
Abstract
Mesenchymal stromal cells (MSCs) are multipotent cells that can be differentiated in vitro into a variety of cell types, including adipocytes or osteoblasts. Our recent studies indicated that a high expression of CD146 on MSCs from bone marrow correlates with their robust osteogenic differentiation potential. We therefore investigated if expression of CD146 on MSCs from the placenta correlates with a similar osteogenic differentiation potential. The MSCs were isolated specifically from the endometrial and fetal parts of human term placenta and expanded in separate cultures and compared with MSCs from bone marrow as controls. The expression of cell surface antigens was investigated by flow cytometry. Differentiation of MSCs was documented by cytochemistry and analysis of typical lineage marker genes. CD146-positive MSCs were separated from CD146-negative cells by magnet-assisted cell sorts (MACS). We report that the expression of CD146 is associated with a higher osteogenic differentiation potential in human placenta-derived MSCs (pMSCs) and the CD146pos pMSCs generated a mineralized extracellular matrix, whereas the CD146neg pMSCs failed to do so. In contrast, adipogenic and chondrogenic differentiation of pMSCs was not different in CD146pos compared with CD146neg pMSCs. Upon enrichment of pMSCs by MACS, the CD146neg and CD146pos populations maintained their expression levels for this antigen for several passages in vitro. We conclude that CD146pos pMSCs either respond to osteogenic stimuli more vividly or, alternatively, CD146pos pMSCs present a pMSC subset that is predetermined to differentiate into osteoblasts.
Introduction
H
Some reagents, including monoclonal antibodies (mAb) W3D5 and W5C5 to SUSD2 [18], W8B2 (=MSCA-1 antigen) to TNAP [19], or to the molecules detectable by mAb clones W5C4, W12D1, or 58B1, generated distinct histogram patterns, indicating that bmMSCs ex vivo present as a complex blend of different cells [18,20,21]. These bmMSC subsets were further separated ex vivo by FACS, and the CD271+CD56+ subset had a more prominent chondrogenic potential compared with the CD271+CD56− fraction, whereas this CD271+CD56− population yielded more adipogenic cells, and the osteogenic potential of bmMSCs was high and not different in these fractions [15]. The CD271+ fraction of periosteum-derived progenitor cells contained the osteoblast precursor cells, which deposited a mineralized matrix, whereas the CD271− subset failed to do so [22]. For pMSCs, functionally different subsets of cells were not defined or separated to a comparable extent.
Compared with bmMSCs, bulk MSCs or MSC subsets isolated from sources other than the bone marrow may differ in their regenerative potential [8]. We recently provided evidence that bmMSCs express CD146, alkaline phosphatase, and Runt-related transcription factor 2 (Runx2) significantly higher than pMSCs and this correlated with a better osteogenic differentiation capacity of bmMSCs [23,24]. CD146, also referred to as melanoma adhesion molecule (MCAM), cell surface glycoprotein MUC18, or S-endo1 antigen, is a cell adhesion molecule expressed on endothelial cells [25], B and T lymphocytes [26], smooth muscle cells [27,28], pericytes [29], and its expression has been described on MSCs from different sources [4,7,30 –34]. It binds to laminin α4 and thereby contributes to the interaction of cells to the basal lamina of vessels [35] and to the evasation of cells [36].
CD146 has been considered a key marker for differentiation-competent MSCs [4,33], but it has not been described as an antigen that can facilitate enrichment or depletion of MSCs based on their osteogenic differentiation capacity. We therefore extended our recent studies [23,24] and investigated if expression of CD146 on pMSCs also correlated with a higher osteogenic potential. In this study, we report that the CD146+ pMSCs generated osteoblasts in vitro upon osteogenic induction, whereas CD146− pMSCs failed to do so. This finding may be important for tissue engineering of soft tissue to reduce unwanted pro-osteogenic cells.
Materials and Methods
Preparation of MSCs
MSCs were isolated from normal, healthy human term placenta (from n>10 donors) as described recently [23,24]. The endometrial maternal part of the placenta was separated from the fetal part to enrich for maternal (pmMSC) and fetal (pfMSC) subsets of placenta MSCs (pMSCs). The tissue was minced and digested by proteolysis for 1h at 37°C (12 U/mL collagenase type XI, SIGMA-Aldrich, Taufkirchen, Germany; 2.4 U/mL Dispase II; Roche, Mannheim, Germany). Proteolysis was stopped by addition of FBS (final concentration 1% vol/vol), and debris was removed by filtration of the cell extract. Live mononuclear cells were enriched by gradient centrifugation (Ficoll, ρ=1.078 g/mL; GE Healthcare, Freiburg, Germany, 20 min 400 g, 20°C), washed twice with PBS, and incubated for 24 h (37°C, 5% CO2, humidified cell incubator) in complete MSC expansion medium (Lonza, Basle, Switzerland) or in a GMP-compliant medium containing 5% pooled human serum and 5% human platelet lysate (both from the Red Cross and Blood Bank at UKT, Tübingen), HEPES, heparin,
Bone marrow-derived MSCs (bmMSCs, from n>10 donors) were prepared as described recently [38] and expanded as illustrated above. All tissue samples were obtained from volunteers after informed consent. The study was approved by the Ethics Committee of the University of Tübingen.
Flow cytometry and cell sorts
Expression of cell surface antigens on MSCs was explored by flow cytometry as described recently [6]. The cells were detached by mild proteolysis (Accutase; Sigma-Aldrich) and counted with the aid of a hematocytometer, and 2×105 MSCs were incubated with diluted preimmune IgG (Gamunex (50 μL, 4°C, 20 min; Grifols, Barcelona, Spain) to reduce unspecific binding of antibodies [6], washed, and resuspended in 10 μL PFEA buffer (PBS containing 2% FBS, 2 mM EDTA, and 0.01% sodium azide). Then, the following mAbs were added to the MSCs according to the supplier's recommendations: anti-CD14-FITC (clone MΦP9; BD Bioscience, Franklin Lakes, NJ); anti-CD34-PE (clone 4H11; Biolegend, San Diego, CA); anti-CD45-APC (clone 2D1; R&D Systems, Minneapolis, MN); anti-CD73-PE (clone: AD2; BD Biosciences); anti-CD90-PE (clone: Thy-1A1; R&D Systems); anti-CD105-FITC (clone: SN6; AdD Serotec, Martinsried, FRG), anti-CD146 (clone: 128018; R&D Systems), or anti-SUSD2 (clone W5D5, generous gift of Dr. Bühring) [18]. After incubation for 30 min on ice, the cells were washed twice with cold PFEA buffer, and binding of unlabeled primary antibodies to the MSCs was visualized by counterstaining of the cells with fluorochrome-labeled goat anti-mouse IgG (30 min on ice, 1:100; Jackson ImmunoResearch, Newmarket, Suffolk, GB). Staining of cells with this secondary antibody alone served as the control.
