
Abstract
The molecular mechanisms that orchestrate the exit from pluripotency, cell cycle progression, and lineage-specific differentiation in human pluripotent stem cells (hPSCs) are poorly understood. RELB, a key protein in the noncanonical nuclear factor-kappaB (NFκB) signaling pathway, was previously implicated in controlling the switch between human embryonic stem cell (hESC) proliferation and differentiation. Here, we show that RELB enhances the proliferation of hESCs and human-induced pluripotent stem cells (hiPSCs) without affecting their pluripotency. We demonstrate that RELB does this by interacting with two RNA-binding proteins LIN28A and IMP3 (IGF2 mRNA-binding protein 3); further, these interactions control mRNA levels and protein expression of insulin-like growth factor 2 (IGF2) and key cell-cycle genes. Finally, after stress, these proteins co-localize in stress granules in hESCs and iPSCs. Our data identify RELB as a novel regulator of hPSC proliferation, and suggest a new function for RELB, in addition to its widely accepted role as a transcription factor, that involves recruitment of IMP3 and LIN28 to the cytosolic mRNA translation-control domains for post-transcriptional modulation of IGF2 and cell-cycle gene expression.
Introduction
U
The mammalian nuclear factor-kappaB (NFκB) transcription factor family is one of the most well-studied transcription factor families and is known to play important roles in tumor promotion and suppression, apoptosis, proliferation, cell cycle, differentiation, inflammation, and regulation of the immune system [34 –36]. NFκB is composed of five members: RELA (p65), RELB, C-REL, NFκB1 (p50 and its precursor p105), and NFκB2 (p52 and its precursor p100), which can form a collection of homodimeric and heterodimeric complexes [37,38] that shuttle between the cytosol and nucleus in response to IκB phosphorylation and modulation of the nuclear export regulators' activities. It was previously shown that canonical NFκB signaling enhances cell survival in hESCs [39,40] and that the noncanonical NFκB protein RELB plays a role in maintenance of pluripotency [40]. The binding of RELB to the regulatory regions of SOX2, BRACHYURY, PAX6, and CDX2 implicated RELB in controlling the switch between hPSC proliferation and differentiation [40]. In this paper, we show that RELB interacts with LIN28A and IMP3 and regulates proliferation and cell-cycle kinetics in hPSCs without affecting pluripotency by regulating the expression of IGF2 and other cell cycle genes. We further show that RELB, LIN28, and IMP3 co-localize in TIA-1-positive stress granules after stress. We conclude that RELB can act as a regulator of the LIN28/IMP3 mRNA translation control machinery present in the cytosol, and that this novel function of RELB likely modulates the cell-cycle gene expression and proliferation of hPSCs.
Materials and Methods
Cell culture
For undifferentiated self-renewing conditions, HES3, H9 hESCs, and C11 iPSCs were cultured using NutriStem™ XF/FF Culture Medium (Stemgent) with additional 10 ng/mL basic fibroblast growth factor (b-FGF) and passaged every 6–7 days onto plates precoated with Matrigel (Invitrogen) at a density of 0.0347 mg/cm2 to culture cells feeder-free.
Plasmids and generation of doxycycline-inducible RELB-knockdown cells and HA-tagged RELB overexpression cells
HES3, H9, and C11 lines cultured as described earlier were seeded onto a six-well plate and transduced with the appropriate virus. All RELB shRNA sequences targeting the 3′ UTR or the CDS region of the RELB mRNA (Supplementary Table S1; Supplementary Data are available online at
HA-tag immunoprecipitation and mass spectrophotometry
HA-tagged RELB over-expressing cells or untagged-over-expression vector control (OE V/C)-expressing cells were quickly washed twice with cold phosphate-buffered saline (PBS)+ (Invitrogen) and 200 μL of 1× Cell Lysis Buffer (Cell Signaling Technology) containing 1 mM PMSF (Sigma-Aldrich) and 1× phosphatase and protease inhibitors (Roche) was added per well of a six-well plate to at least four wells. Two duplicate wells were used for RNA and protein collection. The plate was incubated on ice for 5 min, cells were harvested by scraping, and the resulting lysate was passed through a 30G syringe multiple times, centrifuged at 14,000 g for 15 min twice at 4°C, and the supernatant was collected. One hundred microliters was used for western blot analysis, and 600 μL was used for immunoprecipitation using the Pierce® HA Tag IP/Co-IP Kit (Thermo Scientific) following the manufacturer's instructions. Thirty microliters of pooled eluate was immediately neutralized with 1.5 μL of 1 M Tris, pH 9.5 and mass-spectrophotometric analysis was subsequently carried out.
Sample processing for liquid chromatography-tandem mass spectrometry
Twenty microliters of the affinity eluate was reduced by the addition of NH4HCO3 (Sigma-Aldrich) to 50 mM, sodium dodecyl sulphate (SDS; Bio-Rad) to 2% and dithiothreitol (GE Healthcare) to 20 mM. Samples were incubated for 16 h at 4°C followed by 2 h at 22°C. The reduced proteins were then alkylated by the addition of iodoacetamide (GE Healthcare) to 50 mM and incubated at 22°C in the dark for 2 h. Protein was precipitated by acid precipitation (2-D Clean-Up Kit; GE Healthcare) with an overnight final wash step to ensure the removal of all acid. Precipitated proteins were re-suspended in 30 μL of 10% acetonitrile (J.T. Baker), 40 mM NH4HCO3 and digested with 0.5 μg of modified sequencing-grade trypsin, bovine pancreas (Roche) for 2 h at 37°C, followed by the addition of a further 0.5 μg trypsin and incubation for 6 h at 37°C. The digest liquor was removed in vacuo and the residue was reconstituted in 30 μL of 1% formic acid, 2% acetonitrile.
