
Abstract
Hematopoietic stem and progenitor cells (HSPCs) reside in bone marrow (BM) in an environment rich in CXCL12, the ligand for CXCR4, which is constitutively expressed on all immature hematopoietic cells in BM. This ligand-receptor pair critically controls HSPC retention and (relative) quiescence in BM. Interestingly, in a chemokine-abundant environment, CXCR4 surface expression and CXCL12 sensitivity of BM-residing HSPCs are continuously maintained. The mechanisms underlying this peculiar pattern of G-protein signal integration by BM-HSPCs are unknown. G-protein receptor kinases (GRKs) control receptor function by phosphorylating the intracellular domains upon ligand-induced activation, which results in receptor internalization and transient refractoriness. Using, therefore, a GRK6-deficient (GRK6−/−) mouse, we sought to address how perturbed ligand-induced CXCR4 (in)activation affects HSPC behavior in vitro and in vivo. In vitro, GRK6−/− HSPCs were characterized by hyper-responsiveness to CXCL12, as expected. In vivo, GRK6−/− immature hematopoiesis was characterized by a marked expansion of immature hematopoiesis in spleens and a modest repopulation defect in serial competitive transplantation. Enforced mobilization with granulocyte colony-stimulating factor (G-CSF) and AMD3100 was normal, as was hematopoietic regeneration after noncompetitive transplantation or pharmacological myelosuppression. These observations illustrate that GRK-mediated restriction of CXCR4 signal input after ligand engagement is largely dispensable for BM-resident HSPCs, which may explain how continuous CXCL12 responsiveness of BM-HSPCs can be maintained.
Introduction
M
As a member of the family of seven-transmembrane receptors, CXCR4 is coupled to G-protein-mediated signaling pathways [6 –15]. Cell surface expression of G-protein-coupled receptors (GPCRs) is typically regulated by ambient ligand concentrations. Ligand binding induces intracellular signaling events, including activation of members of Src and MAP family kinases, activation of Phospholipase Cβ (PLC-β) and Ca2+ Flux [7]. Receptor activation is typically followed by rapid phosphorylation of its intracellular C-terminus by one of several G-protein-receptor kinases (GRKs), facilitating recruitment of ß-arrestins and subsequent much slower receptor internalization [7,16]. These molecular events render the cell transiently insensitive to the GPCR ligand until ligand concentrations in the environment change. Rapid desensitization of GPCRs appears to be functionally important to allow cells to sense minute ligand gradients along the length of the cell. Chemotaxis itself likely also requires rapid receptor signal termination for cells to establish reversible adhesion and polarization/elongation, repeated cycles of which add up to movement toward a chemoattractant. Thus in many cells, CXCL12-induced signals are terminated within seconds, a function in mature hematopoietic cells shown by the Lefkowitz group to hinge on GRK6-mediated phosphorylation of GPCRs. In GKR6−/− mature hematopoietic cells, abnormal signaling and in vitro migration were thus predicted and observed [17,18], with functional consequences for immune function in vivo [19,20]. In agreement with the paradigm of need-adapted shuttling between cell surface and intracellular location, chemokine receptors have been reported to be predominantly located intracellularly [21,22]. Given the physiology of GPCRs, it is surprising that in contrast to leukocytes immature hematopoietic cells in BM continuously express CXCR4 on their surface and indefinitely retain their sensitivity to CXCR4, even though CXCL12 is presented more or less continuously and at high concentrations (albeit reportedly with some diurnal variation in concentration [23,24]). The continued responsiveness of HSPCs to CXCL12 was earlier shown to hinge significantly on the presence of TGF-β [25], but further information about the intracellular processing of CXCR4 signaling input on immature hematopoietic cells has not been reported. Seeking to explore this conundrum, we formally analyzed GRK6-deficient immature hematopoiesis using a genetic model of GRK6 deletion.
Materials and Methods
Mice
GRK6−/− mice were previously described [17,26]. GRK6−/− and wild-type (WT) control littermates were generated from GRK6+/− heterozygous breedings. GRK6+/− mice, gifted by Robert Lefkowitz (Duke, Durham, NC), were previously described [26,27]. Fetal liver (FL, E14.5), young (8–12 weeks) and old (60–110 weeks) mice were studied. CD45.1 (B6.SJL-Ptprca Pep3b /BoyJ; Charles River Laboratories, Sulzfeld, Germany) mice served as competitor cell donors and CD45.1/.2 hybrids served as recipients in competitive engraftment experiments where indicated. Splenectomy was aseptically performed on anesthesized mice, as described [1]; mice were allowed a minimum of 5 weeks to recover before undergoing any experiments. Mice were housed under conventional (non-specific pathogen-free) conditions in open-top cages, with water and chow ad libitum. All animal experiments were performed with permission of the local IACUC and municipal government (permit # F27/13).
Genotyping
GRK6-deficient mice were genotyped by polymerase chain reaction (PCR) on DNA samples prepared from tail tips using DirectPCR® Lysis Reagent Tail (Peqlab, Erlangen, Germany). The following genotyping primers were used: forward 5′-GGG AAG TCA GGT TTG GAA CA-3′; reverse WT: 5′-AGA ACC CTC CCT GCA ATC TT-3′ reverse GRK6−/−: 5′-GGG AGA GAA ACA CAG GGT CA-3′.