The samples were analyzed by flow cytometry (LSRII; BD Biosciences). Before each experiment, mouse or hamster/rat κ-chain Comp Beads (BD Biosciences) were reacted with the corresponding fluorochrome-labeled antibodies and incubated for 20 min at room temperature in the dark. Negative Comp Beads were used as an unstained negative control (BD Biosciences). After washing with PFEA, the beads were resuspended in 200 μL PFEA to allow automatic compensation with the BD FACSDiva acquisition software. The MSCs were then measured accordingly. All data were processed and analyzed using FACSDiva and FlowJo 7.2.2 (Treestar, Inc., Ashland, OR) following recently updated guidelines [39]. Flow cytometry data were computed as geometrical means of fluorescence intensity (MFI).
Separation of MSCs by magnetic-activated cell sort
The MSCs were detached by Accutase as described above and washed in MACS® running buffer (0.5% BSA, 2 mM EDTA in PBS; Miltenyi Biotech, Bergisch Gladbach, FRG), and 107 MSCs were blocked by incubation of the cells in Gamunex (50 μL, 4°C, 20 min; Grifols). The MSCs were rinsed in MACS running buffer (Miltenyi Biotech) and reacted with the PE-labeled anti-CD146 mAB (clone: 128018; 10 μL mAB per 107 MSCs; R&D Systems). After washing, MSCs were reacted with 20 μL anti-PE magnetic beads, rinsed again, resuspended in MACS running buffer, and separated in an autoMACS separator (Miltenyi Biotech) as described by the supplier utilizing the programs, possel and possel s, for enrichment of the CD146pos MSCs. After magnetic separation, the MSCs were analyzed by flow cytometry for enrichment of CD146pos cells. Depending on the experimental setup, MSCs were then expanded in GMP-compliant medium in separate cultures (CD146high, CD146dim, CD146low) for later analyses or were incubated in differentiation medium to investigate their trilineage differentiation potential.
Determination of telomere lengths
The lengths of telomeres of pmMSCs and pfMSCs were determined by hybridization of FITC-labeled DNA oligonucleotides to the repetitive telomere motive [.TTAGGG(n).] with the aid of the Telomere PNA Kit following the instructions of the supplier (Dako, Glostrup, Denmark). As a reference, telomeres of T lymphoma 1301 were included. The nuclei of all cells were counterstained by propidium iodide. The mean of fluorescence intensity of FITC-labeled telomeres of MSCs was determined relative to the telomere length of T lymphoma 1301 (ie, 30 kb) by flow cytometry as described above.
Differentiation of MSCs in vitro and cytochemical staining
The differentiation of MSCs after expansion in the GMP-compliant medium was investigated as described recently [1]. Adipogenic differentiation of MSCs was induced by an adipogenic differentiation medium supplemented with 10 μg/mL insulin, 100 μM indomethacin, 0.5 mM isobutyl xanthine, and 1 μM dexamethasone. Osteogenic differentiation was achieved by an osteogenic differentiation medium containing 0.1 μM dexamethasone, 10 mM β-glycerophosphate, and 50 μM ascorbic acid, and chondrogenic differentiation was performed in micromass pellet cultures in a serum-free chondrogenic differentiation medium containing 50 mM ascorbic acid, 0.1 M L-proline, 1 mM dexamethasone, and 10 ng/mL TGF-β3. Unless stated otherwise, the MSCs were incubated in the respective differentiation media for 4 weeks. The differentiation media were refreshed twice a week, and progress of differentiation was monitored as described [40,41]; adipocytes were stained utilizing the Oil Red O method, mineralization by osteoblasts was detected by von Kossa staining, and generation of proteoglycans by chondrocytes was documented in sections of the micromass cultures by Alcian Blue dye.
Immunocytochemistry and immunohistochemistry
To detect expression of CD146 on human MSCs, immunocytochemistry (ICC) was performed. The MSCs were seeded in eight-well PCA chamber slides (Sarstedt, Nümbrecht, Germany) at a starting density of 15,000 MCSs/well and inoculated for 18 h to reach the optimal density. Then, the cells were washed twice with cold PBS, fixed (cold ethanol, 15 min), and washed twice with PBS again. Then, cells were incubated for 1 h at ambient temperature with blocking solution (1% BSA in PBS), followed by incubation in antibody staining solution to detect CD146 (anti-CD146-PE, mouse mAB IgG1, clone 128018, 1:100 in PBS/BSA; R&D Systems) or Thy-1 (anti-CD90-FITC, mouse mAb IgG2a, clone Thy1A1, 1:100 in PBS/BSA; Dianova, Hamburg, Germany), for 4 h at ambient temperature in a humidified chamber in the dark.
To detect CD146+ cells in the placenta, in situ immunohistochemistry (IHC) was performed. The tissue samples were prepared, soaked in Tissue Tec (Sakura Finetek, Alphen aan den Rijn, The Netherlands), shock-frozen in liquid nitrogen, and stored in airtight tubes for later use at −70°C. Cryosections were prepared (7 mm, −15°C) and the samples were mounted on SuperFrost slides (Menzel, Braunschweig, Germany), fixed with cold acetone (−20°C), air-dried, and stained. The samples were washed twice with PBS, preincubated with 1% BSA in PBS, and then stained with PE-labeled anti-CD146 mAb (clone 128018, 1:100; R&D Systems) and FITC-labeled anti-NG2 mAb (mouse mAB IgG1, clone LHM2, 1: 200 in 1% BSA/PBS; Santa Cruz Biotechnology, Dallas, TX). After incubation (16 h, 4°C, humidified chamber), unbound primary antibodies were rinsed away and the samples were incubated for 30 min in BSA/PBS enriched with DAPI (1:500) to visualize the nuclei. Staining with PE-conjugated goat anti-mouse IgG (1:100 in 1%BSA/PBS; Jackson ImmunoResearch) alone served as the control. The slides were covered, sealed with clear nail polish, evaluated, and recorded microscopically (Zeiss, LSM, Oberkochen, Germany) utilizing the AxioVision and Zen software (Zeiss).
Statistics
To obtain the mean values of results from individual experiments, all data were processed by spreadsheet software (Excel®, Microsoft). To evaluate differences between experimental groups, a two-sided t-test was performed, and probability values (P) below 0.05 were considered to indicate statistical significance.
