater the manipulation), then the harder it will be to control the process such that the resulting unintended changes are minimized. Also the more steps there are in a process, the more opportunities there are for unintended changes to accrue, and the more work required to optimize (maximize intended and minimize unintended changes) and control those steps. An attempt to represent this graphically is provided in Fig. 1; even at a high level it is clear that a CTP based on iPSCs will typically (necessary generalization) have a more complex manufacturing process involving steps that bring a greater chance of unintended cell changes.
One of the challenges when developing CTP is identifying the critical quality attributes (CQAs); these are dependent on the pathology of the disease and how the product exerts its effect (mechanism/s of action) to treat the disease (efficacy)—both typically bring significant uncertainties. Whether all or even some of the actual CQAs are identified may never be certain, and consequently the developer must at least strive to achieve manufacturing consistency; this brings significant uncertainty when changes are made. Ultimately clinical safety and efficacy are demonstrated in well-controlled clinical trials; however, these do not necessarily remove the inherent uncertainties identified for product quality. Postapproval changes are therefore more challenging than during development, and will be treated with more caution by regulators because of these uncertain links between quality and clinical safety and efficacy.
For these reasons, iPSC lines not prepared by the same optimized and validated process are very unlikely to be suitable to manufacture the same licensed CTP, whether that CTP is allogeneic or autologous. To use a privately banked autologous iPSC will require a CTP developer to work with the iPSC banking organization to ensure that all batches of CTP are manufactured from iPSCs prepared by the same process; these limitations suggest that private banking of autologous iPSCs may not be a good investment. A more logical strategy would be to bank fresh, unexpanded donor tissue that could be reprogrammed by any autologous iPSC CTP developer in the future.
Manufacturing Challenges for HLA-Matched CTPs
Manufacturing complexity escalates if more than one cell line is required to manufacture the same CTP, either because it is envisaged that a single line would be insufficient to supply the product lifecycle or because there is a wish to match HLA to minimize allogenicity. As already described, the first challenge for HLA-matched CTPs based on iPSCs will be to validate a reprogramming process that can consistently reprogram cells that are sufficiently comparable, allowing for inherent donor variability. Once a series of suitable HLA-typed pre-MCB are available, for each new product, which may only treat a single indication, a series of product MCB of a suitable size will need to be prepared taking into account the considerations identified in Table 3A. The difficulties associated with estimating the likely MCB capacity are such that it seems unrealistic to imagine that these could take into account the needs beyond a single CTP. As discussed previously, the difficulties in establishing comparability of both a new MCB and the resulting CTP suggest that the most realistic approach would be an MCB that can supply the anticipated product lifecycle.
MCB, master cell bank; WCB, working cell bank.
Each HLA-typed MCB will eventually need to be shown to result in comparable CTP from the commercial process; typically, at least three batches are required to do this. It is hard to envisage how this work could be condensed, confirming each bank is comparable is essential regulatory data. The bigger question of whether clinical data will be required for each bank will be touched on later. Following later-manufacturing changes, for example, to increase scale during development and eventual commercial scale manufacture, comparability data will also be needed, again quite possibly for every iPSC line to be used. As discussed elsewhere, such changes will need to factor-in all scales of manufacture, dosage forms, and stability reconfirmation. It also seems possible that more than one production line would be required such that several batches of product could be manufactured in parallel; this too would add additional validation and comparability work. Further, the need to make changes never ends, so even once authorized any significant change (from a critical biological raw material through to improved manufacturing processes) requires appropriate comparability data, and may require revalidation. Depending on the change and the extent of existing evidence that the iPSCs are comparable, this might entail new comparability data from several or even all iPSC lines used to make the CTPs.
CTP manufacturing is still in its relative infancy, and others have written extensively on the challenges [27,28]. Attempting to develop a product based on multiple iPSC lines would add considerable complexity and uncertainty. Where using HLA-typed iPSC early development could be simplified by selecting those iPSC lines that will be used most frequently, potentially even a single line for first-in-human studies, and increasing the number of lines utilized in subsequent studies. However even this poses challenges, since this approach assumes that equivalent safety can be demonstrated for all iPSC lines without clinical evidence. Again this comes back to how robust any comparability data, supported where possible with in vivo animal safety studies (where useful models exist).
