
Abstract
After organ transplantation, recipient T cells contribute to graft rejection. Mesenchymal stromal cells from the bone marrow (BM-MSCs) are known to suppress allogeneic T-cell responses, suggesting a possible clinical application of MSCs in organ transplantation. Human liver grafts harbor resident populations of MSCs (L-MSCs). We aimed to determine the immunosuppressive effects of these graft-derived MSCs on allogeneic T-cell responses and to compare these with the effects of BM-MSCs. BM-MSCs were harvested from aspirates and L-MSCs from liver graft perfusates. We cultured them for 21 days and compared their suppressive effects with the effects of BM-MSCs on allogeneic T-cell responses. Proliferation, cytotoxic degranulation, and interferon-gamma production of alloreactive T cells were more potently suppressed by L-MSCs than BM-MSCs. Suppression was mediated by both cell–cell contact and secreted factors. In addition, L-MSCs showed ex vivo a higher expression of PD-L1 than BM-MSCs, which was associated with inhibition of T-cell proliferation and cytotoxic degranulation in vitro. Blocking PD-L1 partly abrogated the inhibition of cytotoxic degranulation by L-MSCs. In addition, blocking indoleamine 2,3-dioxygenase partly abrogated the inhibitive effects of L-MSCs, but not BM-MSCs, on T-cell proliferation. In conclusion, liver graft-derived MSC suppression of allogeneic T-cell responses is stronger than BM-MSCs, which may be related to in situ priming and mobilization from the graft. These graft-derived MSCs may therefore be relevant in transplantation by promoting allohyporesponsiveness.
Introduction
M
It has been demonstrated that resting or naive MSCs are inherently capable of suppressing T-cell responses [9 –11]. Bone marrow MSCs (BM-MSCs) can suppress recipient alloreactive T-cell responses and thereby prevent graft rejection [12], suggesting a possible clinical application of MSCs in organ transplantation. Human liver grafts also contain MSCs (L-MSCs) [13,14] and these are mobilized from the liver graft during the transplantation procedure. MSCs express several cell surface receptors that enable them to sense the microenvironment and alter their phenotype accordingly [6,15]. Many studies have shown that it is the nature of the environmental cues that dictates the plasticity and—in the end—immunosuppressive capacity of MSCs (14–27). Most of the data that support this concept of MSC mobilization and immune priming are based on experimental mouse studies. However, because humans and mice differ in immunomodulatory pathways used by MSCs, there is an urgent need to assess the immunosuppressive function of human MSCs in the clinical setting of liver transplantation [16].
In addition, it is still unknown whether human graft-derived MSCs are able to suppress alloreactive T-cell responses as potently as their BM counterparts. Although the liver is an immunologically tolerogenic organ [17 –19], it is unknown whether MSCs in the healthy liver contribute to this tolerogenicity. The organ donation process is associated with a wide range of hemodynamic and inflammatory changes throughout the body [20,21] that may well affect the inherent immunosuppressive properties of MSCs in the liver graft. Inflammation affects pathways that can inhibit T-cell responses, such as the PD-L1/PD-1 pathway. PD-1 is an inhibitory receptor expressed on T cells, which inhibits T-cell responses after interaction with its ligand PD-L1 [22,23], which is expressed on antigen-presenting cells and MSCs. PD-L1 is known to be upregulated after exposure to inflammatory cytokines, such as interferon-gamma (IFN-γ) [24]. Another factor that is upregulated in MSCs in response to IFN-γ is indoleamine 2,3-dioxygenase (IDO) [25,26], which has been associated with the immunosuppressive capacity of MSCs. Therefore, these mechanisms may well contribute to the inhibition of alloreactive T cells by MSCs after liver transplantation. In addition, soluble factors secreted by MSCs, such as transforming growth factor-beta, hepatocyte growth factor [27], and prostaglandin E2 (PGE2) [9] may contribute to their capacity to inhibit alloreactive T-cell responses as well.
In the present study, we aimed to assess whether graft-derived MSCs can suppress alloreactive T-cell responses and, if so, whether they suppress more potently than their BM counterparts. To assess this, we isolated L-MSCs from liver perfusates obtained from donor livers and added these cells to mixed lymphocyte reactions (MLRs). We measured proliferation and effector function of alloreactive T cells in these cocultures in the presence or absence of L-MSCs. These outcomes were compared with those of cocultures with BM-MSCs.
Materials and Methods
Ethics statement
Liver perfusates and splenocytes were obtained from deceased donors following organ donation. The use of all human materials was approved by the Medical Ethical Committee of the Erasmus MC-University Medical Center, Rotterdam, and the study was performed in accordance with the amended Declaration of Helsinki.
Isolation and culture of MSCs
BM-MSCs were harvested from aspirated BM as previously described [28]. L-MSCs were isolated from perfusates of human liver grafts. Liver perfusates were collected from liver graft donors during the organ donation procedure by flushing the transplant liver with 1 L of University of Wisconsin preservation solution, followed by a second flush with 400 mL human albumin during the back table bench procedure, before implantation, as previously described [14]. Liver perfusates were collected and centrifuged (1500 rpm, 4°C; 10 min) to pellet cells. The cell pellet was resuspended in phosphate-buffered saline (PBS) and mononuclear cells (MNCs) were isolated using Ficoll-Hypaque density gradient separation. MNCs were counted and resuspended in culture medium consisting of alpha-minimum essential medium (MEM)/GLUTAMAX (Gibco), 2% fetal bovine serum (FBS), 1% antibiotic–antimycotic (Gibco) supplemented with 20 ng/mL of recombinant human epidermal growth factor (eBioscience) and 10 ng/mL of recombinant human fibroblast growth factor 2 (eBioscience), plated at 1.0×105 cells/cm2 in 10-cm dishes, and cultured at 37°C, 5% CO2. After 3 days, the nonadherent fraction was removed and the medium was replaced by fresh medium. Cells were cultured until colonies became confluent and were harvested using a nonenzymatic solution (TrypLE; Gibco) and further expanded for later use. Early passage cells from BM were cultured and expanded in the same culture conditions as described for L-MSCs.