Mobilization
HSPCs were mobilized to peripheral blood with granulocyte colony-stimulating factor (G-CSF) (50 μg/kg i.p. q12 h*9 doses; Chugai Pharma, Tokyo, Japan) or AMD3100 (4 mg/kg i.p. as a single dose; Sigma-Aldrich, St. Louis, MO), as previously described [1,28]. To assess HSPC [colony-forming units culture (CFU-C)] mobilization efficiency, blood was drawn 3 h after the last injection of G-CSF, or 1 h after injection of AMD3100, followed by CFU-C enumeration (see below under “Functional in vitro cell assays”).
Transplantation experiments
Lethally irradiated recipient mice (1×9.5 Gy for engraftment, or 1×12.5 Gy for homing assays, using a cesium source with a dose rate of 1 Gy/min), received i.v. grafts of BM or spleen cells from WT and/or GRK6−/− donors, and homing or engraftment/engraftment kinetics [1,29,30] were analyzed as previously described at indicated time points.
Mouse cells
Blood was drawn from the facial vein without anesthesia. EDTA-anticoagulated samples (complete blood count, CBC) were analyzed with an automatic hemocytometer (Hemavet HV950FS; Drew Scientific, Barrow in Furness, Cumbria, United Kingdom). Splenocytes were recovered from spleens by gentle extrusion, BM cells were flushed from femurs, tibias, or pelvic bones. Surface and intracellular markers were studied by flow cytometry (FACS Calibur™ and FACS Canto™II; BD, Heidelberg, Germany) using established multicolor staining panels [31]. CXCL12-induced intracellular signal transduction pathways were analyzed by western blot and/or flow cytometry.
Signaling assays
CD117+ (c-kit+) cells were isolated by immunomagnetic selection as described [32], subsequently serum-starved for 2 h at 37°C. Cells were stimulated with CXCL12 (100 ng/mL) for the indicated times. For western blot analysis aliquots were immediately lysed with ice-cold 2×Laemmli buffer. Proteins were separated by 7.5% SDS-PAGE gels, blotted, blocked, and then stained with fluorescence-coupled antibodies. Results were visualized by Odyssey CLx (LI-COR, Bad Homburg, Germany). Bands were quantified using Image Studio 2.0 software (LI-COR) and subsequently normalized to β-actin expression. Erk1/2 phosphorylation was further corroborated using a flow cytometry-based assay, following the manufacturer's instructions (BD).
Functional in vitro cell assays
After erythrocyte lysis in hypotonic buffer, BM, blood, or spleen cells were plated into growth factor-supplemented methylcellulose medium (Stemcell Technologies SARL, Grenoble, France). Cultures were incubated at 37°C and 5% CO2 for 1 week and CFU-Cs were enumerated by inverse microscopy with 2.5×magnification [29]. Spontaneous and CXCL12-directed transwell migration was performed using 5 μm pore size transwell inserts (Corning Costar; Corning Incorporated, Corning, NY) and CXCL12 (Peprotech, Rocky Hill, NJ) as described previously [30]. Migrated and total cells were enumerated after 4 h by flow cytometry or by CFU-C assay [30,33].
Quantitative real-time polymerase chain reaction
GRK mRNA expression was analyzed on whole BM or sorted lin-/low Sca-1+ c-kit+ (LSK) cells using real-time-PCR (QuantiFast® Probe Assay; Qiagen, Hilden, Germany). RNA from tissues with reported expression of the respective GRK [retina (GRK1), spleen (GRK2, 3), testis (GRK4), lung (GRK5), and BM (GRK6)] served as positive controls, β-actin served as internal control (dual labeled).
Statistics
Descriptive statistics and t-tests were calculated in Excel v.7.0 (Microsoft, Redmond, WA). The n for each experiment is indicated in the figure legends. Unless otherwise indicated, all values are presented as mean±SEM. A P<0.05 was considered statistically significant.
Results
Aberrant CXCL12 signal integration of GRK-deficient cells in vitro
Expression of GRKs was tested in GRK6−/− or WT primary sorted lin-/low Sca-1+ c-kit+ (LSK) BM cells by real-time PCR experiments. Ablation of GRK6 in cells from GRK6−/− mice was confirmed (Fig. 1A). GRKs 2, 4, and 5 were expressed in WT mice at similar levels as GRK6. GRK3 expression levels were slightly increased in GRK6−/− cells, however, the difference did not reach statistical significance.