Liquid chromatography-tandem mass spectrometry
Acidified digests were analyzed by nanoHPLC-MS/MS using a Prominence nano HPLC system (Shiumadsu) interfaced with an Orbitrap Elite hybrid mass spectrometer (Thermo Fischer Scientific). Digests were loaded onto a Reprosil aq C18 3 μm, 120 Å, 300 μm trap (SGE pn-2222066) at 30 μL/min in 2% acetonitrile 0.1% (v/v) aqueous formic acid for 3.5 min at 50°C, then switched in-line with an analytical column (15 cm×75 μm fused silica, self packed) reprosil aq C18 2.4 μm (pn-r124.aq batch-9756; Dr. Maisch GmbH) using a flow rate of 0.3 μL/min and 98% solvent A [0.1% (v/v) aqueous formic acid], 2% solvent B [80% (v/v) ACN, 0.1% (v/v) aqueous formic acid]. Peptides were separated at 50°C using a sequence of linear gradients: to 15% B over 5 min; to 47% B over 90 min; to 95% B over 10 min; and then holding the column at 95% B for 5 min. Eluate from the analytical column was introduced into the Orbitrap Elite throughout the entire run via a Nanospray Flex Ion Source (Thermo Fisher Scientific) and a 10 μm inner diameter uncoated silica emitter (New Objective). Typical spray voltage was 1.4 kV with no sheath, sweep, or auxiliary gases used. The heated capillary temperature was set to 285°C. The Orbitrap Elite was controlled using Xcalibur 2.2 software (Thermo Fisher Scientific) and operated in a data-dependent acquisition mode to automatically switch between Orbitrap-MS and ion trap-tandem mass spectrometry (MS/MS). The survey full scan mass spectra (from m/z 380–1,700) were acquired in the Orbitrap with resolving power set to 240,000 (at 400 m/z) after accumulating ions to an automatic gain control (AGC) target value of 1.0×106 charges in the linear trap quadrupole (LTQ). MS/MS spectra were concurrently acquired on the 15 most intense ions from the survey scan in the LTQ filled to an AGC target value of 3.0×104. Charge state filtering, where unassigned and charge state 1 precursor ions were not selected for fragmentation, and dynamic exclusion (repeat count 1, repeat duration 30 s, exclusion list size 500, exclusion duration 90 s) were used. Fragmentation conditions in the LTQ were as follows: 35% normalized collision energy; activation q of 0.25; 10 ms activation time; and minimum ion selection intensity 5,000 counts. Maximum ion injection times were 200 ms for survey full scans and 50 ms for MS/MS.
Data processing
Tandem mass spectra were processed using Proteome Discoverer (version 1.3; Thermo Fisher Scientific) and submitted to Mascot (version 2.2.06; Matrix Science) with the following parameters specified: fixed modifications; carbamidomethyl-cysteine, Variable modifications; deamidation (asparagine, glutamine); and oxidation (methionine). Enzyme: trypsin, two missed cleavages. Database: Uniprot human reference proteome (20120228) and bovine trypsin (65837 entries); and MS tolerance 10 ppm, MS/MS tolerance 0.8 Da.
Scaffold (version 3.6.4; Proteome Software) [41] was used to validate Mascot protein identifications and to make a comparison across samples. Scaffold verifies these Mascot peptide identifications using the X!Tandem database searching program. Scaffold then probabilistically validates peptide identifications using PeptideProphet [42] and derives corresponding protein probabilities using ProteinProphet [43]. Two peptides per protein with a 95% or greater probability and an overall protein probability of 99% or greater was required to assign a protein or protein group.
Western blot analysis
Cells were washed twice with PBS and subsequently lysed with 5× loading buffer (130 mM Tris pH 6.8, 4% SDS, 0.02% Bromophenol blue, 20% glycerol, 100 mM DTT) containing 10% PhosSTOP phosphatase inhibitor (Roche), homogenized with 30G syringe needles, heated at 80°C for 10 min, and resolved using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The iBlot® Dry Blotting System (Invitrogen) was used to transfer proteins onto a PVDF membrane. Blots were blocked in blocking buffer [Tris-buffered saline, 0.01% Tween-20 (TBST), 5% bovine serum albumin (BSA)] at room temperature for 2 h, probed with desired primary antibodies in blocking buffer at 4°C overnight with gentle agitation, washed thrice for 15 min each, probed with appropriate secondary antibodies in blocking buffer for 1 h at room temperature, washed as described earlier, and detected using Pierce ECL Western Blotting Detection Kit according to the manufacturer's recommendation (Thermo Scientific) on either Typhoon (GE Healthcare) or V3-Western Workflow (Bio-Rad) for a densitometric representation. The images were, respectively, analyzed using either One-Rad (Bio-Rad) or Image Lab (Bio-Rad) software. All antibodies with working dilutions have been described in Supplementary Table S2.
siRNA knockdown
RELB (siRELB), IMP3 (siIMP3) siRNAs were obtained from Sigma-Aldrich. LIN28A and scrambled (siControl) siRNAs were obtained from ISIS Pharmaceuticals. 7.5 μL lipofectamine RNAiMax (Invitrogen) was mixed in 50 μL Dulbecco's modified Eagle's medium/Ham's F-12 mixture (DMEM-F12; Invitrogen) per well following the manufacturer's instructions to transfect siRNAs. Parental and OE V/C cells were transfected with 60 pmol of siControl siRNAs. RELB OE cells were transfected with either 60 pmol of siControl siRNAs or a mixture of 30 pmol of siLIN28A and 30 pmol of siIMP3. For RELB knockdown, cells were transfected with 60 pmol of siRELB. For LIN28A and IMP3 knockdown, cells were transfected with a mixture of 30 pmol of siLIN28A and 30 pmol of siIMP3. These cells were transfected every 36 h for a total of three times, after which they were assayed for 5-ethynyl-2-deoxyuridine (EdU) incorporation, Phospho-Histone H3 (Ser-10) expression, and RNA and protein extraction.
