
Abstract
A high proliferation rate of malignant cells requires an increased energy production, both by anaerobic glucose metabolism and mitochondrial respiration. Moreover, increased levels of mitochondria-produced reactive oxygen species (ROS) promote survival of transformed cells and contribute to the disease progression both in solid tumors and leukemia. Consequently, interfering with mitochondrial metabolism has been used as a strategy to specifically target leukemic cells. SAM50 is a mitochondrial outer membrane protein involved in the formation of mitochondrial intermembrane space bridging (MIB) complex. Although the importance of SAM50 in maintaining MIB integrity and in the assembly of mitochondrial respiratory chain complexes has been described, its specific role in the normal and leukemic hematopoietic cells remains unknown. We observed that human leukemic cells display a specific dependency on SAM50 expression, as downregulation of SAM50 in BCR-ABL-expressing, but not normal CD34+ human hematopoietic stem and progenitor cells (HSPCs) caused a significant decrease in growth, colony formation, and replating capacity. Mitochondrial functions of BCR-ABL-expressing HSPCs were compromised, as seen by a decreased mitochondrial membrane potential and respiration. This effect of SAM50 downregulation was recapitulated in normal HSPCs exposed to cytokine-rich culture conditions that stimulate proliferation. Both oncogene-transduced and cytokine-stimulated HSPCs showed increased mitochondrial membrane potential and increased ROS levels compared to their normal counterparts. Therefore, we postulate that human leukemic HSPCs are highly dependent on the proper functioning of mitochondria and that disruption of mitochondrial integrity may aid in targeting leukemic cells.
Introduction
M
As a byproduct of their increased metabolism, tumor cells generate high levels of reactive oxygen species (ROS), with mitochondria being one of the main sources [4 –7]. Although elevated ROS levels can be detrimental for both healthy and malignant cells, tumor cells have developed mechanisms to prevent ROS-induced damage.
Cancer cells counteract the overproduction of ROS by upregulating the antioxidant systems to prevent ROS-induced macromolecule damage, senescence, and apoptosis induction [8 –11]. Moreover, large amounts of reduced NADPH that are generated by the upregulated glycolysis pathway scavenge ROS and reduce oxidative stress [12,13]. Consequently, cancer cells can maintain relatively higher ROS levels that are advantageous for tumor progression as they induce a variety of signaling pathways and activate transcription factors that promote growth, survival, and malignant behavior [14 –22].
The important role of mitochondrial metabolism for energy production and redox homeostasis has also been described in hematological malignancies. In acute myeloid leukemia (AML), inhibition of mitochondrial translation disrupted the expression of mitochondrial respiratory chain (MRC) complex proteins and decreased respiration, specifically targeting leukemic cells [23]. In CML, deregulated expression of MRC components has been observed in leukemic stem cells (LSCs), indicating an increased level of oxidative phosphorylation that could reflect their high proliferation rate [24]. Elevated ROS levels have also been described in several leukemia models. Leukemic cells expressing internal tandem duplication of FLT3 (FLT3-ITD), the JAK2 (V617F) activating mutation or the BCR-ABL kinase have shown increased levels of ROS causing DNA double-strand breaks to occur. This in turn has resulted in genomic instability leading to leukemia progression and development of therapy-resistant clones [25 –28]. Moreover, elevated ROS levels increased survival of BCR-ABL-expressing cells through the activation of the PI3K/Akt signaling pathway [29].
Recently, RAC2 has been identified as a new key player in maintaining mitochondrial function in leukemic cells. In BCR-ABL-expressing cells, RAC2 has been shown to alter mitochondrial membrane potential and electron flow through the MRC complex III, generating high levels of ROS and causing DNA damage [30]. Downregulation of RAC2 in human BCR-ABL-transformed cord blood (CB) cells and primary blast crisis CML (BC CML) cells resulted in decreased mitochondrial membrane potential and decreased ROS production. Consequently, RAC2-deficient primitive leukemic cells showed increased apoptosis and diminished proliferation and self-renewal [31].