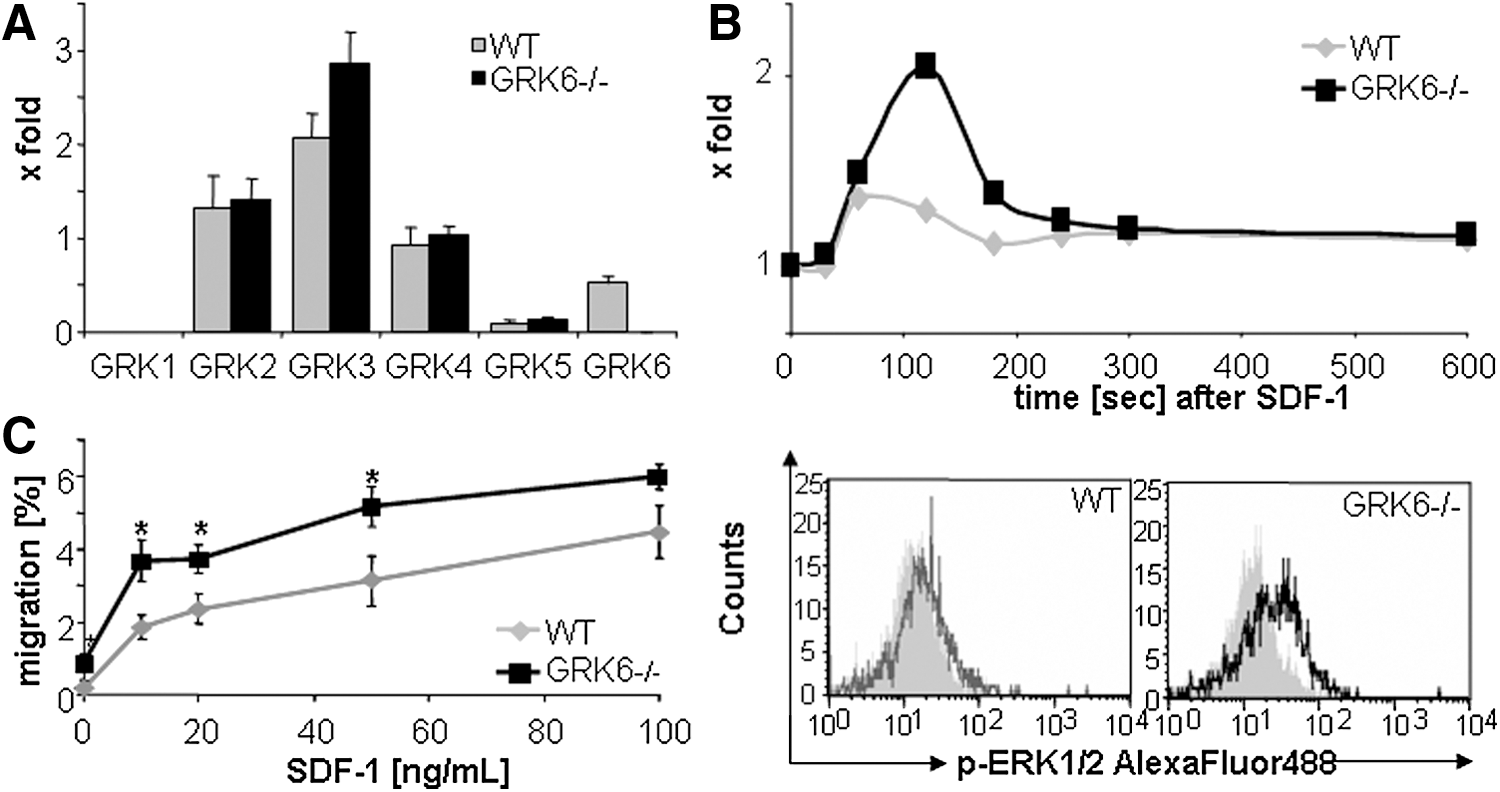
Aberrant CXCL12 signaling in GRK6−/− HSPCs.
Intracellular transduction of CXCL12 signals was tested by assessing magnitude and duration of phosphorylation signals. Ex vivo stimulation of BM c-kit+ cells with CXCL12 resulted in stronger phosphorylation of CXCR4 downstream kinases (p90RSK and Erk1/2) in GRK6−/− compared with WT HSPCs (Fig. 1B and data not shown). Signal integration was further evaluated using functional in vitro assays. In vitro transwell migration assays demonstrated that mature splenocytes reached maximum migration levels at CXCL12 concentrations of 20 ng/mL. In contrast, the percentage of migrating BM CFU-C increased up to the highest dose tested (100 ng/mL). At all concentrations, migration of GRK6−/− CFU-Cs toward CXCL12 was more acute than that of WT CFU-Cs (Fig. 1C).
Immature steady-state GRK6−/− hematopoiesis
Hematopoietic tissues were analyzed for HSPC contents in young adult (8–12 weeks) and old (60–110 weeks) GRK6−/− and WT mice, and during fetal life. Furthermore, splenectomized GRK6−/− or WT mice and stably engrafted recipients of GRK6−/− or WT BM-HSPCs were characterized.
FL hematopoiesis
FL (E14.5) were analyzed for total nucleated cells, c-kit+ cells, LSK cells and CFU-C content (Fig. 2A–C and data not shown). All three subsets were indistinguishable between WT and GRK6−/− FL, indicating redundancy of GRK6 signaling for initiation of fetal hematopoiesis and FL colonization.