Results
Characterization of human term placenta-derived MSCs
The MSCs were isolated from human term placenta as described recently [23,24] and characterized [3,5]. MSCs from the maternal and fetal parts expressed CD73, CD90, and CD105, but not CD14, CD34, or CD45. There was no difference in expression of these MSC marker antigens between placenta-derived maternal MSCs (pmMSC, Fig. 1A–F) versus placenta-derived fetal MSCs (pfMSC, Fig. 1A′–F′). However, the proliferation rates of pfMSCs were higher (20%, P<0.03, n=5 each), telomere lengths were longer (1.29-fold, P<0.008, n=4 each), and expression of the SUSD2 protein was elevated (P<0.04, n=9, not shown) in pfMSCs compared with the corresponding pmMSCs, corroborating that two distinct pMSC populations were prepared from human term placenta.
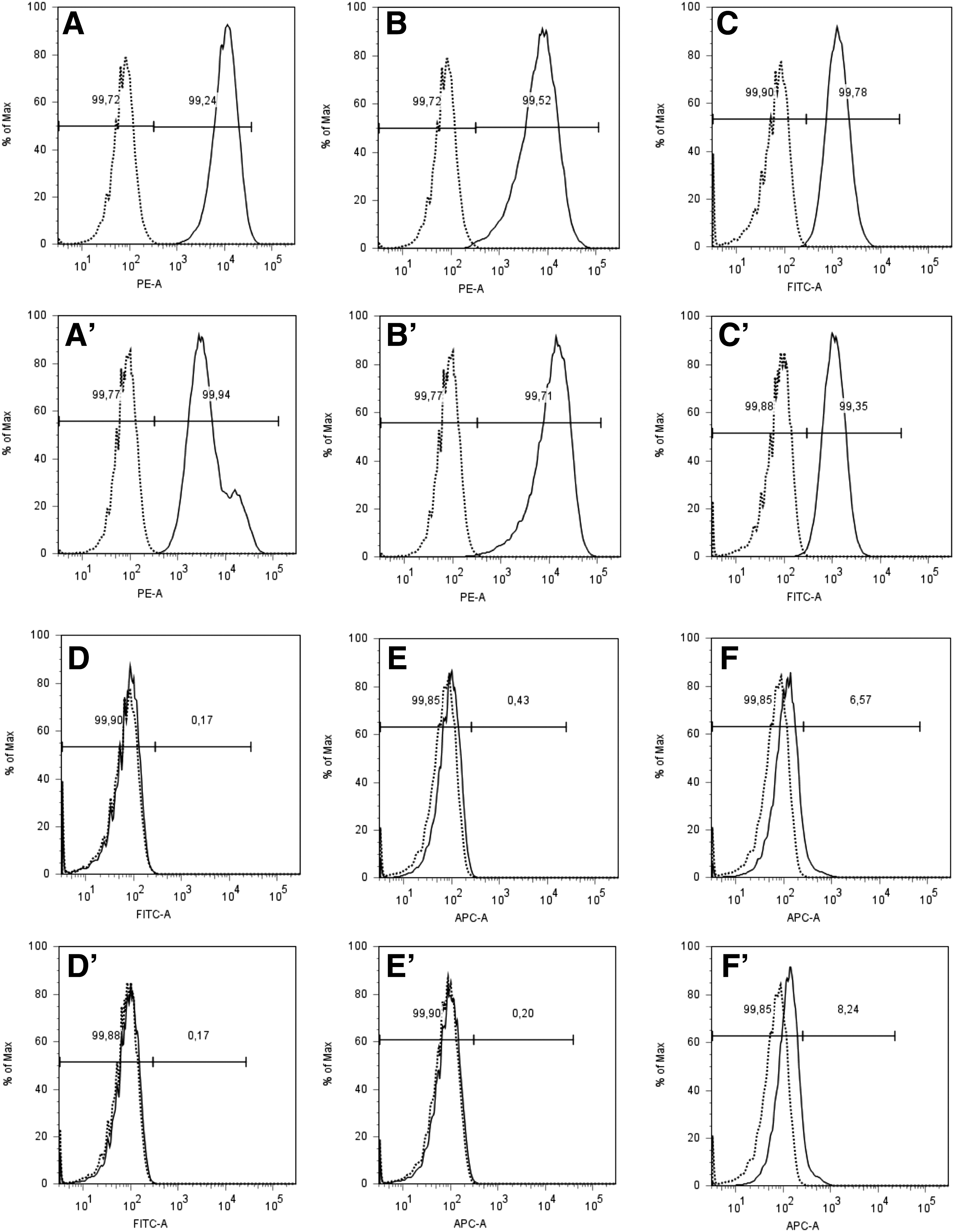
Expression of cell surface antigens on human placenta-derived mesenchymal stromal cells (pMSCs). MSCs were detached and incubated with mAB to CD73
To investigate the differentiation potential of pmMSCs and pfMSCs, adipogenic, osteogenic, and chondrogenic differentiation were induced in vitro. Adipocytic differentiation of pmMSCs (Fig. 2B) and pfMSCs (Fig. 2B′) was visualized by Oil Red O staining, osteogenic differentiation was detected by von Kossa staining (Fig. 2C, C′), and chondrogenesis by Alcian Blue staining of micromasses (Fig. 2D, D′). Differentiation of bmMSCs served as controls (Fig. 2B°–D°). Overall, there was no obvious difference in adipogenic, osteogenic, or chondrogenic differentiation between pmMSCs and pfMSCs. However, the osteogenic differentiation potential of pmMSCs and pfMSCs (Fig. 2C, C′) was rather low compared with bmMSCs (Fig. 2C°), confirming our recent results [23,24].