Another issue for HLA-matched CTPs will also be the differing demands for each haplotype; some will be suitable for a significant portion of the patient population, and others may be used for <1% of patients. Dependent on a range of factors, in particular, the shelf-life of the CTP (regulatory limit 5 years [29]), these differences will likely necessitate different scales of manufacture to ensure continuous supply and limit wastage. Some key considerations for deciding on the eventual manufacturing scale(s) are provided in Table 3B. In addition to different CTP manufacturing scales, depending on the indication(s) that there may be different product strengths, these different manufacturing scales and dosage forms will each need to be incorporated into validation and stability programs. Even assuming arguments could be made to reduce the number of iPSC lines incorporated into these studies, the scale of such an endeavor could be daunting.
Commercial Considerations
Ultimately cost will be a major factor, if not for the initial preparation of the iPSCs to clinical standards and then for the CTPs that will follow. The cost of generating, testing, and expanding research iPSC lines is estimated to be between US$10,000 and US$20,000 per line and requiring 4–6 months from tissue harvest to characterized bank [5,7]. When preparing iPSC lines for clinical use these figures escalate, bringing the likely cost per line in the order of US$0.8 million (G. Stacey, pers. comm.) due to the need for a GMP environment testing and evaluation of tumorigenic hazards. While such cost might be envisaged from public and/or private consortia, developing CTPs from a repository of HLA-typed iPSCs would require commercial funding.
For each CTP it would be necessary to prepare one or a series of product MCB for each iPSC line at a similar cost each, but also to undertake extensive comparability to demonstrate equivalence of each iPSC line used in any CTP process. This likely brings a plausible estimate of the cost per iPSC line to well over US$1 million, and at least some banks would need to be prepared before preclinical development could be started. As the number of iPSC lines increases the additional costs associated with enabling scale-up over development to reach a commercial-scale process become daunting. These figures do not include the likely costs of some of the raw materials to prepare, expand, and differentiate large numbers of iPSCs and/or the differentiated cells of interest. For example, to prepare motor neurons, Boulting et al. [30] mention at least four recombinant proteins; these would each need to be of high quality (and compliant for all licensed regions) from a reliable supplier or custom manufacture. To supply the PAP-CM-CSF fusion protein used in Provenge®, Dendreon signed a deal worth US$8.3 million with GlaxoSmithKline [31]. This was just a single protein, and to be used for a product with a small market. It would therefore likely make a significant impact on costs if reprogramming and differentiation processes could employ small molecules since these are generally cheaper to manufacture to GMP standards.
Further, on-going regulatory requirements such as pharmacovigilance will be highly complicated for a product with so many subvariants, as would risk-management plans and traceability requirements. Regulatory submissions would also be unprecedented; in contrast to what is proposed, Geron, Inc., reportedly spent US$45 million to get to approval of their first IND of 22,000 pages, reportedly the largest the FDA had ever seen [32]. That was for a product derived from a single human embryonic stem cell line, although it also included a complex novel medical device. Once authorized, there will be an obligation on the license holder to ensure continuous supply of the CTPs; product shortages could mean that patients do not get treated, or worse still, patients undergoing treatment cannot continue. As discussed previously, scheduling manufacturing for such a complex product would be a formidable task. Clinical trials for HLA-matched CTPs would also pose significant challenges both in their design and likely size. Whether it would be necessary to use all iPSC lines during clinical development is a significant uncertainty, and would be dependent on the quality of the comparability data and require extensive discussion and negotiation with regulators. These factors suggest that late-stage trials at least would be considerably more expensive than for a single iPSC-based CTP.