Immunophenotypic profile of MSCs in liver perfusates and BM-MSCs
To detect the presence of mobilized MSCs and hematopoietic stem/progenitor cells (HSCs) in liver perfusates from deceased donors, MNCs collected from the liver perfusates, as described above, were counted for viability and stained to detect and quantify MSCs. For HSCs, MNCs were stained with the following antibodies: lineage-FITC (BD Pharmingen), CD34-PE (BD Pharmingen), CD38-APC-Cy7 (eBioscience), CD45RA-PB (eBioscience), and CD90-APC (eBioscience). For MSCs, MNCs were stained with the following antibodies: CD45-PB (eBioscience), CD146-PE (eBioscience), CD44-APC-Cy7 (eBioscience), CD73-APC (eBioscience), CD105-PE (eBioscience), CD90-FITC (eBioscience), CD14-PB (eBioscience), CD19-APC-Cy7 (eBioscience), HLA-ABC-FITC (eBioscience), and HLA-DR-APC (eBioscience). The same antibodies were used to stain BM-MSCs to compare the immunophenotypic profile of L-MSCs and BM-MSCs. The immunophenotypic profile of L-MSCs and BM-MSCs was carried out on early passage (P3) cells. Before addition of antibodies, cells were treated with Fc block (Miltenyi). Cells were stained at 4°C in the dark for 30 min. Afterward, cells were washed, centrifuged, and resuspended in PBS before flow cytometric analysis. All cells were incubated with 7-aminoactinomycin D (7-AAD) (eBioscience) to discriminate dead/dying cells and debris. Samples were collected, and a minimum of 1 million cells were measured on a BD FACS Canto II and analyzed using FlowJo software (Tree Star). In the analyses, gates were based on fluorescence-minus-one controls.
Differentiation assays
Early passage (P3) L-MSCs were placed in defined culture conditions and compared with early passage (P3) BM-MSCs. For osteogenic differentiation, cells were plated at 4,000 cells/cm2 per well in a six-well dish (Corning) in regular culture medium as described above and placed in a humidified chamber with 5% CO2 at 37°C. After 24 h, the medium was changed to osteogenic induction medium [alpha-MEM/GLUTAMAX, 10% FBS, 1% penicillin–streptomycin, 50 μM ascorbic acid (Sigma), 10 mM β-glycerophosphate (Sigma), 100 nM dexamethasone (Calbiochem)]. To induce differentiation, half of the medium was replaced by fresh medium every 3 days for a total of 21 days. After 21 days, cell cultures were fixed with 4% paraformaldehyde (Sigma) for 15 min, washed twice with PBS, and stained with 40 mM Alizarin Red S stain at pH=4.1 (Sigma) for 30 min to detect mineralization.
For adipogenic induction, 1×105 cells were plated per well in a six-well dish in regular culture medium and placed in a humidified chamber with 5% CO2 at 37°C. After 24 h, the wells were washed with PBS and fresh adipogenic maintenance medium [Dulbecco modified Eagle medium (DMEM)/low glucose (Gibco), 10 μg/mL human insulin (Invitrogen), 10% FBS, and 1% penicillin/streptomycin] was added. After 3 days, the medium was changed to adipogenic induction medium [DMEM/low glucose, 10 μg/mL human insulin, 100 μM indomethacin (Sigma), 0.5 mM 3-isobutyl-l-methylxanthine (Sigma), and 1 μM dexamethasone]. After 3 days, the medium was changed back to adipogenic maintenance medium. After an additional two rounds of maintenance–induction media changes, cells were incubated for an additional 7 days in adipogenic maintenance medium. After this, cells were fixed with 4% paraformaldehyde and stained with Oil Red O (Sigma) to detect formation of lipid droplets.
Immune priming of MSCs with IFN-γ plus tumor necrosis factor alpha
Passage 3 culture-expanded L-MSCs and BM-MSCs were plated at 1.0×105 cells per well in a six-well dish in regular culture medium. Cells were grown for 48 h and afterward were growth arrested in serum-free media (RPMI 1640; Gibco) for 24 h. Following this, cells were treated with 10 ng/mL of recombinant human tumor necrosis factor alpha (TNF-α) and 10 ng/mL of recombinant human IFN-γ. After 24 h, the cell media were collected, frozen, and stored at −80°C until further use. The cells were stained with the following primary fluorescently conjugated antibodies: PD-L1-APC (eBioscience), CD73-FITC (eBioscience), CD45-PB (eBioscience), and HLA-DR-PE (eBioscience) in the presence of Fc blocking reagent (Miltenyi) at 4°C in the dark for 30 min. Cells were analyzed using a BD FACS Canto II Flow cytometer (BD Biosciences); 7-AAD was included to discriminate dead/dying cells and debris. A minimum of 10,000 events were collected and analyzed using FlowJo software (Tree Star).
MLRs in the presence or absence of MSCs
To test the suppressive capacity of MSCs on allogeneic T-cell responses, we performed MLRs, in which we stimulated carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled peripheral blood mononuclear cells (PBMCs) with allogeneic CD40 ligand-stimulated (CD40)-B cells in the presence or absence of L-MSCs or BM-MSCs. PBMCs were isolated from the blood of healthy blood bank donors using Ficoll-Hypaque density gradient centrifugation and cryopreserved at −135°C until further use. CD40-B cells, which were used as allogeneic stimulator cells, were expanded from organ donor splenocytes, as previously described [29]. For different experiments, different responder and stimulator combinations were used. Both PBMCs and CD40-B cells were thawed and recuperated overnight in B-cell medium [Iscove's modified Dulbecco's medium+10% human serum+1% penicillin/streptomycin (Gibco)+1% insulin/transferrin/selenium (Gibco)] at 37°C and 5% CO2. PBMCs were labeled with 0.5 μM CFSE (Invitrogen) according to the manufacturer's instructions, and CFSE-labeled PBMCs (1×105) were stimulated with 2×105 irradiated (30 Gy) donor CD40-B cells in 96-well U-bottom plates in a final volume of 250 μL B-cell medium. In separate wells, 1×104 or 2×104 irradiated (30 Gy) liver graft-derived L-MSCs or BM-MSCs were added to the cocultures. PBMCs stimulated with 5 μg/mL phytohemagglutinin (Murex) were included as positive controls to assess their proliferative capacity. Each culture condition was performed in duplicate.