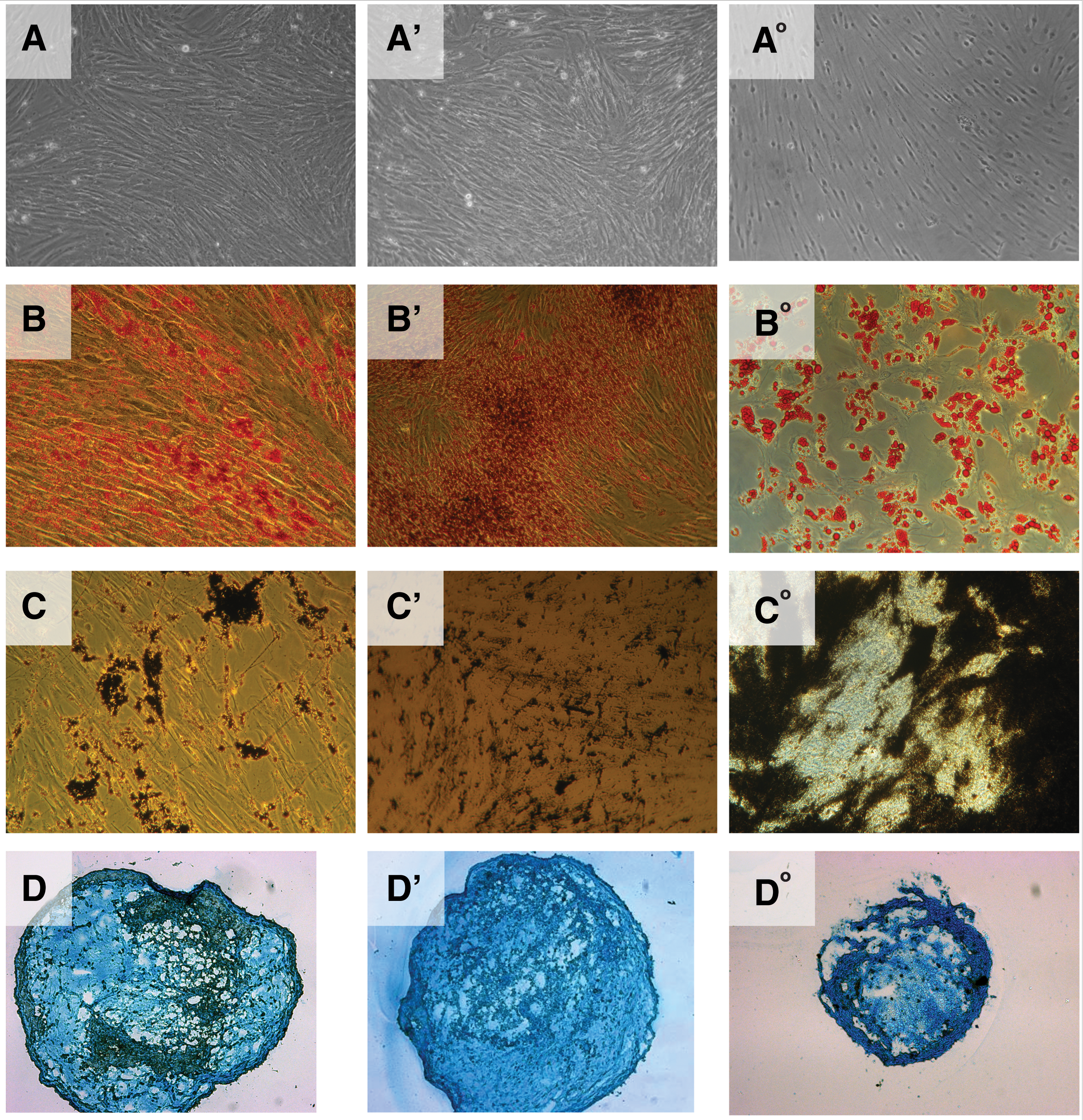
Exploring the trilineage differentiation capacity of human pMSCs. MSCs were harvested and incubated in six-well plates (osteogenesis, adipogenesis) or round-bottom 96-well plates to induce differentiation by addition of the respective differentiation media for 4 weeks. MSCs incubated in expansion medium served as control cells
Expression of CD146 on placenta-derived MSCs
Next, we investigated the expression of CD146 on pMSCs over time. Early passage pmMSCs and pfMSCs were separated by magnetic sort (MACS) to generate CD146high, CD146dim, and CD146low fractions, and the efficacy of the separation was confirmed by flow cytometry immediately after cell sorting (Fig. 3). The pmMSCs and pfMSCs, which were not retained in the MACS columns by magnetic anti-CD146 mAb, were confirmed not to display a robust CD146 signal by flow cytometry (Fig. 3A, A′). The CD146dim and CD146high fractions of pmMSCs (Fig. 3B, C) and pfMSCs (Fig. 3B′, C′) yielded higher fluorescence intensities, indicating that the MACS technology enriched CD146pos and CD146low subsets from suitable pMSC populations with sufficient efficacy.
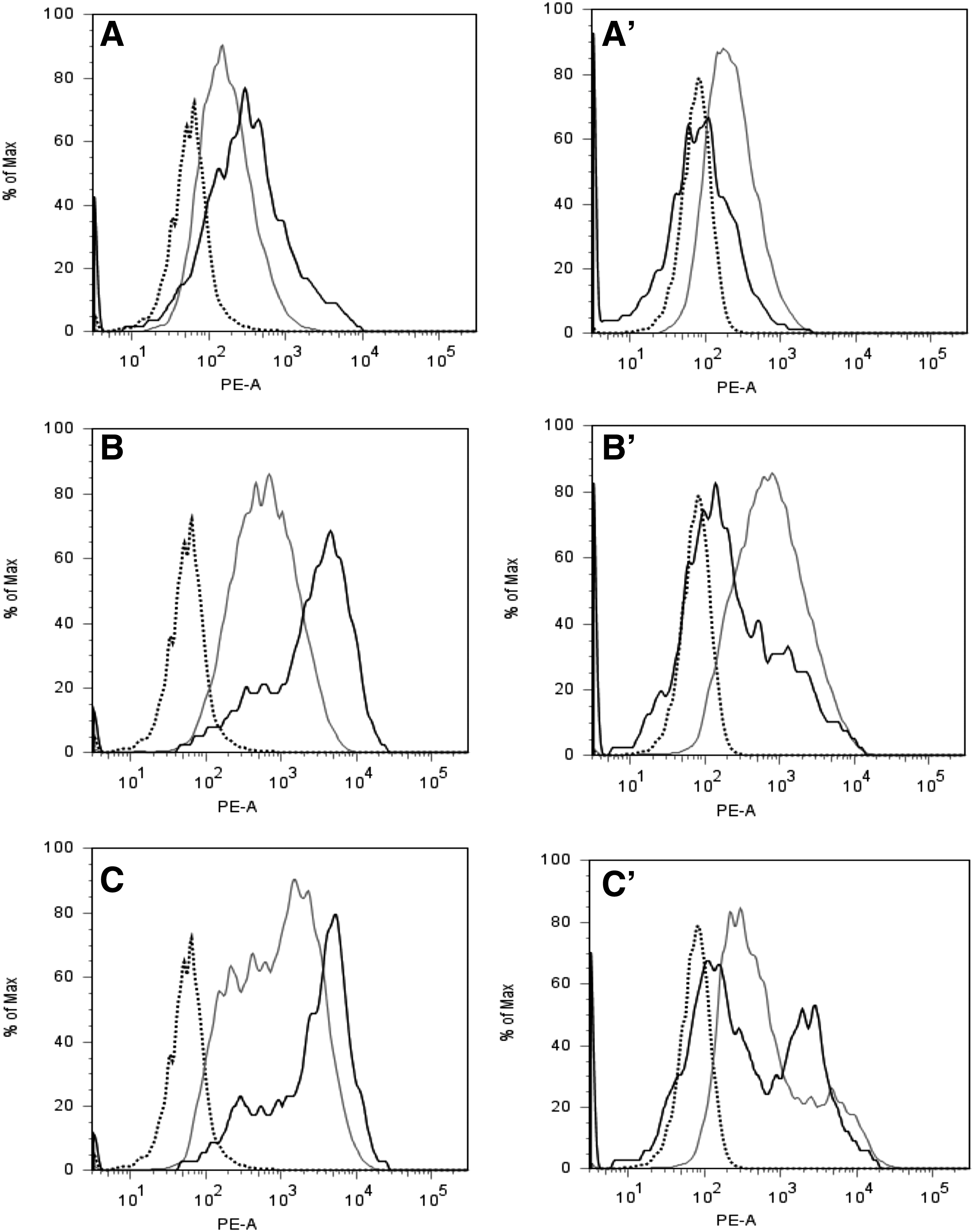
Separation of pMSC subsets by a monoclonal antibody to CD146. Human pmMSCs
As expression of CD146 was possibly changing in time during continued culture of MSCs in vitro, the CD146high, CD146dim, and CD146low fractions of pmMSCs and pfMSCs were expanded over four more passages in individual cultures to investigate if the expression of CD146 changed significantly over time. The results indicated that CD146low MSCs did not acquire a high expression of this cell surface antigen, whereas the CD146dim or CD146high pMSCs maintained expression of CD146 (Fig. 3).