We have identified that RAC2 may be required for the proper functioning of mitochondria due to its direct interaction with mitochondrial transport proteins, such as SAM50 and Metaxin 1 [31]. These proteins, together with Metaxin 2, constitute the sorting and assembly machinery (SAM) in the mitochondrial outer membrane [32 –34]. The SAM complex, by interacting with the mitochondrial inner membrane proteins mitofilin and CHCHD3, forms the mitochondrial intermembrane space bridging (MIB) complex involved in the assembly of MRC complexes. Consequently, downregulation of SAM50 was shown to destabilize the MIB complex, causing defective assembly of respiratory complexes and structurally abnormal cristae [35]. The specific role of SAM50 in normal and leukemic hematopoietic cells, however, remains unknown.
In this study, we report that human leukemic cells are specifically dependent on SAM50 expression. Downregulation of SAM50 in BCR-ABL-expressing, but not normal human hematopoietic stem and progenitor cells (HSPCs) cause a dramatic decrease in growth, colony formation, and replating capacity, as well as a decreased mitochondrial membrane potential and respiration. This effect of SAM50 downregulation can be recapitulated in normal HSPCs when exposed to cytokine-rich culture conditions that strongly enhance proliferation. Both oncogene-transduced and cytokine-stimulated HSPCs show increased mitochondrial membrane potential and increased ROS levels compared to their normal counterparts. Therefore, we postulate that human leukemic HSPCs are highly dependent on the proper functioning of mitochondria and that disruption of mitochondrial integrity may aid in targeting leukemic cells.
Materials and Methods
Primary cell isolation and culture conditions
Neonatal CB was obtained from healthy full-term pregnancies after informed consent in accordance with the Declaration of Helsinki from the obstetrics departments of the University Medical Center Groningen (UMCG) and Martini Hospital, Groningen, The Netherlands. All protocols were approved by the Medical Ethics Committee of the UMCG. After separation of mononuclear cells with the lymphocyte separation medium (PAA Laboratories, Coble, Germany), CD34+ cells were isolated using a Magnetically Activated Cell Sorting (MACS) CD34 Progenitor Kit (Miltenyi Biotech, Amsterdam, The Netherlands). For the MS5 coculture experiments, cells were grown in Gartner's medium consisting of α-modified essential medium (α–MEM; Fisher Scientific Europe, Emergo, The Netherlands) supplemented with 12.5% heat-inactivated fetal calf serum (FCS; Lonza, Leusden, The Netherlands), 12.5% heat-inactivated horse serum (Invitrogen, Breda, The Netherlands), 1% penicillin and streptomycin, 2 mM glutamine (all from PAA Laboratories), 57.2 μM β-mercaptoethanol (Merck Sharp & Dohme BV, Haarlem, The Netherlands), and 1 μM hydrocortisone (Sigma-Aldrich Chemie B.V., Zwijndrecht, The Netherlands). Alternatively, cocultures were expanded in Gartner's medium supplemented with 20 ng/mL interleukin-3 (IL-3; Gist-Brocades, Delft, The Netherlands) and stem cell factor (SCF; Amgen, Thousand Oaks, CA).
Cell lines and culture conditions
The 293T embryonic kidney cells (ACC-635; DSMZ, Braunschweig, Germany) and PG13 packaging cells (ATCC CRL-10686, Wesel, Germany) were grown in Dulbecco's modified Eagle's medium (DMEM) with 200 mM glutamine (BioWhittaker, Veries, Belgium) supplemented with 10% FCS and 1% penicillin and streptomycin. K562 myelogenous leukemia cells (ACC-10; DSMZ) were grown in Roswell Park Memorial Institute medium with 200 mM glutamine (BioWhittaker) supplemented with 10% FCS, and 1% penicillin and streptomycin. MS5 murine stromal cells (ACC-441; DSMZ) were grown in αMEM with 200 mM glutamine (BioWhittaker) supplemented with 10% FCS and 1% penicillin and streptomycin.