In separate experiments, we studied the roles of PD-L1, IDO, and PGE2 in inhibition of allogeneic T-cell responses by MSCs by adding anti-PD-L1 mAb (10 μg/mL; eBioscience) to block PD-L1; 1-methyl-
After 5 days of culture at 37°C and 5% CO2, cell-free supernatant was collected, frozen, and stored at −20°C for later analysis, and cells were stained for cell viability, using the LIVE/DEAD® Fixable Dead Cell Stain Kit (Invitrogen), according to the manufacturer's protocol. Cells were then stained with anti-CD3-PerCP-Cy5.5 (BD Biosciences), anti-CD4-APC-H7 (BD Biosciences), anti-CD8-efluor450 (eBioscience) to distinguish T-cell subsets, and anti-CD19-horizonV500 (BD Biosciences) to exclude B cells from analysis. Cytotoxic degranulation was detected by addition of CD107a-APC (eBioscience) during the last 15 h of the cocultures. Cells were analyzed for proliferation, using CFSE dilution patterns, and for the phenotype on a BD FACS Canto II Flow cytometer (BD Biosciences). For analysis of phenotypic markers, we used FACSDiva software (Becton Dickinson) and precursor frequencies (PFs) of alloreactive CD4+, and CD8+ T cells were calculated using ModFit LT® software (Verity Software House), as previously described [29]. From duplicate assays, average PFs were calculated.
IFN-γ production was measured in the culture supernatants by standard enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (Human IFNγ CytoSet™; Invitrogen). TNF-α production was also measured in the culture supernatants by ELISA (human TNF-α Ready-Set-Go!; eBioscience).
Polymerase chain reaction
Early passage (P3) liver perfusate-derived and BM-MSCs were plated at 1.0×105 cells per well in a six-well dish (Corning) in full growth medium. After 24 h, cells were growth arrested in serum-free media (RPMI) for 24 h. Following this, 700 μL of Qiazol lysis buffer (Qiagen) was added to each well and cells were harvested. RNA was isolated using a Qiagen miRNeasy mini kit (Qiagen) and quantified using a Nanodrop ND-1000. A total of 300 ng was used to make cDNA using an iScript cDNA synthesis kit from Bio-Rad (Bio-Rad Laboratories), and 15 ng cDNA was used per real-time quantitative polymerase chain reaction (RT-qPCR). The expression of prostaglandin-endoperoxide synthase 1 and 2 (PTGS1 and PTGS2) was quantified using RT-qPCR with a SensiMix Plus SYBR Kit (BioLine) according to the manufacturer's instructions. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a household gene for normalization of gene expression. RT-qPCR was performed using the following primers: PTGS1-forward CGCCAGTGAATCCCTGTTGTT, PTGS1-reverse AAGGTGGCATTGACAAACTCC; PTGS2-forward CTGGCGCTCAGCCATACAG, PTGS2-reverse CGCACTTATACTGGTCAAATCCC; GAPDH-forward: AAGGTCGGAGTCAACGGATTT, GAPDH-reverse: ACCAGAGTTAAAAGCAGCCCTG. The fold change in mRNA was determined using the ΔCt method.
Statistics
Data were analyzed with GraphPad Prism 5.0 software and expressed as mean±standard error of the means. To test whether data were normally distributed, the Kolmogorov–Smirnov test was used. Differences between groups were analyzed using the paired t-test (normally distributed paired data), Wilcoxon signed rank test (nonnormally distributed paired data), or Mann–Whitney test (unpaired data). One-sample t-test was performed to test differences between experimental conditions and control conditions consisting of one sample. P-values<0.05 were considered significant.
Results
Immunophenotypic and differentiation potential of MSC-like cells mobilized from liver grafts are similar to those of BM-MSCs
To explore whether MSCs mobilize from human liver grafts after tissue injury in vivo, we collected liver graft perfusates during flushing of liver grafts at the time of transplantation. These livers have been exposed to hemodynamic, ischemic, and inflammatory changes during the donation process. From these liver perfusates, we recovered a population of cells that adhered to plastic and were amenable to expansion, such as BM-MSCs (Fig. 1A, B). In addition, these graft-derived cells could well differentiate into adipocytes and osteocytes (Supplementary Fig. S1; Supplementary Data are available online at
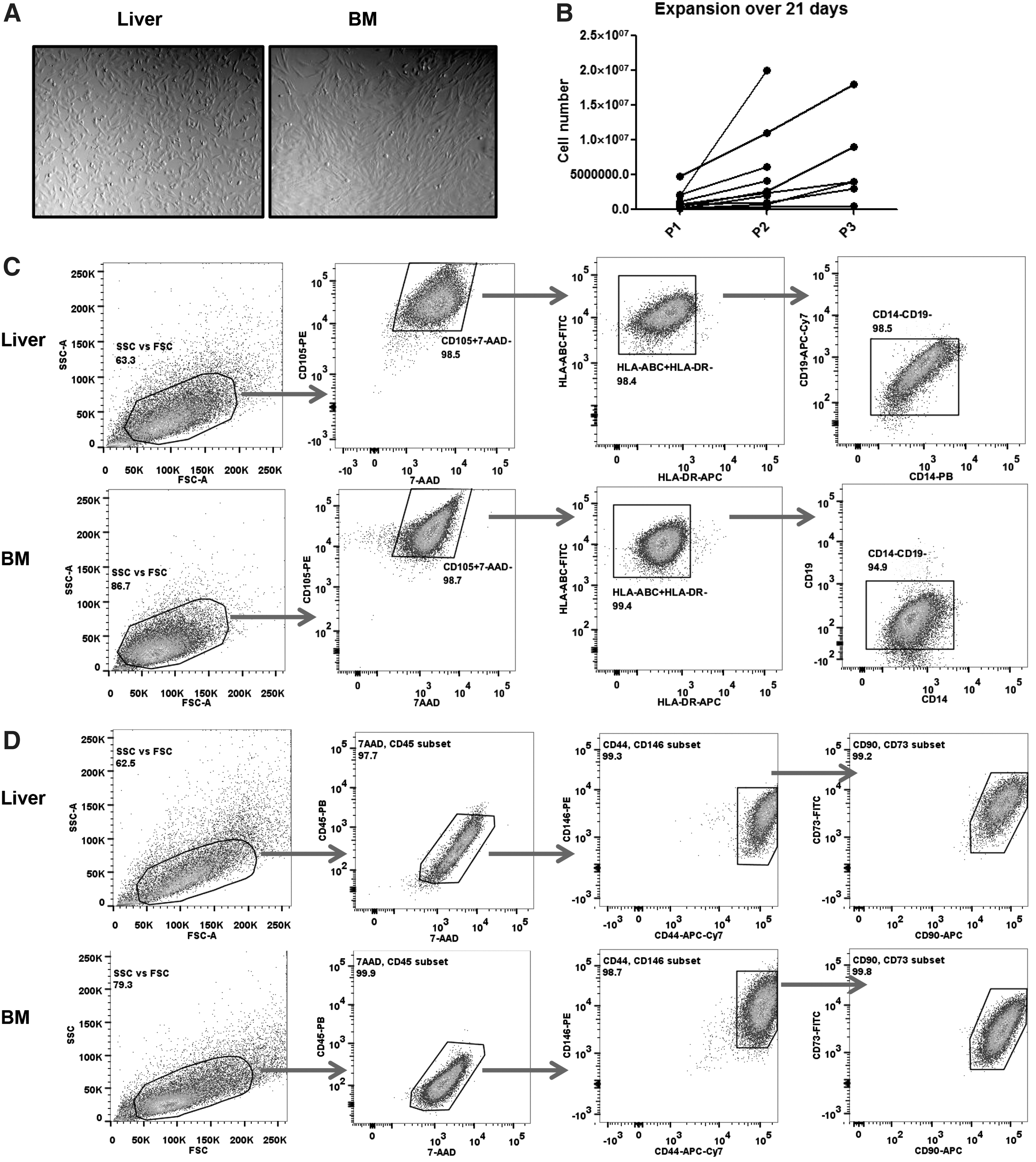
Detection of mesenchymal stromal-like cells (MSCs) in the perfusates from human liver grafts.