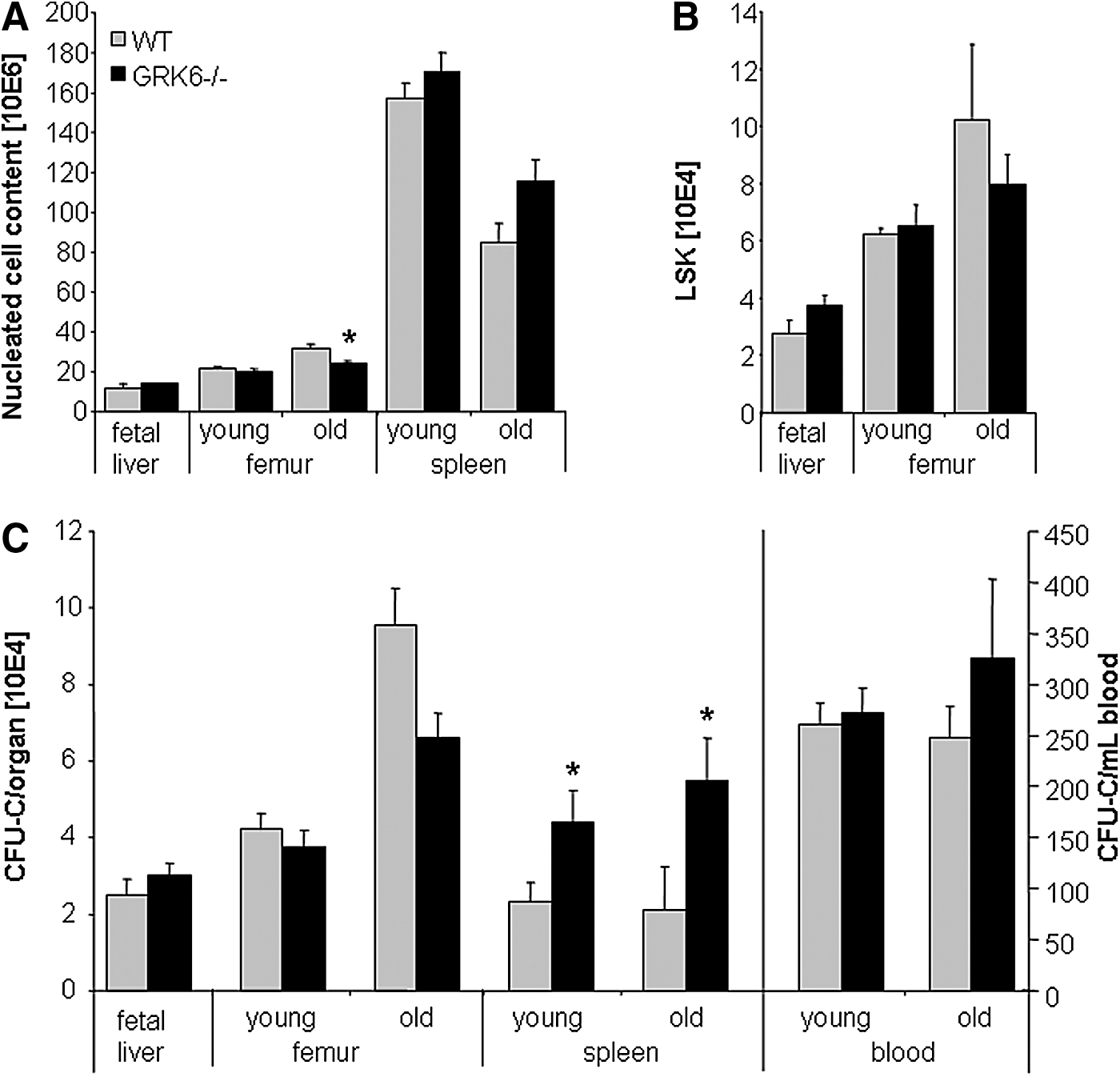
Leukocyte (white blood cells, WBC), LSK and colony (CFU-C) content in hematopoietic organs of WT and GRK6−/− mice.
Immature hematopoiesis in young adults
BM hematopoiesis in young adult GRK6−/− and WT mice was the same with respect to mature and immature cell content (Fig. 2A–C). Similarly, the relative distribution of myeloid lineages within the BM progenitor cell population was unperturbed in GRK6−/− mice (Supplementary Fig. S1; Supplementary Data are available online at
Data are given as mean±SEM from a minimum of three analyses for each group.
MFI, mean fluorescence intensity; WT, wild-type.
A remarkable two-fold increase of immature cells (Figs. 2C and 3A) including long-term repopulating cells (Fig. 3A–C) was observed in spleens of young adult GRK6−/− mice. The number of stress BFU-Es (BFU-E grown in methylcellulose media containing erythropoietin as the only growth factor [34]) was increased in GRK6−/− spleens to the same degree as CFU-Cs (not shown). Despite increased CFU-C content in the spleen, the number of circulating CFU-Cs was unchanged (Fig. 2C).

HSPC frequency is increased in spleens of GRK6−/− mice. Lethally irradiated CD45.1/.2 F1 hybrids received transplants of 2.5×10E5 CD45.1 BM cells mixed with 2×10E6 spleen cells from WT (CD45.2) or GRK6−/− (CD45.2) donors (n=6 per group).
Immature hematopoiesis in old mice
As mice age, immature hematopoiesis changes in a reproducible manner [35 –37]. In both WT and GRK6−/− aged (60–110 weeks old) mice, immature hematopoiesis significantly differed from that of young mice. Furthermore, differences between WT and GRK6−/− mice became apparent. The number of immature cells in BM of GRK6−/− mice was slightly reduced (Fig. 2C, n.s.), whereas the number of CFU-Cs in circulation was modestly increased (Fig. 2C, n.s.). The substantial increase in spleen CFU-C content observed in young adult mice was even more pronounced in aged GRK6−/− as compared to WT spleens (Fig. 2C).