Retro- and lentivirus generation and transduction
Stable PG13 producer cell lines of BCR-ABL retroviral constructs were generated and used as published previously [36]. Supernatants from the PG13 cells were harvested after 8–12 h of incubation in human progenitor growth medium (HPGM; Cambrex, Verviers, Belgium) before the retroviral transduction rounds and passed through 0.45-μm filters (Sigma-Aldrich Chemie B.V.). Before the first transduction round, CD34+ CB cells were prestimulated for 48 h in HPGM supplemented with 100 ng/mL SCF, FLT3 Ligand (Flt3L; both from Amgen), and thrombopoietin (TPO; Kirin, Tokyo, Japan). Three rounds of transduction were performed on retronectin-coated 24-well plates in the presence of the same cytokines as for prestimulation and 4 μg/mL polybrene (Sigma-Aldrich Chemie B.V.). During the last round of retroviral transduction with BCR-ABL, lentiviral short hairpin RNA (shRNA) particles were also added as described below.
shRNA sequences targeting SAM50 were ligated into pHR'trip vector using AcsI and SbfI restriction sites as previously described [31]. For the control, scrambled (SCR) shRNA sequence was used. The 293T embryonic kidney cells were transfected using FuGENE6 (Roche Diagnostics, Almere, The Netherlands) with 3 μg pCMV Δ8.91, 0.7 μg VSV-G, and 3 μg of pHR'trip vector constructs (shSCR or shSAM50). After 24 h, the medium was changed to HPGM and after 12 h the supernatant containing lentiviral particles was harvested and either stored at −80°C or used fresh for transduction of target cells. CD34+ CB cells were subjected to three rounds of transduction with lentiviral particles, and BCR-ABL-transduced CD34+ CB cells were subjected to one round of transduction with lentiviral particles, together with the last round of retroviral transduction, in the presence of prestimulation cytokines and 4 μg/mL polybrene (Sigma-Aldrich Chemie B.V.) on retronectin-coated 24-well plates (TaKaRa, Tokyo, Japan). K562 cells were subjected to one round of transduction with lentiviral particles in the presence of 4 μg/mL polybrene, on retronectin-coated 24-well plates. After transduction, transduced green fluorescent protein-positive, truncated nerve growth factor receptor (NGFR)-positive, or double-positive cells were sorted on a MoFlo sorter (Dako Cytomation, Carpinteria, CA).
Long-term cultures on stroma, colony forming cell, and long term culture-initiating cell assay
After sorting, 5 × 104 CB cells or 5 × 103 BCR-ABL cells were plated onto a T25 flask precoated with MS5 stromal cells in 5 mL of Gartner's medium in duplicate. Cocultures were kept at 37°C and 5% CO2 and cells were demi-depopulated weekly for analysis. Images of cobblestones were acquired on Leica DMIL inverted phase microscope (Leica Microsystems, Eindhoven, The Netherlands). CFC assays were performed as previously described [37]. For the LTC-IC assay, CB cells were plated in the range of 6–1,458 cells per well in a 96-well plate using Gartner's medium. Methylcellulose (StemCell Technologies, Grenoble, France) supplemented with 20 ng/mL of IL-3, 20 ng/mL of interleukin-6 (IL-6; Gist-Brocades), 20 ng/mL of granulocyte-colony stimulating factor, 20 ng/mL of c-kit ligand (Amgen), and 6 U/mL of erythropoietin (Epo; Cilag, Eprex, Brussels, Belgium) was added at week 5. Two weeks later, wells containing CFCs were scored as positive and the LTC-IC frequency was calculated using L-Calc software (StemCell Technologies). May-Grünwald–Giemsa staining was used to stain cytospins. Cytospin preparations were evaluated and photographed using a Leica DM3000 microscope equipped with a Leica DFC420C digital camera at a total magnification of ×400.
Flow cytometry analyses and sorting
All fluorescence-activated cell sorter (FACS) analyses were performed on a FACSCalibur [Becton-Dickinson (BD), Alpen a/d Rijn, The Netherlands] and data were analyzed using WinList 3D (Verity Software House, Topsham, ME). Cells were sorted on a MoFlo sorter. Antibodies NGFR-APC, CD14-PE, and CD15-APC were obtained from BD.