L-MSCs suppress alloreactive T cells better than BM-MSCs
Previously, it has been shown that BM-MSCs can suppress proliferation of alloprimed T cells [9]. We compared the suppressive capacity of L-MSCs with that of BM-MSCs in MLRs, in which CFSE-labeled PBMCs were stimulated with allogeneic splenocyte-derived CD40-B cells as strong antigen-presenting cells, as described previously [29]. The level of T-cell proliferation was determined using CFSE dilution patterns, from which we calculated PFs of responding T cells, as shown in Fig. 2A. We found that both CD4+ and CD8+ T cells proliferated in response to stimulation with allogeneic CD40-B cells over a 5-day period. PFs in the control conditions varied from ∼2% to 10% because different responder and stimulator cell combinations were used for different experiments. To test the suppressive capacity of L-MSCs and BM-MSCs, we added L-MSCs or BM-MSCs to the cocultures at an MSC/PBMC ratio of 1:10 or 1:5. With addition of L-MSCs (n=12, different donors) at a ratio of 1:5, proliferation of alloreactive CD4+ T cells was nine-fold lower and of CD8+ T cells was 10-fold lower than in control culture without L-MSCs. This difference was significantly different (P<0.0001). The antiproliferative effect of BM-MSCs (n=6, different donors) was also evident, causing a four-fold lower proliferation of both CD4+ and CD8+ T cells than in control cultures without BM-MSCs (P<0.0001). Interestingly, L-MSCs suppressed CD8+ T-cell proliferation significantly better than BM-MSCs (Fig. 2B; P<0.05). This superior suppressive effect of L-MSCs was also observed for CD4+ T cells, although not statistically significantly (Fig. 2B, P=0.068). With the addition of MSCs at the MSC/PBMC ratio of 1:10, we found the same trend toward a stronger antiproliferative effect of L-MSCs than BM-MSCs, although the suppression was less strong and differences between L-MSCs and BM-MSCs were stronger at a ratio of 1:5. Apparently, there is a dose-dependent inhibitive effect of MSCs on T-cell proliferation. In addition, L-MSCs inhibited cytotoxic degranulation, as measured with surface expression of CD107a on proliferated T cells, of both alloreactive CD4+ and CD8+ T cells significantly better than BM-MSCs (P<0.01, Fig. 2C, D). Finally, L-MSCs reduced IFN-γ production significantly (P<0.01), while BM-MSCs did not. This difference between BM-MSCs and L-MSCs was statistically significant (Fig. 2E, P<0.05). TNF-α production was strongly inhibited by both L-MSCs and BM-MSCs with no significant difference between L-MSCs and BM-MSCs (data not shown).
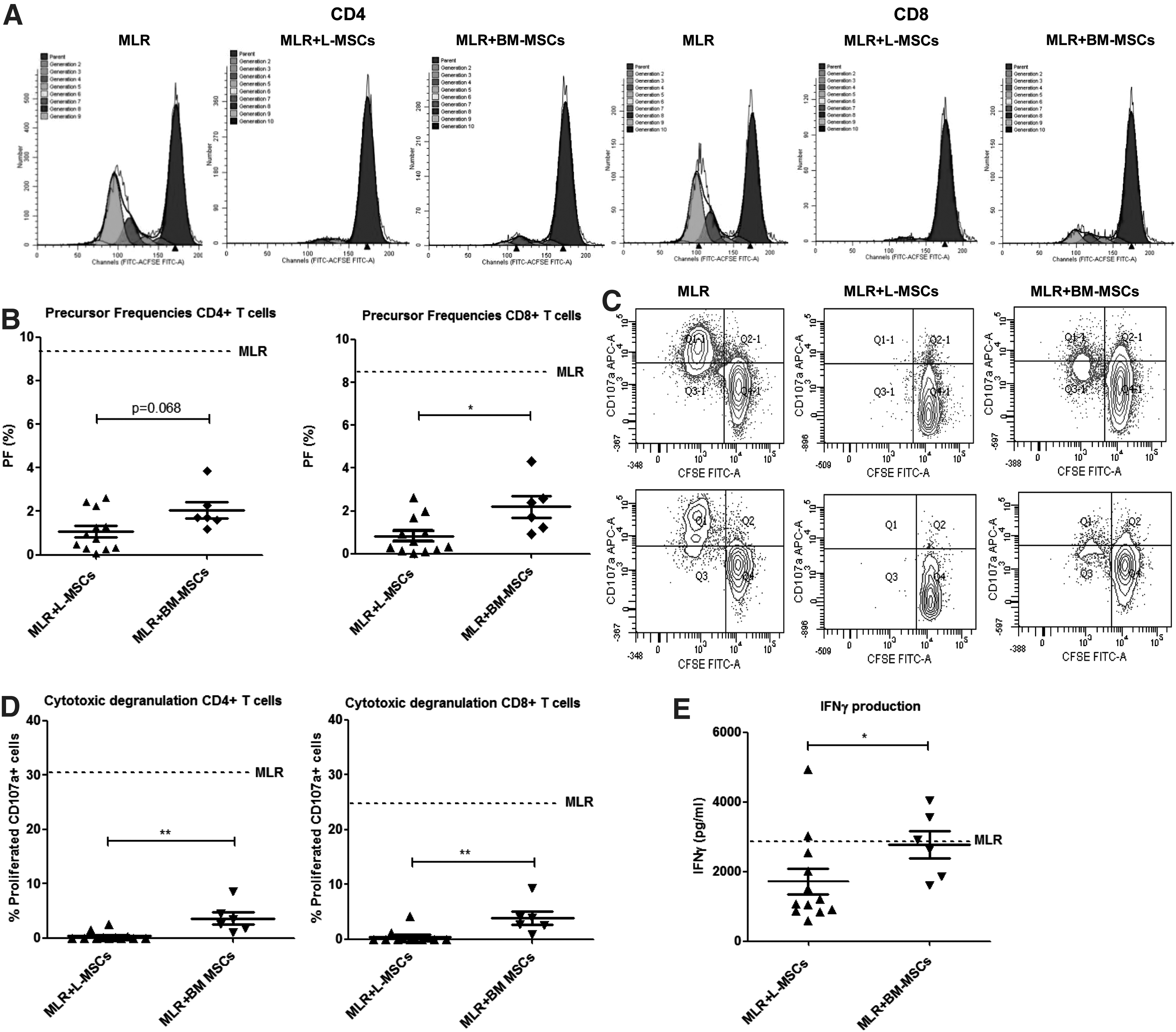