Cell proliferation and cell-cycle analysis
Cells were grown under pluripotency conditions or transfected with indicated siRNAs as described earlier until they reached about 60% confluence, and media was changed on cells 8 h before commencement of the assay to ensure minimal external factors affected cell cycle. These cells were next assayed for EdU incorporation, Phospho-Histone H3 (Ser-10) expression, and RNA and protein extraction. For EdU incorporation, 20 mM EdU was added to hPSCs for 2 h and EdU incorporation was then measured via flow cytometry using Click-iT® EdU Alexa Fluor® 647 Flow Cytometry Assay Kit (Invitrogen) as per the manufacturer's instructions. Non-EdU-treated cells were used as a negative control. Propidium iodide (PI) (1 μg/mL, Invitrogen) and 0.5 mg/mL RNAase A was added to all samples before analysis. Phospho-Histone H3 (Ser-10) (1:50; Cell Signaling Technology) expression was measured using flow cytometry to determine the percentage of cells undergoing mitosis. RNA and protein was also collected from these cells using procedures described in relevant sections.
Cell counts
Cell counts were also performed for assessing proliferation. Equal numbers of cells were seeded into each of the six wells of a 24-well plate per condition (RELB OE and OE V/C). Each subsequent day, cells were dissociated from one well in each condition using TrypleE (Invitrogen) and counted subsequently using TC10™ Automated Cell Counter (Bio-Rad) as per the manufacturer's instructions.
Immunofluorescence assays
Cells were washed thrice with PBS, fixed with 3% PFA, permeabilized with chilled 100% methanol at −20°C for 10 min, blocked in blocking buffer (5% BSA, 5% goat serum, 0.2% sodium azide, 0.3% Triton X-100 in PBS) for 1 h at room temperature, and incubated overnight at 4°C with desired primary antibodies. Next, cells were washed thrice with PBS and incubated for 2 h at room temperature in the dark with species and isotype-matched AlexaFluor-conjugated secondary antibodies (1:1,000; Invitrogen), washed as described earlier, mounted with ProLong® Gold antifade reagent with DAPI (Invitrogen), coverslipped, and sealed using nail-polish. The cells were cured for 24 h at room temperature and then examined for staining using either using QImaging-Retiga-EXi Fast 1394 fluorescence microscope or Carl Zeiss LSM 710 for confocal microscopy at room temperature. Images were acquired with equal exposure and gain settings and were equally adjusted for brightness or contrast postacquisition. Secondary-antibody only treated cells were used as a negative control. All antibodies with working dilutions have been described in Supplementary Table S2. Stress Granule formation was induced by treatment of hPSCs with 0.5 mM sodium arsenite (NaAs) for 45 min.
RNA isolation, cDNA synthesis, and quantitative reverse-transcriptase–polymerase chain reaction analysis
Total RNA was prepared using the RNA Purification Kit (Macherey Nagel), and 1 μg RNA was converted to cDNA in a 10 μL reaction mix with iScript cDNA Synthesis Kit (Bio-Rad), both of which were according to the manufacturer's protocol. Quantitative reverse-transcriptase–polymerase chain reaction (qPCR) reactions used SsoFast™ EvaGreen® Supermix (Bio-Rad) with cDNA template (from ∼5 ng RNA equivalent/reaction) according to the manufacturer's instructions using a C1000 Thermal Cycler (Bio-Rad). All primer sequences are listed in Supplementary Table S1. The expression value of each gene was normalized to a human β-actin control (Applied Biosystems). Relative gene expression was analyzed using software C1000 (Bio-Rad), ΔΔCt method as described in [44,45].
Flow cytometry analysis
Flow cytometry was performed on hPSCs cultures as previously described [45], using appropriate primary antibodies as described in Supplementary Table S2. Species and isotype-matched AlexaFluor-conjugated secondary antibodies (1:1,000; Invitrogen) were used. Appropriate isotype control antibodies diluted at an identical final concentration as the primary antibodies were used as a negative control for the experiments. Stained cells were analyzed by flow cytometry using either BD™ LSR II or BD Accuri™ C6 Flow Cytometers (both from BD Biosciences). Data were subsequently analyzed using either BD Accuri® C6 CFlow SAMPLER (BD Biosciences) or FCS EXPRESS 4 image cytometry analysis software (DeNovo).
Statistical analysis
Data represent mean±standard deviation of the number (n) of independent experiments unless indicated otherwise. Statistical significance has been calculated by a two-way ANOVA with Bonferroni post-tests to compare replicate means between the indicated groups for all experiments.
Results
RELB expression modulates hPSC proliferation but does not affect expression of pluripotency or differentiation genes in hPSCs cultured under pluripotency-maintaining conditions
To address the role of RELB in hPSCs, we established HES3, H9 hESCs, and nonviral integration-free C11 iPSCs [45] that stably express doxycycline (dox)-inducible RELB-shRNA or targetless control shRNA. Multiple lentivirally transduced and puromycin-selected clones from each of the three lines displayed robust reductions of the RELB transcript levels as measured by qPCR (Fig. 1A) and RELB protein expression measured by western blotting (Fig. 1B) after 4 days of doxycycline addition. This shRNA-mediated knockdown of RELB was maintained for more than 21 days of doxycycline treatment (Supplementary Fig. S1A), whereas targetless shRNA-expressing cells did not exhibit significant RELB knockdown (Fig. 1A and Supplementary Fig. S1B). Both control and RELB-knockdown hPSCs were passaged weekly.