Immature hematopoiesis of WT recipients of GRK6−/− HSPC transplants
WT recipients of GRK6−/− HSPCs displayed a phenotype similar to that observed in aged mice. Specifically, increased numbers of circulating HSPCs (Fig. 5C) and markedly (four-fold) increased numbers of HSPCs in spleens (Fig. 5C), but normal CFU-C numbers in BM (Fig. 5C) were observed.
Extramedullary hematopoiesis in GRK6-deficiency—role of the spleen
To rule out a BM retention defect with subsequent siphoning off of immature cells by the spleen as the cause for the increased CFU-C contents in the latter, we splenectomized WT and GRK6−/− mice and enumerated HSPC in BM and peripheral blood (PB). As in nonsplenectomized mice, the numbers of circulating HSPCs in WT and GRK6−/− mice were indistinguishable (Fig. 4D). Moreover, BM hematopoiesis was fully competent (Fig. 4A–C).
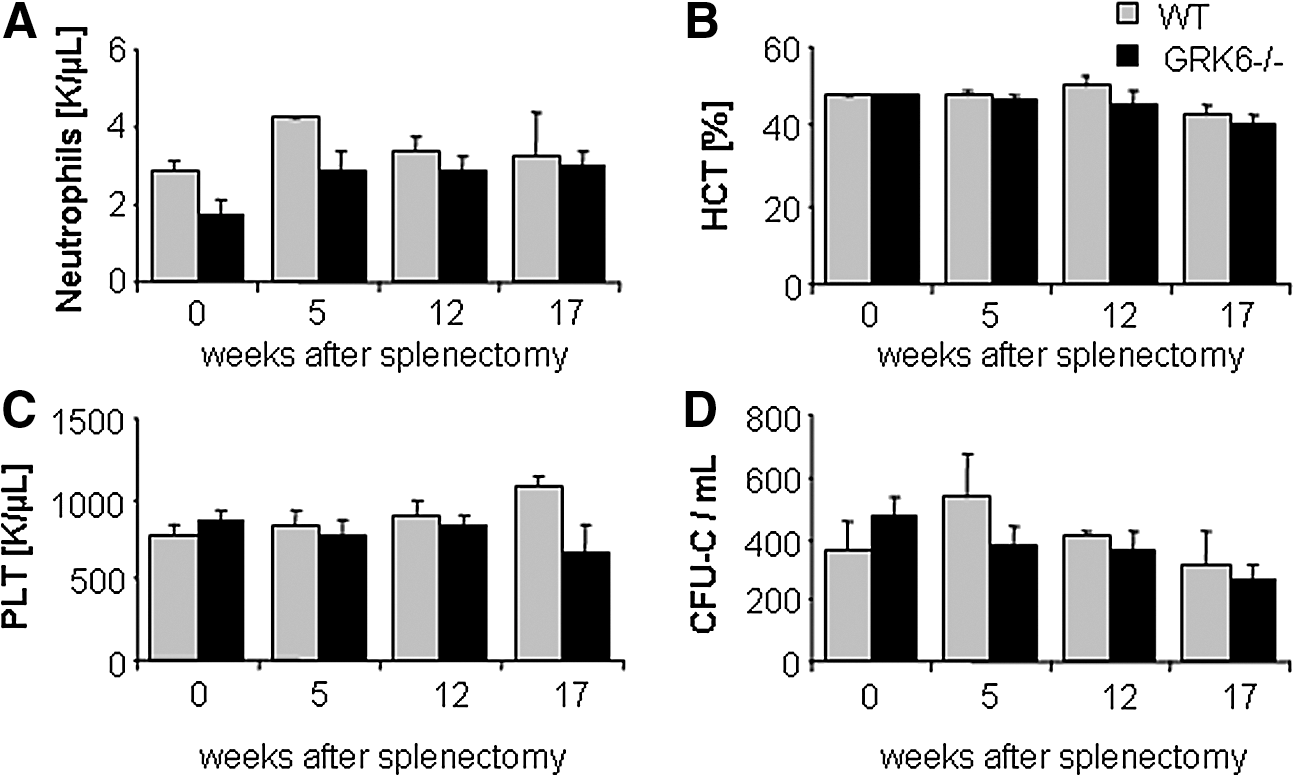
Mature cell and CFU-C content of splenectomized WT or GRK6−/− mice. Neutrophils
Homing and engraftment of GRK6−/− HSPCs
To provide engraftment, HSPCs must seed to BM, migrate to stem cell supportive (presumably endosteal) BM regions, and establish hematopoiesis [38,39]. Redundancy of CXCR4 for homing of immature hematopoietic cells was previously demonstrated by several independent approaches [4,40]. Nevertheless, formal functional in vivo homing assays were performed. Twenty hours after transplantation, similar numbers of CFU-Cs were recovered from BM, spleen, and blood of recipients of GRK6−/− or WT HSPCs, indicating normal clearance of HSPCs from circulation and dispensability of GRK6 for BM and spleen homing of HSPCs (Fig. 5A).