Differentiation potential of CD146high and CD146low placenta-derived MSCs
To investigate if the expression of CD146 on pMSCs correlated with their osteogenic differentiation potential, pmMSCs and pfMSCs were separated by MACS to generate CD146high and CD146low subsets (Fig. 3) and expanded in individual cultures. Spontaneous osteogenesis was not observed in the CD146high subsets of pmMSCs (Fig. 4A, B) and pfMSCs (Fig. 4A′, B′), but the CD146high subsets of pmMSCs (Fig. 4C, D) and pfMSCs (Fig. 4C′, D′) underwent moderate osteogenic differentiation in the osteogenic differentiation medium and mineralization was observed. However, CD146high pfMSCs and pmMSCs did not deposit the significant amounts of mineralized extracellular matrix after induction of osteogenesis to yield the intensities and patterns of von Kossa staining that were observed with bmMSCs (Fig. 2C°). Similar to CD146high pMSC subsets, spontaneous osteogenesis was not recorded in the CD146low subsets of pmMSCs (Fig. 4E, F) and pfMSCs (Fig. 4E′, F′). In contrast to the CD146high pMSCs, mineralization following incubation in osteogenic differentiation medium was also not observed in the CD146low subsets of pmMSCs (Fig. 4G, H) and pfMSCs Fig. 4G′, H′).
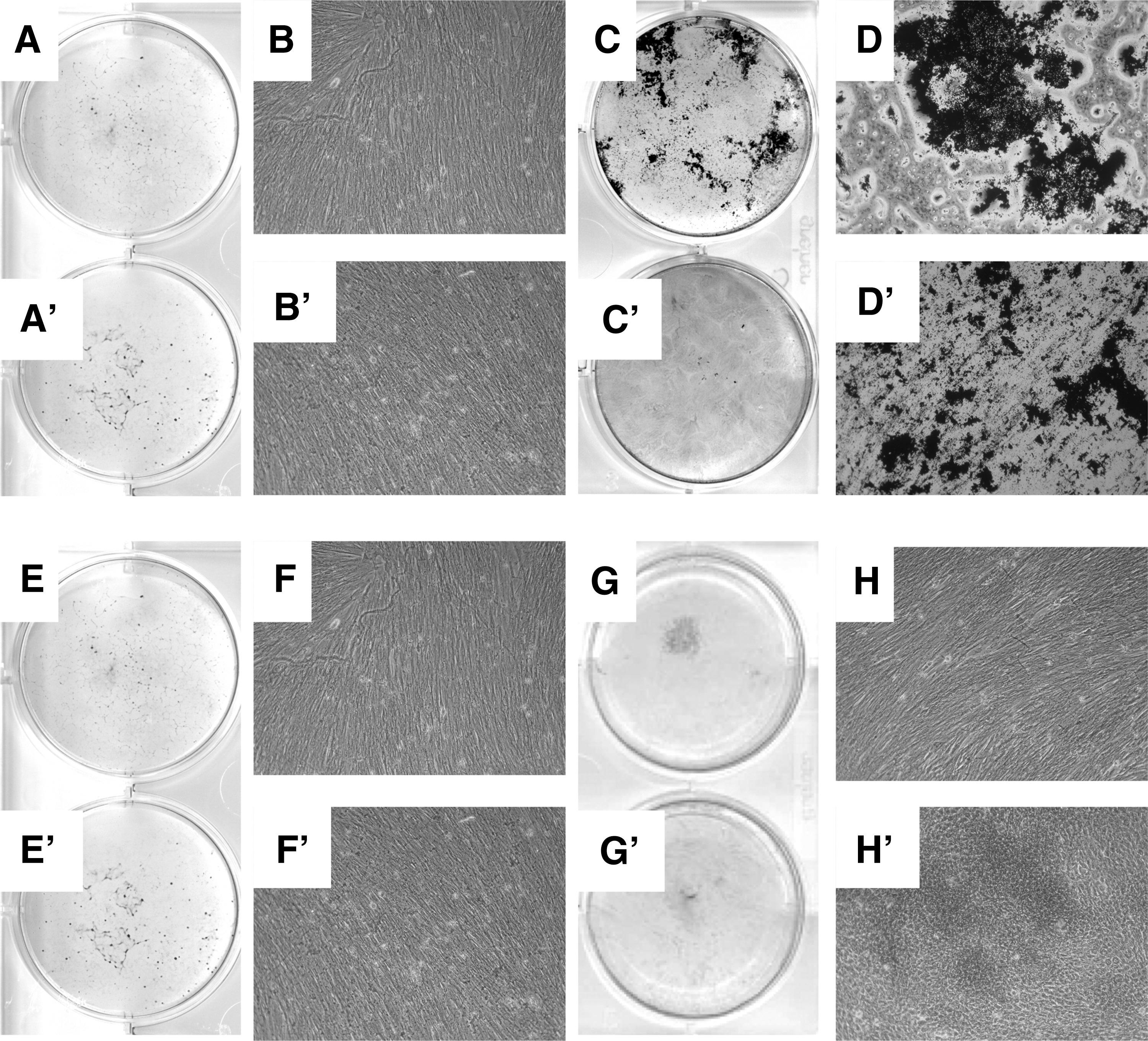
Exploring the osteogenic differentiaton in CD146high and CD146low subsets of pMSCs. The pmMSCs
To test if the general differentiation capacity of the CD146high versus the CD146low subsets of pMSCs was distinct, we next explored their adipogenic and chondrogenic differentiation capacities as well. Significant differences in adipogenesis (Fig. 5A–C) and chondrogenesis (Fig. 5D–F) were not seen in bulk pMSCs (Fig. A, D) compared with both the CD146high (Fig. B, E) and CD146low (Fig. D, F) subsets of pMSCs. We conclude that the expression of CD146 on pMSCs correlates specifically with an elevated osteogenic differentiation potential.