Graft-derived MSC, from human liver grafts (L-MSCs), suppression of alloreactive T-cell responses is stronger than BM-MSCs. Carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled peripheral blood mononuclear cells (PBMCs) were cultured for 5 days with allogeneic splenocyte-derived CD40-ligand-stimulated-B cells (CD40-B cells). Mixed lymphocyte reactions (MLRs) were performed in the presence and absence of either L-MSCs (n=12) or BM-MSCs (n=6). CFSE dilution patterns of CD4+ and CD8+ T cells were analyzed and precursor frequencies (PFs) were calculated with ModFit® software.
Soluble factors secreted by L-MSCs suppress CD4+, but not CD8+, T-cell proliferation, and CD4+ and CD8+ cytotoxic degranulation
To assess whether the inhibition of L-MSCs on T-cell responses was cell–cell contact independent, MLRs were performed in the presence of conditioned medium (CM) derived from MSC cultures. We found that the CM from L-MSCs suppressed the proliferation of alloreactive CD4+ T cells, while CM from BM-MSCs did not (Fig. 3A, P<0.001). CM from L-MSCs also suppressed proliferation of CD8+ T cells significantly (P<0.01), but this effect was not significantly different from the suppressive effects of CM from BM-MSCs (Fig. 3A). In addition, CM from L-MSCs suppressed the cytotoxic degranulation capacity of alloreactive CD4+ T cells and CD8+ T cells significantly better than CM from BM-MSCs (Fig. 3B, P<0.001 and P<0.01, respectively). CM from L-MSCs was not able to suppress IFN-γ production or TNF-α production (data not shown). Taken together, these data suggest that the immunomodulatory potential of L-MSCs is at least partially mediated by secreted factors.

Graft-derived L-MSC suppression of alloreactive T-cell responses is stronger by soluble factors than BM-MSCs. Supernatants from MSCs [conditioned medium (CM)] were added to the MLR, and at the end of 5 days, alloreactive T-cell responses were measured.
L-MSCs express higher levels of PD-L1 than BM-MSCs, which is associated with suppression of alloreactive T-cell responses
We wondered which mechanism could cause the superior immunosuppressive capacity of L-MSCs in comparison with their BM-MSC counterparts. An important coinhibitory pathway in (alloreactive) T-cell responses is the PD-1/PD-L1 pathway [31]. We found that L-MSCs have higher baseline expression levels of PD-L1 than BM-MSCs (Fig. 4A, C, P<0.05), which could be a reason for the higher suppressive capacity of L-MSCs. Proinflammatory cytokines have been described to upregulate PD-L1. Since proinflammatory cytokines increase during the organ donation process, it is possible that during donation, MSCs in the liver become primed and thereby upregulate PD-L1. To assess the effect of proinflammatory cytokine priming on PD-L1 expression and suppressive capacity, we exposed L-MSCs and BM-MSCs to TNF-α and IFN-γ ex vivo and measured their PD-L1 expression and suppressive function. After priming, we indeed found a strongly upregulated PD-L1 expression on both L-MSCs and BM-MSCs (Fig. 4B, C).