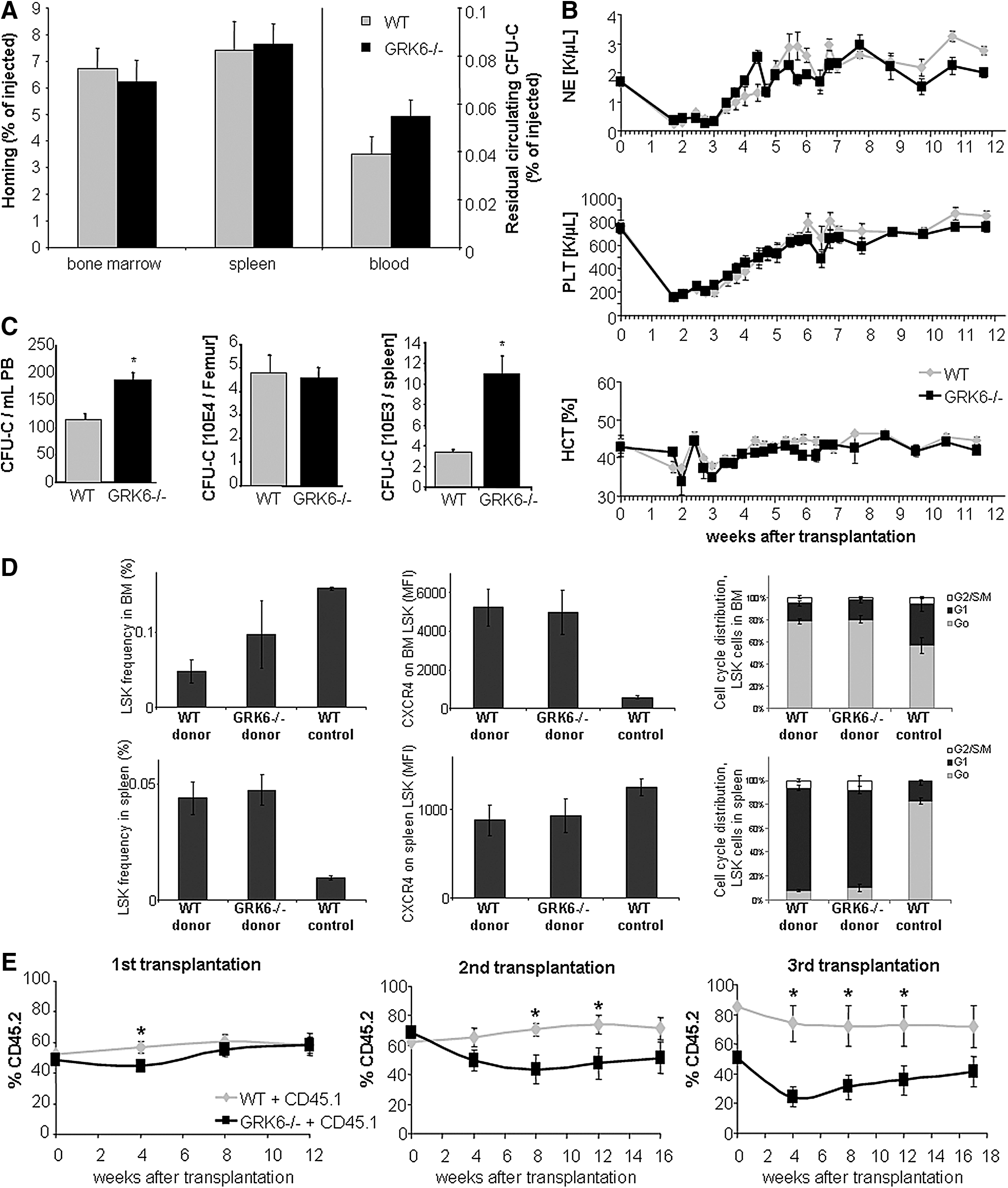
Homing, short-term noncompetitive engraftment and serial competitive engraftment in WT recipients.
Next, we compared the kinetics of hematopoietic repopulation in lethal irradiation-conditioned WT mice transplanted with limiting doses (250,000) of BM cells. Regeneration kinetics for all myeloid lineages were similar for recipients of GRK6−/− and WT cells (Fig. 5B). To gain additional insight into immature hematopoiesis at early times after transplantation, that is, at a time of dramatic expansion and differentiation stress, reconstitution of BM and spleen was additionally analyzed 8 days after transplantation. We observed BM-CFU-C content of ∼10% of steady-state values in both recipient groups, and spleen CFU-C content approaching homeostatic values. Relevant numeric differences for LSK cells or CFU-Cs between recipients of WT versus GRK6−/− transplants were not apparent in any of the hematopoietic organs (Fig. 5D and data not shown). LSK cells in BM were almost exclusively in G0 phase of the cell cycle, in contrast to the steady-state situation, where less than half the HSCs resided in this state (Fig. 5D). Several-fold increased CXCR4 expression on BM-resident HSCs on day 8 after transplantation was observed (Fig. 5D). In contrast, the LSK population in spleens was actively cycling, unlike the homeostatic situation, and these LSK cells were characterized by reduced CXCR4 expression compared to steady-state spleen LSK cells (Fig. 5D). Significant differences in cell cycle activity between GRK6−/− or WT derived grafts were not observed.