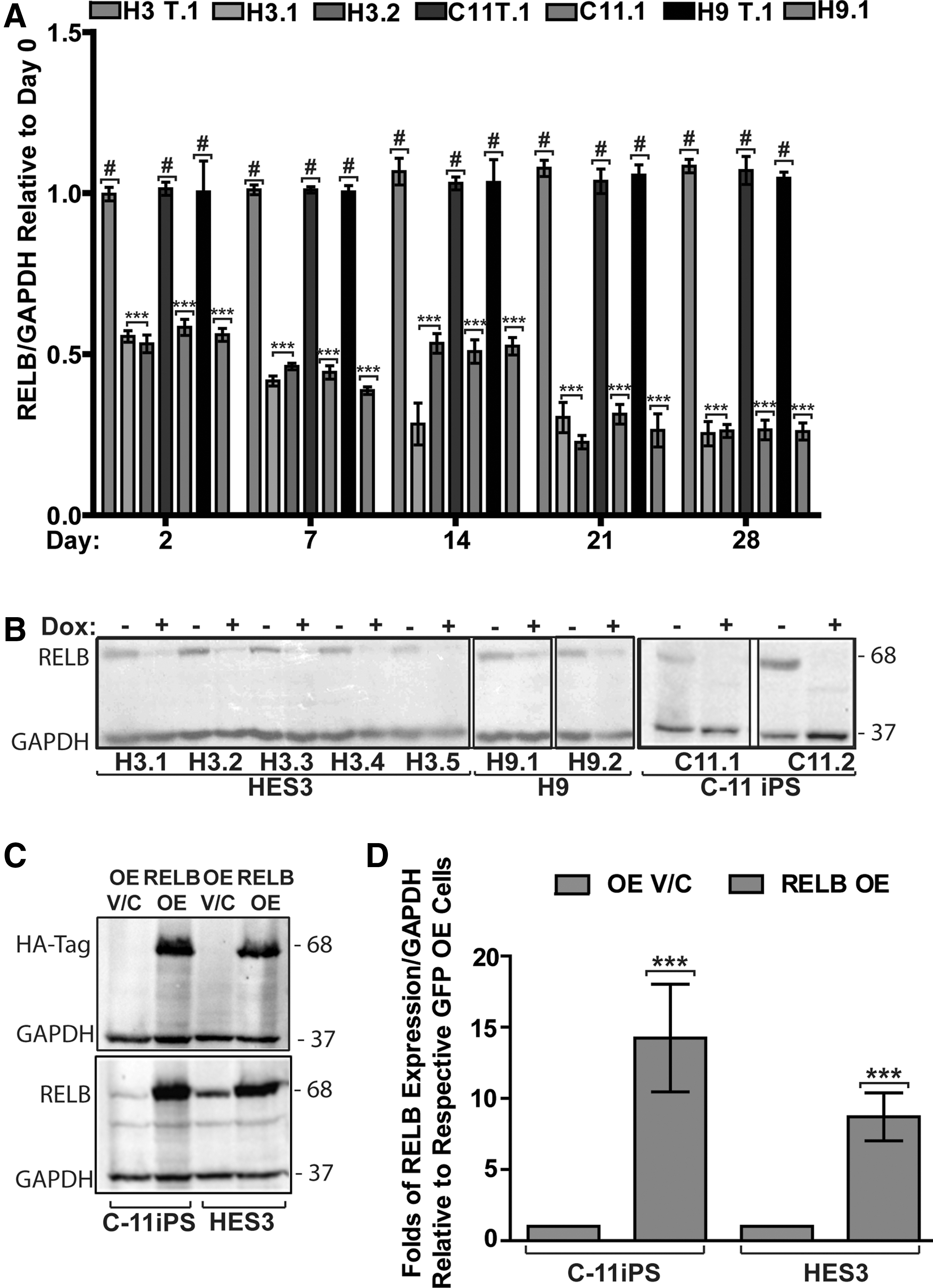
Establishment of human embryonic stem cells (hESCs; HES3 and H9) and induced pluripotent stem cells (iPSCs; C11) that either stably express doxycycline-inducible shRNAs against RELB or overexpress HA-tagged RELB.
We next investigated the impact of RELB knockdown on pluripotency in the RELB-shRNA and targetless-shRNA hESC and iPSC lines cultured under pluripotency-maintenance conditions after 7, 14, and 21 days of exposure to doxycycline. Despite robust RELB knockdown, no differences in expression levels of the pluripotency transcription factor OCT4 were observed in any of the hPSC lines tested by either immunofluorescence (Supplementary Figs. S1C and S2) or western-blot analysis (Supplementary Fig. S1D). In agreement with these data, no significant upregulation of protein expression of lineage markers such as PAX6 (ectoderm), ISL1 (mesodermal), or AFP and FOXA2 (endodermal) was observed after prolonged RELB knockdown (Supplementary Fig. S1E).
To assess the effects of RELB overexpression, we transduced HES3 hES and C11 iPS cell lines with a lentivirus carrying an HA-tagged RELB overexpression construct (RELB OE). Western blot analysis at 10 days post-transduction and puromycin selection shows that these hESCs and iPSCs display 8- to 14-fold increased RELB protein expression (Fig. 1C, D) as compared with identically generated OE V/C HES3 or C11 cells.
Similar to RELB knockdown, RELB overexpression did not affect the expression of pluripotency-associated proteins OCT4 and NANOG under pluripotency-maintaining cell culture conditions (Supplementary Fig. S3A, B). Our data further show that RELB overexpression did not affect expression of the canonical NFκB proteins RELA and NFκB1, while expression of the noncanonical NFκB proteins NFκB2 and NIK was reduced in RELB OE as compared with OE V/C hPSCs, perhaps indicative of a negative-feedback mechanism (Supplementary Fig. S3A, B). Based on these data, we conclude that modulation of RELB expression does not affect pluripotent growth of hESCs and hiPSCs. In both hESC and iPSC lines that overexpress RELB, we consistently observed an obvious increase in cell number, resulting in 2-fold higher cell numbers after only 5 days of culture under pluripotency conditions (Fig. 2A). This appears to be due to enhanced cell-cycle transit rate rather than due to reduced cell death, since we did not detect any effect of RELB over- or under-expression on apoptosis as assessed by activated caspase-3, the number of PI-positive sub-G1/G0 cells, or TUNEL labeling (results not shown).