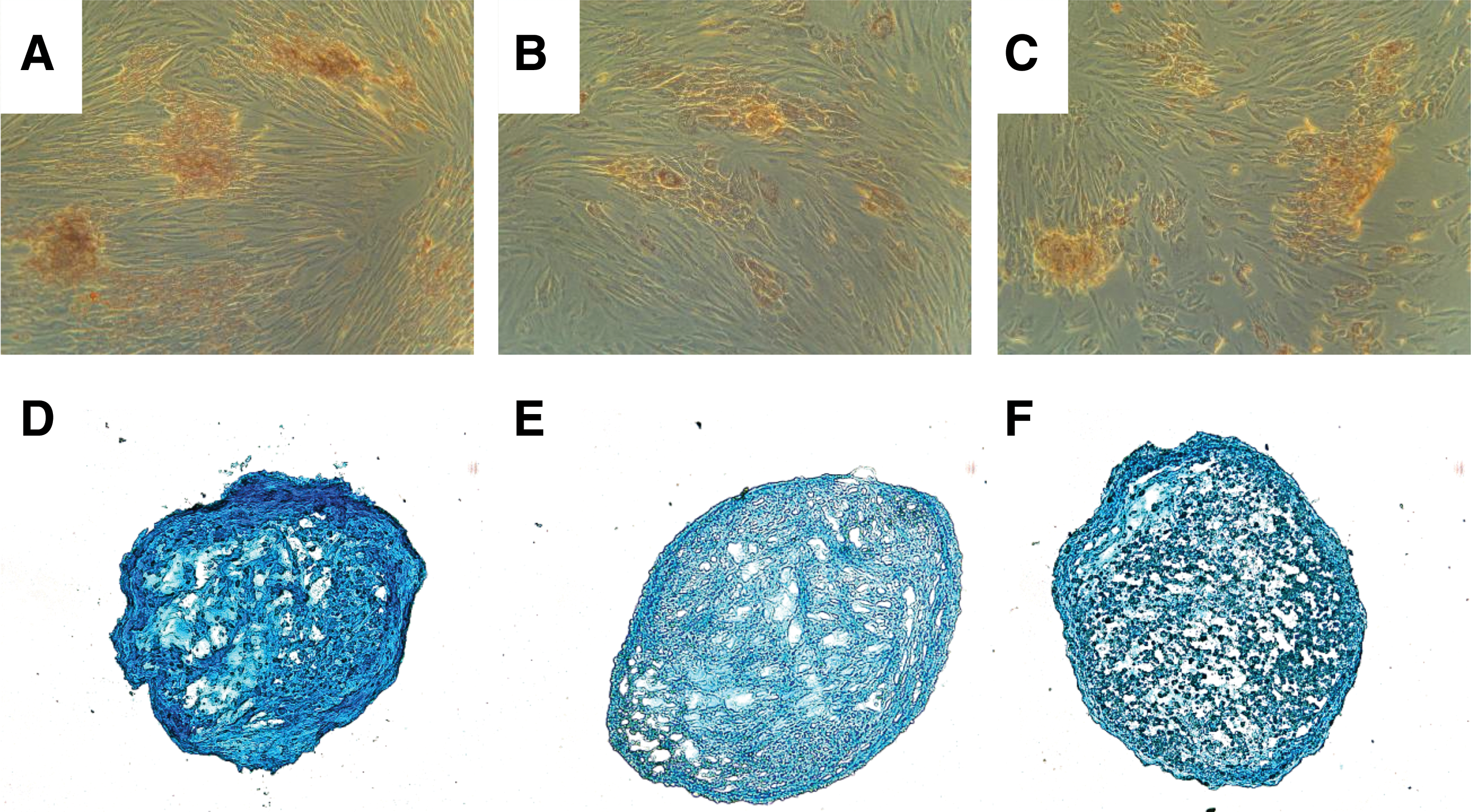
Exploring the adipogenic and chondrogenic differentiaton potential in CD146high and CD146low pMSCs. pMSCs were expanded in vitro and either separated by MACS to generate CD146high and CD146low subsets or maintained as unseparated bulk cultures. Adipogenic or chondrogenic differentiation was induced by addition of the specific differentiation media for 4 weeks, and success of differentiation of unseparated bulk pMSCs
Detection of CD146 on human MSCs in vitro and its localization in situ
The expression of CD146 on characterized human pMSCs in vitro was visualized by ICC on cells in the second passage of in vitro culture (Fig. 6). However, ICC is a qualitative method and signal intensities obtained by ICC do not allow an exact quantification of protein expression. Expression of CD146 in placenta tissue was also explored by IHC, and cells surrounding small vessels were stained with NG2, an antibody reactive with pericytes, MSCs, and related mesenchymal cells [4,28,31]. Expression of CD146 was confirmed in placenta tissue in close proximity of NG2 signals, suggesting that perivascular cells expressed CD146 in situ (Fig. 6).