Further issues not addressed in detail include freedom to operate or the need to pay royalties to use intellectual property, and whether the cost effectiveness of resulting CTPs can justify adequate reimbursement to provide a return on investment. Developing any medicinal product comes with significant risks, one being the risk that others identify safety concerns that could relate to the product under development. A public repository of HLA-typed iPSCs would likely be seen by some as a significant business threat if access to the same iPSC lines by others was possible. Clinical or research findings suggesting a potential safety issue could have a significant impact on all developers using the same iPSC line, whether founded on good science or not. Further, negative findings could force regulatory authorities to place clinical development of all products using the same iPSC line on hold; for both privately funded and publicly listed companies such outcomes could be disastrous. Thus, commercial developers would likely want to control access to their iPSC lines, suggesting a public repository would not be an attractive source.
Conclusions
By 2011 at least 14 strategies to reprogram cells had been identified [3], and more have followed since. Further, understanding the biology of cell reprogramming is also in a rapid state of development, with accruing knowledge of how to successfully switch cell differentiation states while bypassing pluripotency [33,34]. Whether HLA typing is required and whether iPSCs and CTPs made from them express unique antigens that will be immunogenic will ultimately need to be determined for each clinical application, as recently acknowledged by Kaneko and Yamanaka [35]. It may be that HLA typing alone is not sufficient and alternative approaches, such as immunosuppression or tolerance-inducing regimens, are a necessary part of the therapy, as they have been for transplantation. Until the scientific community is able to determine and justify unequivocally which of these strategies is appropriate to deploy, even leaving aside the considerable level of funding required to generate even a single bank, it seems premature to consider HLA-typed iPSC banks. In principle most of the issues identified here that would also apply to HLA-matched CTPs based on human embryonic stem cells, and even HLA-matched adult stem cell CTPs, would be significantly more complicated.
Developing any medicinal product brings significant commercial risks; based on a range of small-molecule and biotech products developed by the top 50 pharma companies entering clinical development between 1994 and 2004, the likelihood of eventual approval by the FDA ranged from 8% to 24% (average 19%) [36] depending on the indication. Whether CTPs will be more or less successful is unknown, but based on their increased complexity over therapeutic proteins and the relative immaturity of the field, it seems unlikely that they will be more successful. Prescott [7] speculated that the use of tissue-matched iPSCs might overcome the high costs of autologous use of iPSCs; however, this synthesis of the issues associated with this approach makes it clear that these cost savings may not be as significant as hoped. Further, the sheer complexity of developing such products and maintaining a continuous supply once authorized brings unprecedented uncertainties and risks. The number of uncertainties suggests that it would be prudent to await evidence that CTPs based on single iPSC line can be safe and effective in the clinic before embarking on such an ambitious project. This combination of scientific and commercial uncertainties combined with investment costs suggests that it is unlikely that any investor would consider the risks acceptable.
In conclusion, there are a range of scientific, practical, and financial barriers to developing HLA-matched iPSC–based CTPs at this time. While it is true that the field is progressing rapidly, the paucity of currently authorized, yet less complex, CTPs suggests that the enormous effort required to undertake such a task would be premature at this time. Even if a suitably consistent method of preparing HLA-typed iPSCs was available now, it would be very risky, not least because better and safer methods of iPSC production may be developed in the near future, potentially undermining the effort. Further, the cost just to prepare the iPSC banks is likely prohibitive, let alone the cost of developing CTPs from them, and with so much uncertainty around their future utility, these lines would likely have low prospects of returning significant investment from expended public money.
Footnotes
Acknowledgments
The author would like to thank the following for their useful comments during preparation of this article: Tim Allsopp (Neusentis), David Brindley (CASMI), Kim Bure (TAP Biosystems), Alex Denoon and Julian Hitchcock (Lawford Davies Denoon), Anna French (CASMI), Karin Hoogendoorn and Sharon Longhurst (Parexel), and Cathy Prescott (Biolatris). This work was supported by funds from the CASMI Translational Stem Cell Consortium.
Author Disclosure Statement
No competing financial interests exist.