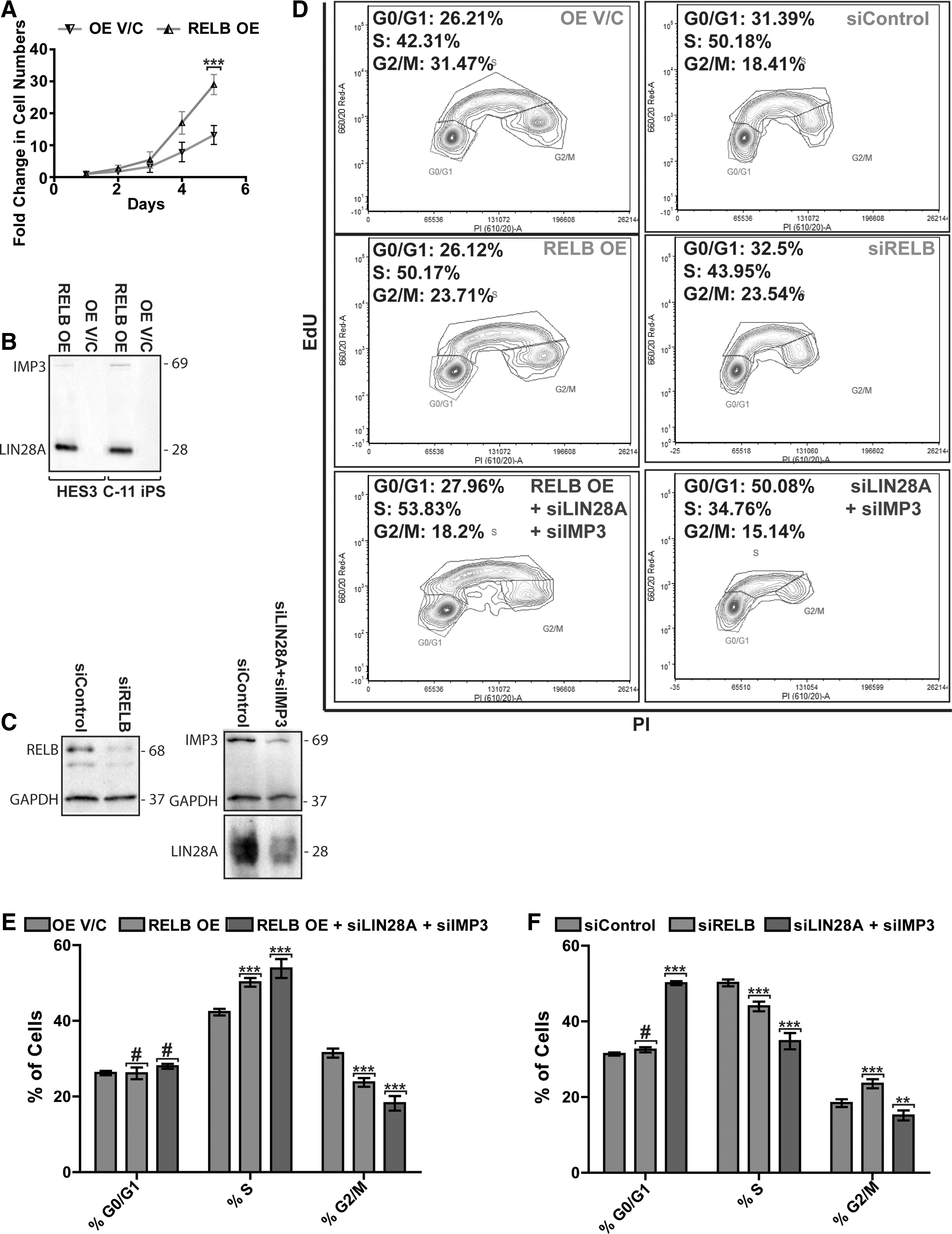
RELB interacts with LIN28A and IMP3 (IGF2 mRNA-binding protein 3) and regulates proliferation of human pluripotent stem cells (hPSCs).
RELB interacts with IMP3 and LIN28
To gain insight into the mechanism underlying RELB's role in hPSC cell proliferation, we decided to identify its interacting partners. To this end, we immuno-precipitated HA-tagged RELB from the HA-tagged RELB-overexpressing lines using the corresponding OE V/C lines to control for nonspecific interactions. Proteins that specifically interact with HA-RELB but not with OE V/C were identified by liquid chromatography-mass spectrometry (LC-MS) in triplicate samples of both hESCs (HES3) and iPSCs (C11). Of the 479 proteins identified by LC-MS, 206 proteins were found only in the HA-RELB and not in the untagged-OE V/C hPSC lines (Supplementary Table S3). IPA Ingenuity software analysis of this set of proteins reveals processes such as protein synthesis, mRNA translation, and cell cycle as significantly over-represented functional categories across both hESCs and hiPSCs (Supplementary Table S4). Of note, within this set of putative RELB-interacting proteins, we find a preponderance of RNA-binding proteins that have been directly or indirectly associated with cytoplasmic post-transcriptional regulation of mRNA translation in polysomes, p-bodies, and stress granules, including RUVB1 [46], RUVB2 [46], the RNA-binding cold shock protein CIRBP [47], and the Poly-(rC) Binding Protein PCBP1 [48,49].