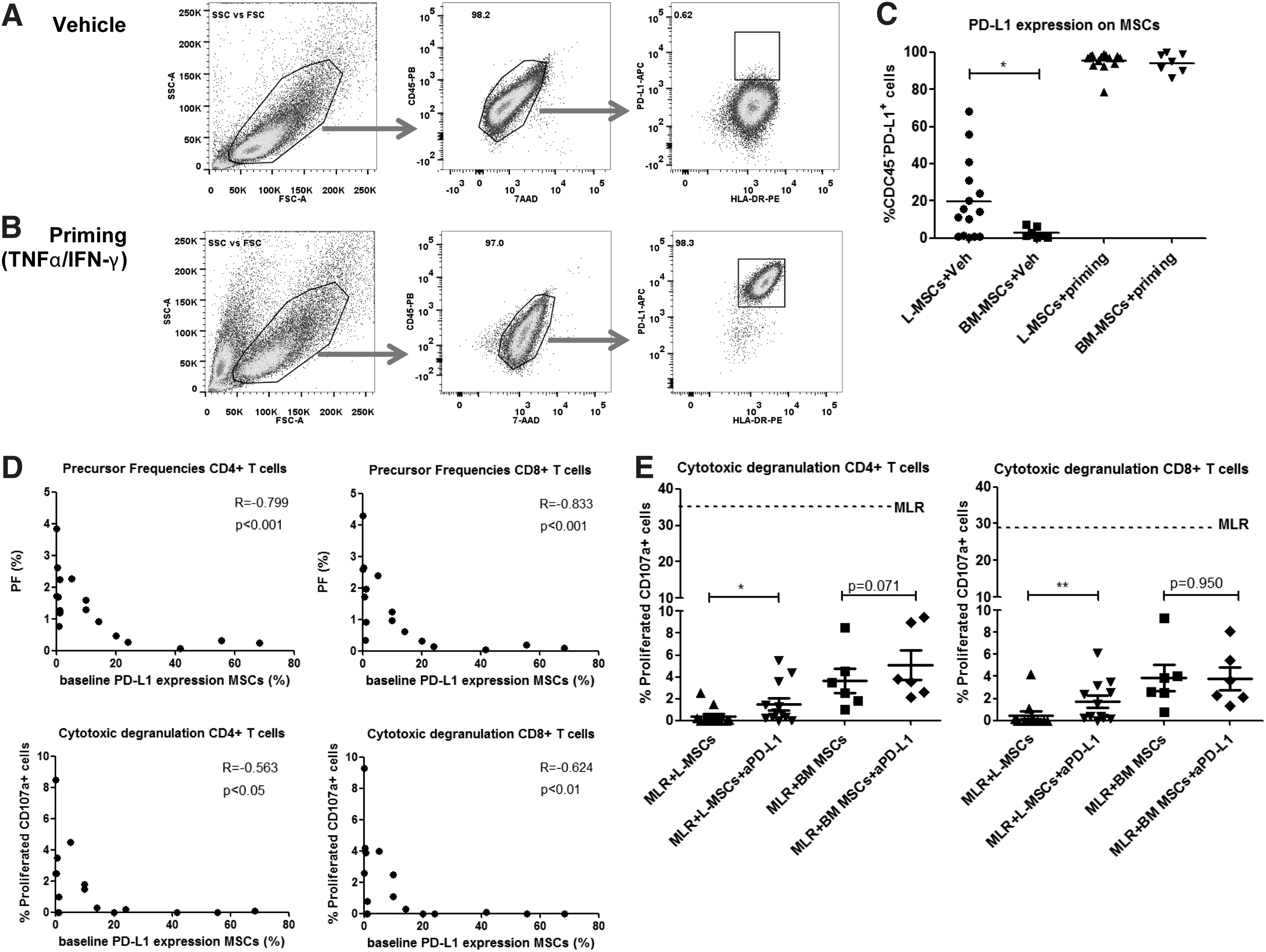
Graft-derived L-MSCs express higher levels of PD-L1 than BM-MSCs; PD-L1 is upregulated after ex vivo priming with proinflammatory cytokines and associated with suppression of alloreactive T-cell responses.
We wondered whether the superior suppressive effects of L-MSCs were mediated by their higher level of PD-L1 expression. We first looked at the correlation between PD-L1 expression on MSCs and the corresponding T-cell responses in the MLRs and found that a higher PD-L1 expression on MSCs was strongly correlated with lower proliferation of both CD4+ and CD8+ T cells in the MLRs. In addition, higher PD-L1 expression levels were also strongly correlated with lower cytotoxic degranulation capacities of both CD4+ and CD8+ T cells in the MLRs (Fig. 4D). We then added an anti-PD-L1 antibody to the MLRs to test whether blockade of the PD-1/PD-L1 interaction abrogated the suppressive effects of the L-MSCs. We found that anti-PD-L1 did not abrogate the suppressive effects of L-MSCs on proliferation and IFN-γ production or TNF-α production of T cells (data not shown), but significantly abrogated the effects on CD4+ and CD8+ T-cell cytotoxic degranulation (Fig. 4E, P<0.05 and P<0.01, respectively).
Immunosuppressive effects of MSCs partly involve IDO, but not PGE2
In a previous study of our research group [14], we performed a gene expression profiling to compare the molecular phenotypes of L-MSCs and BM-MSCs. These data showed that L-MSCs and BM-MSCs were very similar, but a small number of genes were differentially expressed between L-MSCs and BM-MSCs. In the present study, we focused on the genes that were differentially expressed to investigate whether these could provide insight into the differences in immunomodulatory effects of L-MSCs and BM-MSCs. One of the genes that were significantly differentially expressed was PTGS1, which encodes for cyclooxygenase-1 (COX-1). COX-1 is the key enzyme in prostaglandin biosynthesis [32]. Prostaglandins are produced by MSCs and have previously been described to inhibit T-cell responses [9]. Since PTGS1 was expressed 20-fold higher in L-MSCs than in BM-MSCs, differences in prostaglandin production between the two types of MSCs could well explain the immunomodulatory differences that we found. We first confirmed the difference in PTGS1 gene expression between L-MSCs and BM-MSCs using PCR (Fig. 5A, P<0.001). We then investigated whether culturing MSCs in the presence of COX-1 inhibitor, indomethacin, affected the immunomodulatory function of L-MSCs or BM-MSCs in the cocultures with PBMCs. However, we found no effects of this (Fig. 5B). In addition, we determined whether blockers of the prostaglandin receptors, EP1,2,3, and 4, abrogated the inhibitive effects of CM from L-MSCs or BM-MSCs. However, we were not able to show higher rates of T-cell proliferation after addition of the blockers (Fig. 5C).