The most stringent characterization of true stem cells hinges on their ability to serially repopulate hematopoiesis in lethally irradiated recipients. In a competitive transplantation setting, control HSPCs are co-engrafted together with test HSPCs, which controls for host factors that might affect HSPC function [41]. Moreover, because of its competitive nature the assay can discover relatively subtle phenotypes. Serial competitive transplantation assays performed with BM from long-term engrafted (16 weeks) mice that had received CD45.1 competitor BM plus CD45.2 BM from either WT or GRK6−/− donors, revealed that after three serial transplants GRK6−/− transplants contributed less than 40% of mature blood cells in the circulation, despite the inherent engraftment advantage of CD45.2 hematopoietic cells [42,43]. In contrast, WT transplants had almost completely outcompeted the initial CD45.1 cell graft, as expected (Fig. 5E). A corresponding distribution was seen in BM of tertiary recipients. Of note, representation of significantly fewer GRK6−/− cells than expected from the distribution of HSPCs in the transplant mixture was noted at the time point 4 weeks after transplantation (ie, during the engraftment period) in all recipient generations. Thereafter, GRK6−/− contribution showed a relative recovery. Thus, overall a significant defect of GRK6−/− HSC function was observed.
Recovery of GRK6−/− mice from hematopoietic insults
In addition to stem cell transplantation in lethally irradiated hosts, other models of stress hematopoiesis were also tested, including HSPC mobilization (G-CSF, AMD3100), cytoreductive chemotherapy with 5-fluorouracil (1×150 mg/kg i.v. [44]), sub-lethal total-body irradiation (1×450 cGy [45,46]) and severe hemolytic anemia with phenylhydrazine (1×60 mg/kg i.p. [47]).
As mentioned above, baseline circulating HSPC numbers were the same in GRK6−/− and WT mice (Fig. 6A). HSPC mobilization was induced by G-CSF or AMD3100 in GRK6−/− mice or age and sex-matched WT mice, as described [1,48,49]. In both groups, both agents mobilized with the same kinetics and potency mature (notably including neutrophils, Supplementary Fig. S2) and immature (CFU-C) hematopoietic cells (Fig. 6A), total numbers being in agreement with published data [1,2,48,49]. Because of the arguable role of splenic HSPC pools for mobilization with CXCR4 antagonists [1,50], these studies were also corroborated in splenectomized mice, with almost identical outcomes (Fig. 6B and Supplementary Fig. S3).
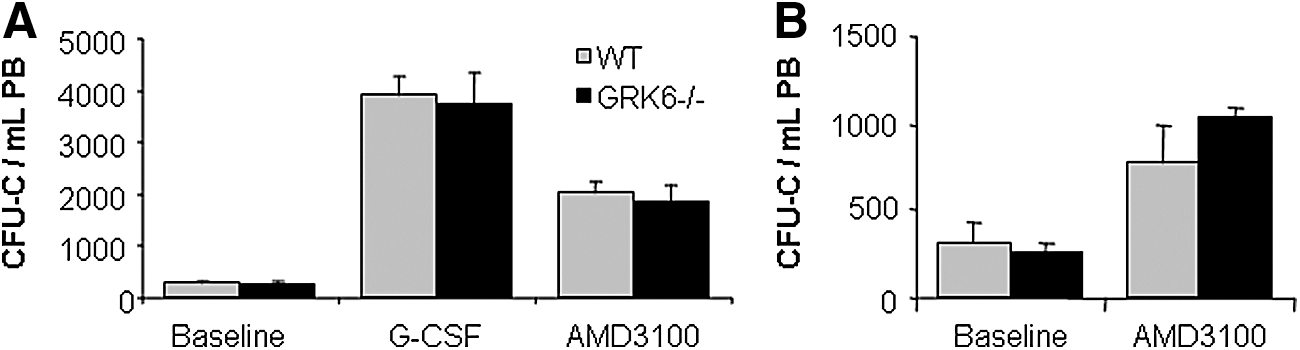
Granulocyte colony-stimulating factor (G-CSF) or AMD3100-induced progenitor cell mobilization.
Following chemotherapy, induced hemolysis or sub-lethal irradiation, GRK6−/− mice became similarly anemic or pan-cytopenic as WT mice, that is, were sensitive toward analyzed hematopoietic insults. Subsequent regeneration of affected lineages occurred in a timely manner and was indistinguishable between GRK6−/− and WT mice (Supplementary Fig. S4 and data not shown).
Discussion
GRK6 had initially been proposed as the predominant GRK in hematopoietic cells [17,51]; the in vitro and in vivo phenotype of mature GRK6−/− hematopoietic cells was in full agreement with such an assessment. Therefore, similar expression levels of GRKs 2, 3, and 4 as of GRK6 in HSPCs were unexpected. Slightly upregulated expression of GRK3 might suggest partial compensatory upregulation given the very recently reported role of GRK3 for immature hematopoiesis [52]. The observed robust signaling perturbance in GRK6−/− HSPCs in vitro indicates, however, that compensation is at best partial. The hypersensitive signaling phenotype is in agreement with the expected behavior of cells with impaired CXCR4 signal termination and is similar to the one observed with the WHIM mutation [53 –56]. Increased in vitro transwell migration toward CXCL12 represents the expected corresponding functional phenotype. Of interest, dose dependency of CXCL12-directed migration was saturable in lymphocytes but not in BM-CFU-C, suggesting different mechanisms of CXCL12 signal integration for these cell types.