We were, however, most intrigued by the observation that in hiPSCs and hESCs RELB protein directly interacts with LIN28A and IMP3 (Supplementary Table S3) since LIN28A and IMP3 were previously shown to bind to mRNA of IGF2, an important mitogen for hPSC that stimulates their proliferation, and were shown to enhance IGF2 translation efficiency during skeletal myogenesis. Indeed, anti-HA tag antibodies were able to immuno-precipitate IMP3 and LIN28A from hPSCs overexpressing HA-RELB but not from OE V/C cells (Fig. 2B), suggesting that RELB may form a multi-protein complex with LIN28A and IMP3 and regulate IGF2 translation in these cells.
RELB regulates the expression of IGF2 and key cell-cycle proteins via a mechanism dependent on LIN28 and IMP3
To explore this possibility, we used LIN28 and IMP3 siRNAs to assess whether such an interaction was required for the RelB-dependent regulation of IGF2 and/or the expression of cell-cycle regulators in hiPSC. First, we verified the efficiency of siRNA-mediated knockdown of RELB, LIN28A, and IMP3 using western-blot analysis (Fig. 2C) and next ascertained that RELB knockdown did not affect LIN28A or IMP3 expression in undifferentiated hPSCs or vice versa (Supplementary Fig. S4A, B).
Next, we assessed the effect of RELB under- and overexpression on cell-cycle regulation in hPSC via EdU incorporation in the absence and presence of LIN28A and IMP3 siRNAs. Cell-cycle analysis shows that RELB overexpressing cells display an increased number of cells residing in the S-phase accompanied by a decline in G2/M population as compared with OE V/C control cells (Fig. 2D, E). In agreement with the observed increased S-phase occupancy, RELB overexpressing hiPSCs display increased levels of IGF2, CYCLIN E, CYCLIN D1, and C-MYC proteins (Fig. 3A, B) and CYCLIN E1, CYCLIN E2, and CYCLIN D1 mRNAs (Supplementary Fig. S4A). Conversely, RELB knockdown in hiPSCs leads to a decrease in cells in the S-phase and increased G2/M population as compared with targetless-siRNA-expressing hiPSCs (Fig. 2D, F). This is accompanied by a decrease in IGF2, CYCLIN E, C-MYC, and CYCLIN D1 protein expression (Fig. 3A, B) and decreased CYCLIN E, C-MYC, C-FOS, and CYCLIN D1 transcript levels (Supplementary Fig. S4B). We conclude that RELB promotes cell-cycle progression of hPSCs by controlling the expression of IGF2 and key cell-cycle regulators that specifically enhance G1-S progression.
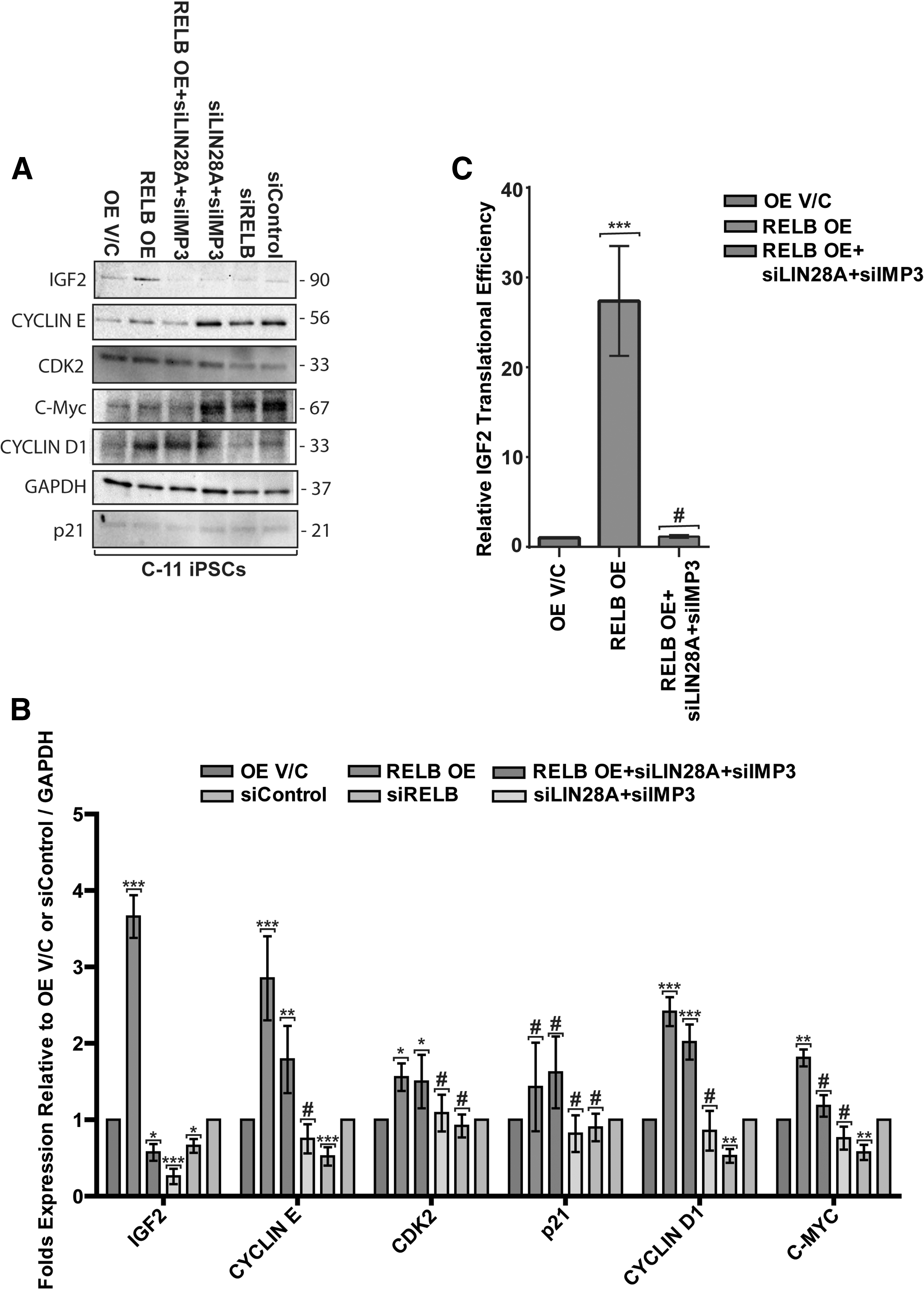
RELB regulates the expression of insulin-like growth factor 2 (IGF2) and other key cell-cycle proteins involved in G1-S transition.