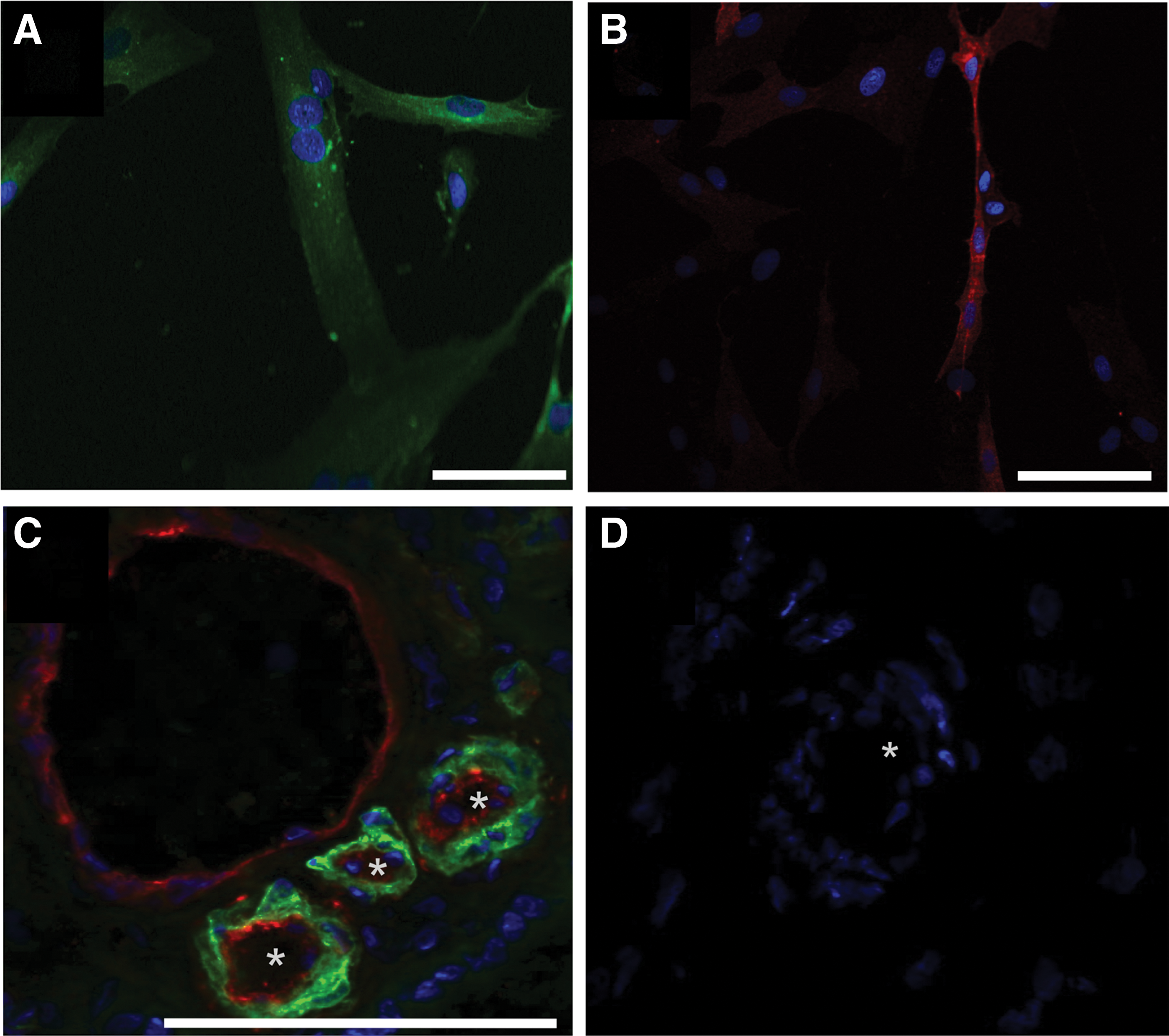
Detection of CD146 on MSCs in vitro and its localization in placenta tissue. Human pMSCs were expanded and seeded in chamber slides (top), stained with FITC-labeled mAb to CD90
Discussion
CD146 was originally described to be primarily expressed at the intercellular junction of endothelial cells and plays a role in cell–cell and cell–matrix interaction and in angiogenesis [42]. Recent evidence indicates that CD146 is a multifaceted molecule actively involved in miscellaneous processes, such as development, signaling transduction, cell migration, and MSC differentiation [43]. Recently, CD146+ periodontal ligament stem cells (PDLCs) showed a greater proliferative and osteogenic potential than CD146− PDLCs [44]. Our previous studies provided evidence on differences in expression of CD146 and correlated this with an osteogenic potential [23]. This study complements these previous reports very well in several aspects. In this study, we confirm that expression of CD146 correlates with the osteogenic differentiation capacity not only in human bmMSCs as shown previously [2,4] but also in MSCs from human term placenta. This includes the MSCs derived from both the maternal and fetal sections of the placenta and is also in line with our recent studies [23,24]. For bmMSCs, CD146 has been considered to be an indicator of stemness since its expression correlated with the trilineage differentiation capacity of bmMSC clones [33]. However, for pMSCs, we did not observe significant differences in the adipogenic or chondrogenic differentiation of CD146pos compared with CD146neg pMSCs. In addition, in our recent experiments, all human bmMSCs expressed CD146 at a bright signal intensity [23,24]. We still attempted to separate human bmMSCs in CD146pos or CD146neg subsets by FACS, but functionally different subsets of bmMSCs were not obtained (data not shown), supporting recent results [28]. Therefore, the default setting for differentiation of bmMSCs tends to go toward the osteogenic lineage [2], and in about a third of samples investigated, spontaneous ossification of bmMSCs was observed upon injection of the cells in muscle tissue [45]. The ostensive conflict of results in these details—correlation of CD146 with osteogenesis, but to a lesser extent adipogenesis and chondrogenesis of MSCs—may be explained by a recent finding, which provided evidence that knockdown of CD146 in MSCs significantly reduced the expression of genes associated with osteogenesis and adipogenesis, but overexpression of CD146 enhanced osteogenesis without facilitating adipogenesis [46]. In this sense, CD146 seems not to describe a distinct lineage of MSCs, but its intensity of expression appears to be an indicator of its osteogenic differentiation potential.
In the present study, expression of CD146 was detected on human MSCs in situ, ex vivo, and after primary culture. Expression of CD146 was previously shown to be elevated on bmMSCs [4] and also expressed on pMSCs [23], but was not detected on fibroblasts [4]. Therefore, this antigen may allow the discrimination of differentiation-competent MSCs from contaminating fibroblasts. Expression of CD146 on bmMSCs and pMSCs does vary among samples from different donors and also depends to some degree on cell culture conditions [23,47]. Enrichment of CD146high bmMSCs yielded cells with a significantly elevated production of proteoglycans compared with the CD146low subset, whereas osteogenesis and adipogenesis remained unchanged [47]. In our hands, separation of CD146high pMSCs from CD146low pMSCs facilitated an enrichment of osteogenic, but not adipogenic or chondrogenic, subsets of pMSCs. Recently, others reported that CD146+ endometrial stem cells gave rise not only to mesenchymal cells such osteoblast and adipocytes but may also have an even wider differentiation capacity [48]. This suggests that CD146 plays a role in defining functional subsets of MSCs at least upon expansion in vitro. At the same time, it also suggests that MSCs from different niches or tissues differ to some degree in their expression of receptors and the processing of such receptor signals.