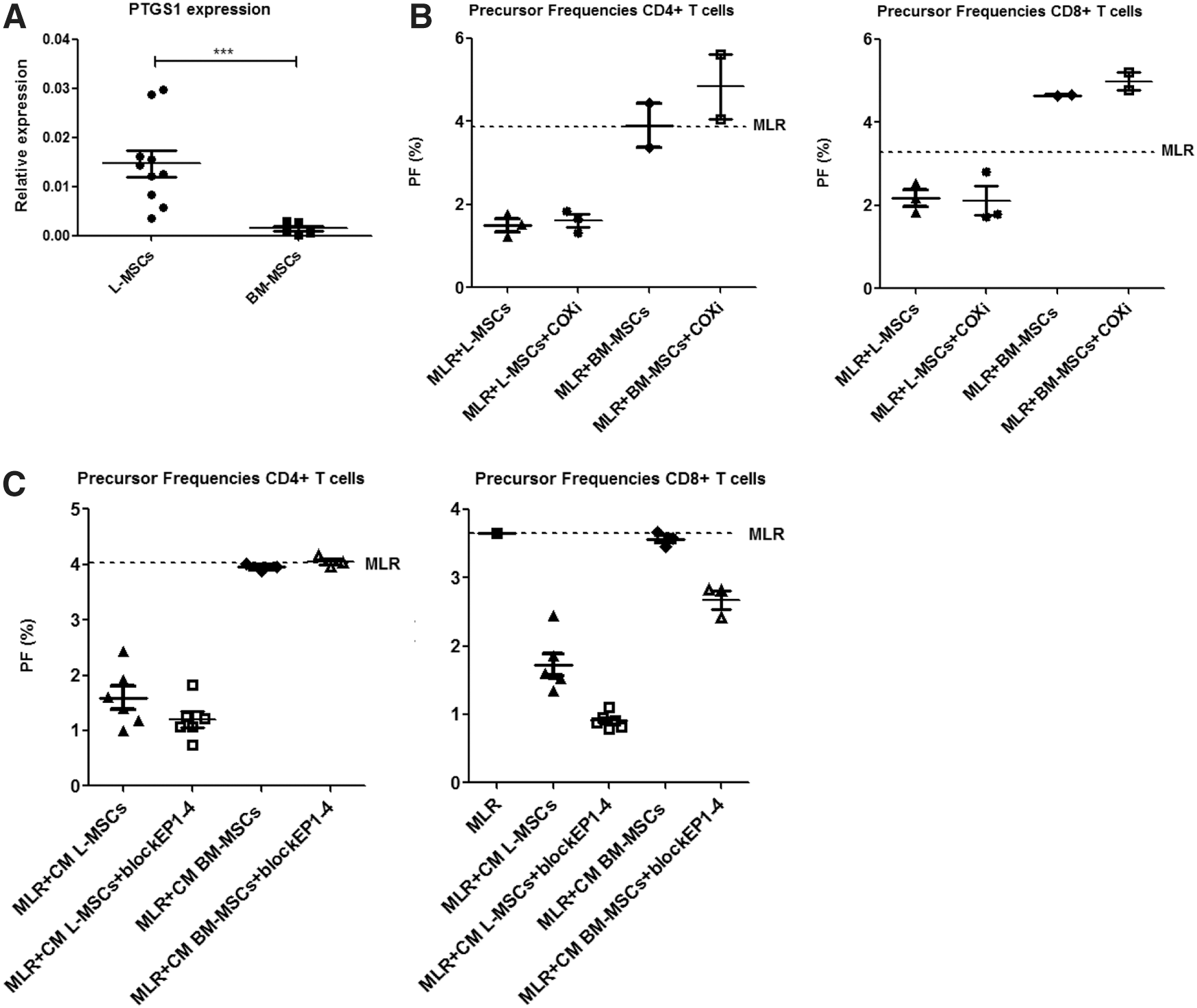
Effects of blocking prostaglandin E2 (PGE2) on the inhibition of T-cell responses by L-MSCs and BM-MSCs. (
Another suppressive mechanism described for MSCs is the production and activity of IDO [25,26]. After addition of 1MT to block IDO, we found significant abrogation of the immunosuppressive effects of L-MSCs, but not BM-MSCs, on T-cell proliferation (Fig. 6, P<0.001). No effect on cytotoxic degranulation was observed (data not shown). In conclusion, L-MSCs inhibit CD4+ and CD8+ T-cell degranulation partly by PD-L1/PD-1 interaction and inhibit T-cell proliferation partly by IDO.
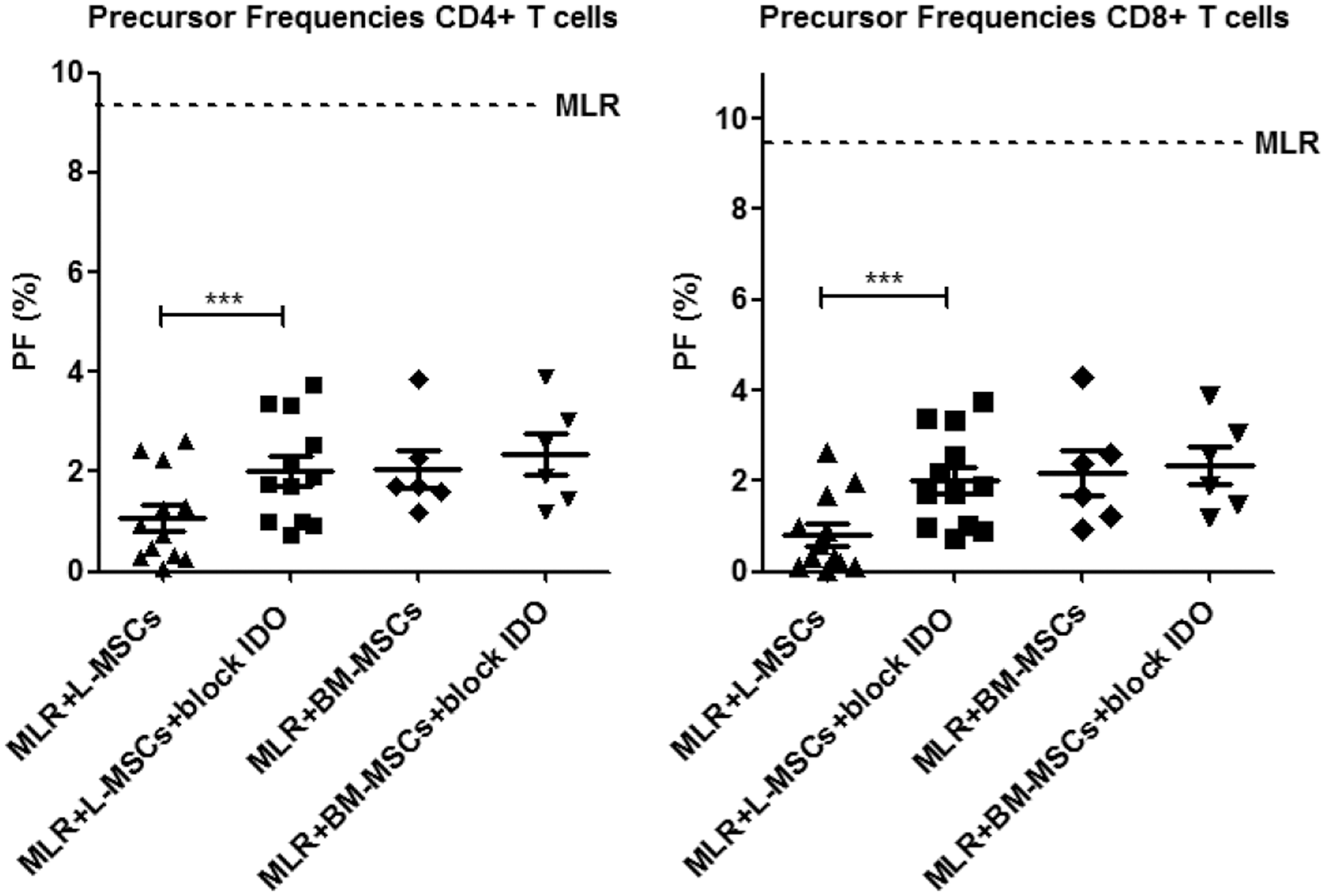
Effects of blocking indoleamine 2,3-dioxygenase (IDO) on the inhibition of T-cell responses by L-MSCs and BM-MSCs. Scatter dot plots showing PFs of alloreactive CD4+ and CD8+ T cells in the MLR without MSCs (control MLR, dotted line) and in the presence of L-MSCs (n=12) versus BM-MSCs (n=6) in the presence and absence of IDO blocker, 1-methyl-
Discussion
In this study, we showed that graft-derived L-MSCs, which are released during transplantation, suppress proliferation, cytotoxic degranulation, and IFN-γ production of alloreactive T cells. This suppressive capacity of L-MSCs was significantly better than that of BM-MSCs even though the phenotype and differentiation potential of L-MSCs and BM-MSCs were very similar. However, L-MSCs expressed higher levels of PD-L1 than BM-MSCs. PD-L1 is the ligand for the coinhibitory receptor, PD-1, on T cells. On L-MSCs, expression of PD-L1 was associated with inhibition of T-cell proliferation and cytotoxic degranulation in vitro. There was a strong correlation between levels of PD-L1 expression on MSCs and corresponding proliferation and cytotoxic degranulation of T cells in the cocultures. The upregulation of PD-L1 on graft-derived MSCs may be a consequence of in vivo priming by proinflammatory cytokines, which are elevated during the donation process [33,34].
Blocking of PD-L1 in vitro partly abrogated the suppressive effects of L-MSCs on CD4+ and CD8+ T-cell cytotoxic degranulation. However, blocking of PD-L1 did not abrogate the effects on proliferation and IFN-γ and TNF-α production. Apparently, the PD-L1 expression represents a surrogate marker of inhibition, but the higher PD-L1 expression on L-MSCs only partly explains that suppressive effects of L-MSCs on T-cell responses were superior to those of BM-MSCs. It is likely that other mechanisms are involved in this.
We showed that, besides cell–cell contact-dependent mechanisms, soluble factors produced by L-MSCs contribute to the superior suppression of alloreactive T-cell responses as well. Since soluble factors have been described to contribute to the immunosuppressive properties of MSCs [35], we further investigated the suppressive function of secreted factors of MSCs present in conditioned culture media on alloreactive T-cell responses. Our finding that CM from L-MSCs suppressed T-cell proliferation, but not T-cell degranulation and IFN-γ and TNF-α production, whereas CM from BM-MSCs exerted no suppressive effects suggests that L-MSCs produce T-cell-suppressive soluble factors that BM-MSCs do not produce. These soluble factors released by graft-derived L-MSCs may directly affect T cells or indirectly through their effect on antigen-presenting cells [12,36]. Soluble factors secreted by L-MSCs could inhibit the stimulator CD40-B cells in the cocultures, which thereby cause inhibition of the T-cell responses.