Next, we analyzed these parameters in iPSC that either under- or overexpressed RELB in the absence or presence of LIN28 and IMP3-targeting siRNAs. Notably, IGF2 protein expression clearly increased in RELB-overexpressing hPSCs (Fig. 3A, B) despite reduced IGF2 mRNA levels in these cells (Supplementary Fig. S4A), suggesting this is due to increased mRNA translation or protein stability. Importantly, increased IGF2 translational efficiency (Fig. 3C) in RELB-overexpressing hPSC was abrogated on siRNA knockdown of LIN28A and IMP3 (Fig. 3A, B), suggesting that IMP3 and LIN28 are required for this effect. Conversely, RELB knockdown leads to a significant reduction in IGF2 protein expression that is further exacerbated with a simultaneous knockdown of LIN28A and IMP3 (Fig. 3A, B) despite identical IGF2 mRNA levels in all of these samples (Supplementary Fig. S4B). Furthermore, the increases in CYCLIN E, CYCLIN D1, and C-MYC proteins brought about by RELB OE were also significantly reduced on knockdown of LIN28A and IMP3 (Fig. 3A, B), strongly suggesting that RELB, along with LIN28A and IMP3, plays a role in the modulation of IGF2 mRNA translation/stability, as well as translation/mRNA stability of key G1-S phase transition regulators such as CYCLIN E, CYCLIN D1, and C-MYC. The protein expression of p21 and CDK2 (Fig. 3A, B), and mRNA levels of NFκB2, p27, CYCLIN D3, and CDC25A remained largely unaffected after modulation of RELB alone or in combination with either LIN28A or IMP3 expression in hPSCs (Supplementary Fig. S4B), indicating that this is not a general effect on all cell-cycle regulators. Collectively, these data reveal that RELB interacts with IMP3 and LIN28A and that this interaction is likely required for modulating the protein expression of IGF2 and selected cell-cycle regulators through a mechanism largely independent from mRNA levels.
RELB, IMP3, and LIN28 co-localize in cytoplasmic processing bodies such as stress granules in hPSCs
In light of the preponderance of cytoplasmic RNA binding and processing proteins that immunoprecipitated with RELB and the apparent synergistic action of RELB, LIN28A and IMP3 in post-translational regulation of the expression of IGF2 and other cell-cycle regulators, we hypothesized that RELB, LIN28, and IMP3 may be a part of a complex that is involved in regulation of mRNA translation. To explore this hypothesis, we treated hPSCs with sodium arsenite, a well-known inducer of cytoplasmic stress granules, structures known to be associated with translational stalling. Indeed, after arsenite treatment of control and RELB-overexpressing hPSCs, a proportion of the RELB protein in the cell is redistributed to stress granules marked by TIA-1 protein, a well-established stress granule marker, and this is accompanied by a prominent redistribution of both IMP3 and LIN28A to such TIA-1-positive structures (Fig. 4 and Supplementary Fig. S5). In arsenite-untreated RELB-overexpressing and control cells, TIA-1 is solely nuclear; RELB displayed a distinctly punctate staining in the nucleus, peri-nuclear border, and sections of the cytoplasm; LIN28A displayed strong punctate peri-nuclear and cytoplasmic staining; and IMP3 displayed strong punctate peri-nuclear and cytoplasmic staining.

RELB, IMP3, and LIN28A co-localize to the cytoplasmic stress-induced granules. Immunofluorescence analysis of RELB, LIN28A, IMP3, and TIA-1 intracellular localization using confocal microscopy in RELB overexpressing cells and control hiPSCs treated with and without sodium arsenite for induction of stress granule formation. Images were acquired at 63×magnification with 2× digital zoom. Nuclear DNA was stained using DAPI. Scale bar, 10 μm. The images are representative of at least three independent experiments. High magnification images of the insets can be found in Supplementary Fig. S5.
To our knowledge, this is the first demonstration that RELB protein localizes to stress granules, and it lends further support to the idea that formation of an RELB-LIN28A-IMP3-containing protein complex regulates expression levels of selected cell-cycle regulators in hPSCs.