Technical differences, such as the composition of the expansion media in the experimental conditions, may also explain the dissimilarities in results obtained. Adult stem cells typically remain in a quiescent stage and do not necessarily proliferate [49 –52]. Cloning of MSCs from primary culture may therefore yield clones of cells different from bulk cultures as overgrowth of mitotically active cells over slow growing cells is avoided. Therefore, by cloning MSCs ex vivo or early on in vitro, different subtypes within the MSC population may be resolved [28,33]. In addition, in many investigations, media containing bovine serum were employed for expansion of bmMSCs [28,32,33], and it is known that the composition of the expansion media influences the phenotype, mitotic activity, and differentiation potential of such cells considerably [37,41,53 –55]. We expanded the MSCs in a fully GMP-compliant medium complemented with human serum and platelet lysate [37]. We therefore may yield primarily trilineage-competent CD146pos bmMSCs and at the same time may lose the less differentiation-competent CD146low bmMSCs or other CD146− mesenchymal cells that possibly contaminated the bulk MSC cultures [4].
Distinct subsets of bmMSCs have been described in ex vivo analyses [12,15,18
–20,56] as well as after expansion in vitro [33,53,57] and by in vivo functional analyses [2]. Expression of CD271, alkaline phosphatase (TNAP/MSCA-1), and CD56 on human bmMSCs ex vivo correlated with higher clonogenic and chondrogenic capacities, whereas the CD271
Specific differences between bulk MSCs isolated from different sources such as bone marrow versus adipose tissue, placenta, and others have been described as well [1,5,13,16,30,32,58 –62], and phenotypic changes during continued in vitro culture were noticed early on [63,64]. Therefore, the correlation of a distinct cell surface marker with a given MSC population may be related to the source of the cell as well, not to mention interindividual differences between human donors or between cells obtained from different species.
In contrast to bmMSCs, which presented as CD146high cells in our hands, MSCs from human term placenta resulted in a larger population of CD146dim or CD146low cells as detected in in vitro cultures [6]. The isolation of MSCs from solid tissues requires several steps of dissection and mincing, followed by enzymatic degradation, filtration, and purification. It is very likely that fibroblast-like cells, smooth muscle cells, or pericytes may be prepared when MSCs are isolated from such sources. By microscopy and flow cytometry, contaminating mesenchymal cells cannot be discriminated from true MSCs when simple criteria are administered to define the stem cells [3]. Sophisticated analyses exploring the expression of several cell surface antigens or lineage markers may help to better define truly differentiation-competent MSCs [31,32]. The CD146low cells investigated in this study seem to be MSC-like cells with a two-lineage differentiation potential as the CD146low pMSCs generated chondrocytes and adipocytes, but not osteoblasts in vitro. Accordingly, the CD146high cells represent the MSCs with a trilineage differentiation capacity. Again, this is in line with the results reported from bmMSCs and PDLCs recently [4,33,44].
A technical aspect not discussed yet also merits critical examination. The preparation of subsets of human MSCs by FACS or MACS clearly differs in the yield of cells obtained by separation, in efficacy and purity of the subsets generated, and the viability of the cells acquired. For this study, the MACS technique was preferred as the yield and viability of pMSCs were higher, but, of course, at the cost of the purity of the cells gained. This decision was mainly made due to the fact that many cells were needed for the subsequent expansion and differentiation experiments. In particular, chondrogenesis required many cells for the micromass cell cultures. Therefore, in this study, the term separation does not mean an absolute distinction between CD146pos and CD146neg cells, but it rather reflects a relative enrichment of one over the other population. In contrast to many other lineage markers, including CD73, CD90, and CD105, expression of CD146 on human pMSCs generated in many samples a somewhat wide histogram [23], indicating that different cells expressed this antigen to a quite variable extent. A stringent sorting by FACS might dissect functional subsets, but this requires prolonged expansion to generate the number of cells needed. Still, to the best of our knowledge, we show for the first time that CD146 can be used to functionally separate pMSCs into an osteogenic subset and a nonosteogenic subset. The expression of CD146 on pMSCs seems to be stable at least over a few passages as seen in this study. This finding grants new aspects for tissue engineering or clinical purposes: if osteogenic cells have to be depleted (eg, in soft tissue engineering/regenerative medicine), the pro-osteogenic cells can be reduced by MACS or FACS using a CD146 mAb without compromising too much on proadipogenic or chondrogenic cells. Whether this applies also for differentiation of pMSCs in other cells, such as smooth muscle cells, for instance, remains to be explored. In animal studies, spontaneous heterotopic ossification was observed upon injection of bulk MSCs [4,45]. Unwanted osteogenesis can possibly be ameliorated by functional enrichment of MSC subsets. However, selection of defined MSC subsets may require extended expansion of the cells in vitro. However, the differentiation potential, and here especially the chondrogenesis, may be significantly compromised in MSCs at later stages of expansion in vitro [63]. This has to be taken into account when planning MSC-based therapies.
Summary and Conclusion
Bone marrow-derived MSCs have been enriched ex vivo to obtain more adipogenic or chondrogenic subsets by mAB to CD271, TNAP1, and CD56 [15]. However, to the best of our knowledge, enrichment or depletion of osteogenic cells from bulk pMSCs by a monoclonal antibody has not been reported. We report on a functional difference in CD146pos compared with CD146neg human pMSCs regarding their osteogenic differentiation potential and show that CD146pos pMSCs generated osteoblasts in vitro upon osteogenic induction, whereas CD146neg pMSCs failed to do so. Furthermore, the CD146low pMSCs did not gain expression of the cell surface antigen, nor did CD146pos pMSCs lose its expression after prolonged expansion in vitro. We therefore conclude that CD146pos and CD146neg pMSCs represent two subsets of cells that share the chondrogenic and adipogenic differentiation capacities and can be functionally separated to enrich or deplete osteogenic pMSCs. Such separations of MSCs into functionally distinct subsets can be advantageous when osteogenesis and mineralization cause an adverse problem in tissue engineering or in regenerative medicine studies aiming at restoration of soft tissue.
Footnotes
Acknowledgments
The authors thank their colleagues in the hospitals for taking the extra time during and after surgery to collect, wrap, and ship biopsy samples, which were key elements for our studies, and Chaim Goziga for help in preparation of the artwork. This project was supported by grants to WKA from the BMBF (ReGiNA TP1) and the DFG (KFO273) and, in part, by institutional funding.
Author Disclosure Statement
In the name of all coauthors, we declare that there are no actual or potential conflicts of interest to be disclosed for any of the authors of this investigation and manuscript.