Clearly, GRK6−/− HSCs initiate quantitatively and qualitatively normal hematopoiesis in FL. The entirely normal BM-HSC contents in young adult GRK6−/− mice further suggests that migration of HSCs from FL to and establishment and expansion in BM are also unaffected by GRK6-deficiency. A quantitative, partially reversible post-transplantation engraftment defect was uncovered in serial competitive transplantation experiments. The engraftment disadvantage of GRK6−/− cells, which was reproducibly observed during the first 4 weeks of each serial transplantation, coincides with the time of proliferative activity in BM [30,57]. We conclude that this engraftment defect represents a modest relative self-renewal defect of GRK6−/− HSCs under strong expansion stress. Because of this observation, we also looked at immature hematopoietic cells very early (day 8) after transplantation, to assess potential differences in HSPC content or cycling between GRK6−/− and WT cells. While in those experiments no difference between WT and GRK6−/− HSPC was observed, we made two striking observations of interest beyond the GRK6 story: HSCs in BM were overwhelmingly quiescent, much in contrast to steady-state BM in which 40% of cells are in non-G0, and characterized by high CXCR4 expression. The opposite phenotype—markedly enhanced cycling of HSCs compared to steady-state—was observed in spleens on day 8 after transplantation. In aggregate, these data suggest that the first burst of mature cells indicating engraftment emanate from spleens, whereas BM regenerates much more slowly. Except for the above-described reduction in competitiveness, BM hematopoiesis was quite normal also under stress conditions, despite the considerable functional impairment of GRK6−/− cells in vitro.
An unusual phenotype was observed with respect to immature hematopoiesis in the spleen: HSCs were considerably expanded in GRK6−/− mice or WT recipients of GRK6−/− HSC transplants. Such phenotypes were previously described in mice with deficient HSC retention in BM, and explained by splenic uptake of circulating HSCs [4,5,47]. In the case of GRK6 ablation, however, this defect was clearly independent of a retention defect, suggesting differential sensitivity of GRK6−/− and WT hematopoietic cells to factors facilitating HSC survival and/or expansion in spleen. The effect was observed both in homeostatic mice and in recipients of stem cell transplants of GRK6−/− cells, indicative of hematopoietic cell-intrinsic functions. Functionally, the expanded HSPC pool in spleens was apparently irrelevant: Stable hematopoiesis in splenectomized hosts provided direct evidence that the large splenic HSPC pool is not critical for normal stable hematopoietic function during homeostasis or stress.
Changes in immature hematopoiesis associated with age have been described previously [35 –37,58] and were similarly observed by us. Differences between WT and GRK6−/− mice became more apparent with age, in that the increase in BM-CFU-C contents compared to young mice was markedly less pronounced in GRK6−/− than in WT mice (Fig. 2C). By contrast, CFU-C contents and the number of spontaneously circulating CFU-Cs were slightly, albeit not significantly, increased in aged GRK6−/− mice, both relative to young GRK6−/− mice and aged WT mice. The relatively less increased BM progenitor cell content of the aged GRK6−/− mice compared with the increase in aged WT mice is compatible with a modestly reduced self-renewal capacity in keeping with the observations in our competitive engraftment experiments (Fig. 5E).
Because of the crucial contribution of the CXCL12/CXCR4 axis in G-CSF-mediated mobilization [48,59] and the possibility to target CXCR4 directly for mobilization [1,2,28], HSPC mobilization was also tested in the GRK6−/− model. Mobilization with G-CSF or the CXCR4 antagonist AMD3100 were equally efficient in GRK6−/− and WT mice. The reportedly impaired neutrophil mobilization [18] could not be reproduced. Observations in normal and splenectomized WT or GRK6−/− mice's response to mobilizing agents are also of relevance with respect to the origin of mobilized HSPCs from spleen versus BM: HSPC mobilization responses were the same in GRK6−/− and normal mice, whether spleen-competent or splenectomized, even though the HSPC content of GRK6−/− spleens was at least twice as high as that of WT spleens. On the weight of the evidence we conclude that the spleen is not a relevant source of mobilized HSPCs at least in response to CXCR4 antagonists, a conclusion that is in agreement with previously published data [1].
In summary, GRK6-deficient primary immature hematopoietic cells are characterized by the expected altered signaling phenotype in vitro. Compared to these, but also to the strong effects of GRK6 ablation on mature leukocyte function [17,27], perturbations of immature hematopoiesis in vivo were relatively modest. Expansion of immature cell pools in spleen were observed during homeostasis and further accentuated after hematopoietic stress in aged mice. Partly reversible deficits in competitiveness in engraftment assays became apparent during the initial weeks after transplantation. These data demonstrate that, while the CXCR4/CXCL12 signaling pathway is of high importance for normal function of immature hematopoiesis, ligand-induced phosphorylation of CXCR4 by GRK6 does not critically contribute to this.
Footnotes
Acknowledgments
Robert Lefkowitz's (Duke University, Durham, NC) generous gift of GRK+/− mice is kindly acknowledged. The studies are part of the Ph.D. thesis of DC. Some of the data were previously presented at the 2011 annual meeting of the American Society of Hematology in San Diego, CA.
Studies were funded by Deutsche Krebshilfe grant 108031 and Deutsche Forschungsgemeinschaft grant BO3553/1-1 to HB. HB is a member of the LOEWE Cell and Gene Therapy Frankfurt faculty, funded by Hessian Ministry of Higher Education, Research and the Arts ref. no.: III L 4-518/17.004 (2010).
Author Disclosure Statement
None of the authors have conflicts of interest to declare.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.