Discussion
An ability to make rapid cell fate decisions and controlling the pluripotent stem cell pool size in the inner cell mass of the blastocyst is critical for achieving correct developmental outcomes. Although an understanding of the drivers of lineage fate decisions and the epigenetic changes that accompany these processes is constantly improving [50 –52], little is known about what controls these intricate and timing-sensitive processes. Considering the short cell cycle of hESCs and hiPSCs and the need to carefully control the number of lineage-committed progenitors in the early embryo, it is difficult to envisage that this is solely dependent on the mitogen-induced mRNA transcription-protein translation cascade or epigenetic regulation alone, given the timescales involved. An emerging mechanism that allows for such rapid responses is prestoring mRNA and regulating its stability, splicing, and translation in cytoplasmic processing compartments such as p-bodies and stress granules. Indeed, these cytoplasmic structures were found to transiently interact with each other and with polysomes to dynamically exchange mRNA and regulate mRNA translation [14,53 –56]. These “molecular handshakes” are governed by a suite of RNA-binding molecules, RNA helicases, and cytoskeletal transport regulatory proteins. LIN28 is a prototypical example of such an RNA-binding protein that is involved in both repression of let-7 miRNA processing and mRNA translation in p-bodies and stress granules, but the processes that direct LIN28 to its different subcellular locations or how it may affect mRNA translation have remained unclear. As a member of the NFκB transcription factor family, RELB is well placed to respond to extracellular signals, for example, being the target of CD40 and BAFF (B cell-activating factor). RELB's cycle of nucleo-cytoplasmic translocation, its stability, positive and negative feedback regulatory loops on its own processing, cross-regulation of, and dimerization with other NFκB family members make RELB a versatile mediator for regulation of the intracellular trafficking [57 –65]. Indeed, our data strongly suggest that RELB, in addition to its role as a classical nuclear transcription factor, can also act as a regulator of mRNA translation in the cytosol. This is supported by our finding that the RELB protein interacts with IMP3 and LIN28A proteins and that after a stress stimulus (eg, treatment with arsenite), IMP3, LIN28, and RELB localize to stress granules that are marked by TIA-1. Our data indicate that in hPSCs, RELB may be involved in coordinating the shuttling of specific mRNAs to such cytoplasmic mRNA regulatory domains in conjunction with IMP3 and LIN28A, and thus affect the translation of IGF2 (an important mitogen for these cells) and cyclins E, D1, and C-MYC. It thus appears plausible that RELB recruits LIN28A and IMP3 to a subset of genes that are transcriptionally controlled by RELB, and that this complex together with the mRNA shuttles to the cytosol where it can next be stored in stress granules until rapid translation is needed. This post-transcriptional regulatory role of RELB is particularly evident in the regulation of IGF2 protein levels, an important mitogen for hPSC that is required for inner cell mass survival and fetal growth. While OE of RELB does not increase IGF2 mRNA expression (it, in fact, lowers IGF2 mRNA levels), it leads to an almost 4-fold upregulation of IGF2 protein expression. Importantly, this effect on IGF2 mRNA translation is entirely abrogated by simultaneous siRNA-mediated knockdown of LIN28A and IMP3, two proteins known to both interact with IGF2 mRNA and regulate its translation [20,66], and both of which interact with RELB protein, as demonstrated in this paper. Given the central role of IGF2 in cell cycle and pluripotency in hESC, the robust decrease in IGF2 protein observed on RELB knockdown caused surprisingly little changes in cell cycle and did not affect maintenance of pluripotency markers. This is possibly because of the fact that hPSCs were cultured under pluripotency-maintenance conditions supplemented with molecules such as FGF2, which may compensate for this decrease in IGF2.
In addition to implicating RELB in gene expression regulation at this novel level, our data also provide further insights into the role of this transcription factor in hPSCs. Previously, complete genetic inactivation of the RelB gene in mice was shown to cause abnormalities from as early as 8 days of development, leading to multi-organ inflammation, impaired cellular immunity, myeloid hyperplasia, splenomegaly, and a reduced population of thymic dendritic cells, indicating an important role for RelB in the immune system. RelB was further shown to control the switch between proliferation of CD34+ hematopoietic progenitors and their differentiation into functional dendritic cells [67] and to inhibit differentiation and promote mitochondrial biogenesis during skeletal myogenesis. To investigate the role of RELB in hESCs, Yang et al. [40] employed siRNA-mediated RELB knockdown in an hESC line to show that RELB knockdown inhibited hESC proliferation, increased BRACHYURY and CDX2 while reducing SOX2 and PAX6 mRNA levels, leading the authors to suggest that RELB may maintain hESC pluripotency by repression of differentiation. To allow repression of RELB over a timescale that is more amenable to investigations into its role in proliferation and differentiation and to circumvent potential difficulties in interpreting effects of short-term knockdown of the long-lived RELB protein, we opted for generation of the hESC and hiPSC lines with doxycycline-inducible RELB shRNA. Our data show that prolonged knockdown or overexpression of RELB in either human ES or iPS cell lines does not affect pluripotency and the expression of OCT4 and NANOG. Our data agree with the previous RELB study, in which RELB knockdown indeed inhibits proliferation, but differs from that study in showing that RELB overexpression increases proliferation of hPSCs without affecting their pluripotency. We further demonstrate that RELB regulates several cell-cycle genes such as CYCLIN D1, C-MYC, CDK2, and CYCLIN E, known to control G1-S phase transition, and that this largely occurs at a post-transcriptional level. In light of the strong protein–protein interaction between RELB and LIN28A/IMP3, their synergistic effect on IGF2 and cell-cycle regulators' mRNA translation, and their co-localization in stress granules in hPSCs (after arsenite-induced stress), we propose that in addition to its role as a transcription factor, RELB is involved in attenuating the mRNA translation in cytoplasmic mRNA-processing bodies. We speculate that this novel function of RELB may play a role similar to its oncogenic role in glioblastomas [68] and pancreatic cancer [69], where both of its binding partners identified in this study, IMP3 and LIN28, have been implicated in regulation of the epithelial-to-mesenchymal transition and metastasis [70]. While much is still to be learned about this novel function of RELB in regulating cytoplasmic mRNA metabolism and translation, and how this relates to early differentiation patterning and proliferation of hPSCs during early development, data from our laboratory show that elevated RELB acts to inhibit early mesodermal and neuro-ectodermal differentiation through a very similar mechanism (Supplementary Figs. S6–S10; Supplementary Materials, Methods and Results are available online at
Footnotes
Acknowledgments
Access to proteomic infrastructure in the QIMR Berghofer Protein Discovery Center was made possible by funding from Bioplatforms Australia and the Queensland State Government provided through the Australian Government National Collaborative Infrastructure Scheme (NCRIS). The authors would like to thank StemCore QLD, ISIS Pharmaceuticals, Prof. Brian Gabrielli, Prof. Martin Lavin, and Prof. Ranjeny Thomas for provision of reagents. They are also extremely grateful to Mr. Owen Hawksworth, Dr. Alejandro Hidalgo, and Ms. Emily Allan for their help with collection and assembly of data.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.