An interesting finding was that L-MSCs showed higher expression of PTGS1, a key enzyme involved in prostaglandin production, than BM-MSCs. Although prostaglandins are produced by MSCs and are able to inhibit T-cell responses [9], we did not find abrogative effects of blocking prostaglandins either by indomethacin or by prostaglandin receptor inhibitors. Clearly, these results show no role for PGE2 in the inhibition by MSCs.
More importantly, we found that blocking IDO partly restored T-cell proliferation that was inhibited by L-MSCs, while this had no effect on the conditions with BM-MSCs. IDO has previously been shown to inhibit T-cell responses by MSCs [25,26,37]. Our finding that blocking IDO only abrogated the inhibition by L-MSCs and not BM-MSCs supports the hypothesis that the immunomodulatory function of MSCs is highly plastic, as published previously [38], since L-MSCs have been exposed to an inflammatory environment.
One of the limitations of comparative studies of MSC populations is the lack of consensus to immunophenotypically define these cells [6]. The current study compared MSC populations from liver grafts and BM. MSCs found in liver graft perfusates adhered to plastic, were amenable to expansion, and expressed cell surface markers that are described to identify MSCs, similar to BM-MSCs [6]. The superior suppressive capacity of L-MSCs may be related to the tissue source. The phenotype of MSCs in the liver may reflect the hyporesponsiveness of the hepatic immune system. Another difference is that L-MSCs have been exposed to extensive ischemia, whereas cells from BM were not exposed to substantial ischemia. Last, a difference between BM-MSCs and L-MSCs is their mobilization. L-MSCs were mobilized and released from the liver parenchyma during the donation process, whereas BM-MSCs were obtained from the stroma without mobilization and release. Which of these differences is most likely to attribute to the T-cell-suppressive capacity of MSCs needs further evaluation.
For future clinical applications, our study provides insights in the superior immunosuppressive capacity of L-MSCs, which may lead to cell-based immunosuppressive therapies using liver graft-derived MSCs. For this therapeutic application, L-MSCs need to be isolated from the donor liver and expanded in vitro, as described previously [39], since L-MSCs are present in low numbers. After expansion, L-MSCs should be infused in patients and may have an important function in the setting after liver transplantation [39] or may be used as a bridging therapy before transplantation in patients with acute liver failure [40,41].
In conclusion, we showed that MSCs mobilized from livers grafts strongly suppress alloreactive T-cell responses. This suppression was stronger than nonmobilized and nonprimed MSCs derived from BM stroma. These differences may not only be related to the intragraft immune priming of L-MSCs in the organ donor or due to mobilization of MSCs from the graft but may also be related to the inherent immunological tolerogenicity of the liver. In the setting of liver transplantation, resident and/or mobilized L-MSCs may therefore regulate immune-mediated liver damage by suppressing pathogenic alloreactive T-cell responses, leading to better acceptance of liver grafts.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.