Measurement of mitochondrial functions
Mitochondrial membrane potential was measured by flow cytometry using hexamethylindodicarbocyanine iodide (DilC1; Life Technologies, Bleiswijk, The Netherlands) as described previously [38]. Briefly, 5 × 105 cells were incubated with 50 ng/mL DilC1 for 30 min at 37°C, washed twice in phosphate-buffered saline (PBS), and DilC1 fluorescence was analyzed by FACS. To measure intracellular ROS levels, staining with CellROX Deep Red reagent was performed according to the manufacturer's protocols (Life Technologies). Fluorescence of the probe was analyzed by FACS. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured on the XFe24 Extracellular Flux Analyzer according to the manufacturer's instructions (Seahorse Bioscience, Billerica, MA). Briefly, XFe24 Cell Culture Microplate was coated with poly-
Immunoblotting
Western blot analysis was performed according to standard protocols. Antibody against SAM50 was kindly provided by Dr. V. Kozjak-Pavlovic (University of Würzburg, Würzburg, Germany). Secondary antibody (goat-anti-rabbit-horseradish peroxidase) was purchased from Dako Cytomation and used in 1:3,000 dilutions. Binding of antibodies was detected by chemiluminescence, according to the manufacturer's instructions (Roche Diagnostics).
Statistical analysis
All values are expressed as mean ± standard deviation (SD). A two-tailed Student's t-test was used for all comparisons. Differences were considered statistically significant at P < 0.05.
Results
SAM50 expression is required for the long-term expansion of human BCR-ABL-expressing HSPCs
We have recently identified that the mitochondrial protein SAM50 is a bona fide interaction partner of RAC2 [31]. To investigate in more detail the effect of SAM50 downregulation in human leukemic cells, we retrovirally transduced CD34+ CB cells to overexpress the BCR-ABL oncogene. Simultaneously, lentiviral transduction with a shRNA-containing construct was performed to downregulate SAM50, and double-transduced cells were sorted and plated in stromal cocultures. The efficiency of SAM50 knockdown was assessed by western blot (Fig. 1A). SAM50-depleted cells reproducibly showed a growth impairment within the first weeks of coculture and a significantly reduced replating capacity (Fig. 1B, C). Although progenitor frequencies assessed by CFC assay directly after sorting and at week 1 of coculture were not significantly lower in SAM50-depleted cocultures, at week 2 they were markedly reduced (Fig. 1D). Importantly, also the replating potential of colony-forming cells was significantly decreased upon SAM50 downregulation (Fig. 1D). Differentiation of SAM50-depleted cells did not differ from the controls and also cell viability, assessed by microscopical evaluation of the cocultures and flow cytometry, was not affected by SAM50 downregulation (data not shown). To further validate these findings, two independent shRNA vectors were generated, and both efficiently downregulated SAM50 at the RNA as well as protein level (Fig. 1E, F). Again, a marked decrease in cumulative cell counts was observed upon knockdown of SAM50 (Fig. 1G), coinciding with a reduction in cobblestone formation indicating that the most primitive cell fraction was affected by SAM50 downregulation (Fig. 1H). Overall, these data suggested that BCR-ABL-transformed cells require SAM50 expression for their long-term proliferation and self-renewal.

Human BCR-ABL-transduced HSPCs depend on SAM50 expression for their long-term expansion.
Depletion of SAM50 results in mitochondrial dysfunction in BCR-ABL-expressing HSPCs
Downregulation of SAM50 has been shown to destabilize the MIB complex, causing defective assembly of respiratory complexes [35]. To further investigate whether, in a human leukemia model, SAM50 depletion would result in mitochondrial impairment, we assessed the mitochondrial membrane potential in BCR-ABL-transduced CD34+ CB cells upon knockdown of SAM50. FACS analysis using the DilC mitochondrial dye revealed that depletion of SAM50 caused a marked decrease in the mitochondrial membrane potential in BCR-ABL-transduced HSPCs, in accordance with our previous findings (Fig. 2A) [31]. The DilC peak was broader than we usually observe, see for instance also Fig. 3F. This was most likely due to the fact that part of the BCR-ABL-expressing cells differentiated along the erythroid lineage as we have seen previously [36]. As a consequence, a heterogeneity of immature cells as well as more mature cells committed toward the erythroid lineage were present in these cultures all displaying different levels of DilC, and we noted that the decrease in DilC staining upon SAM50 downregulation was particularly seen in the more immature cells (data not shown). The level of ROS measured in BCR-ABL HSPCs was not significantly altered upon SAM50 downregulation (Fig. 2B).

Depletion of SAM50 causes mitochondrial dysfunction in BCR-ABL-expressing HSPCs.
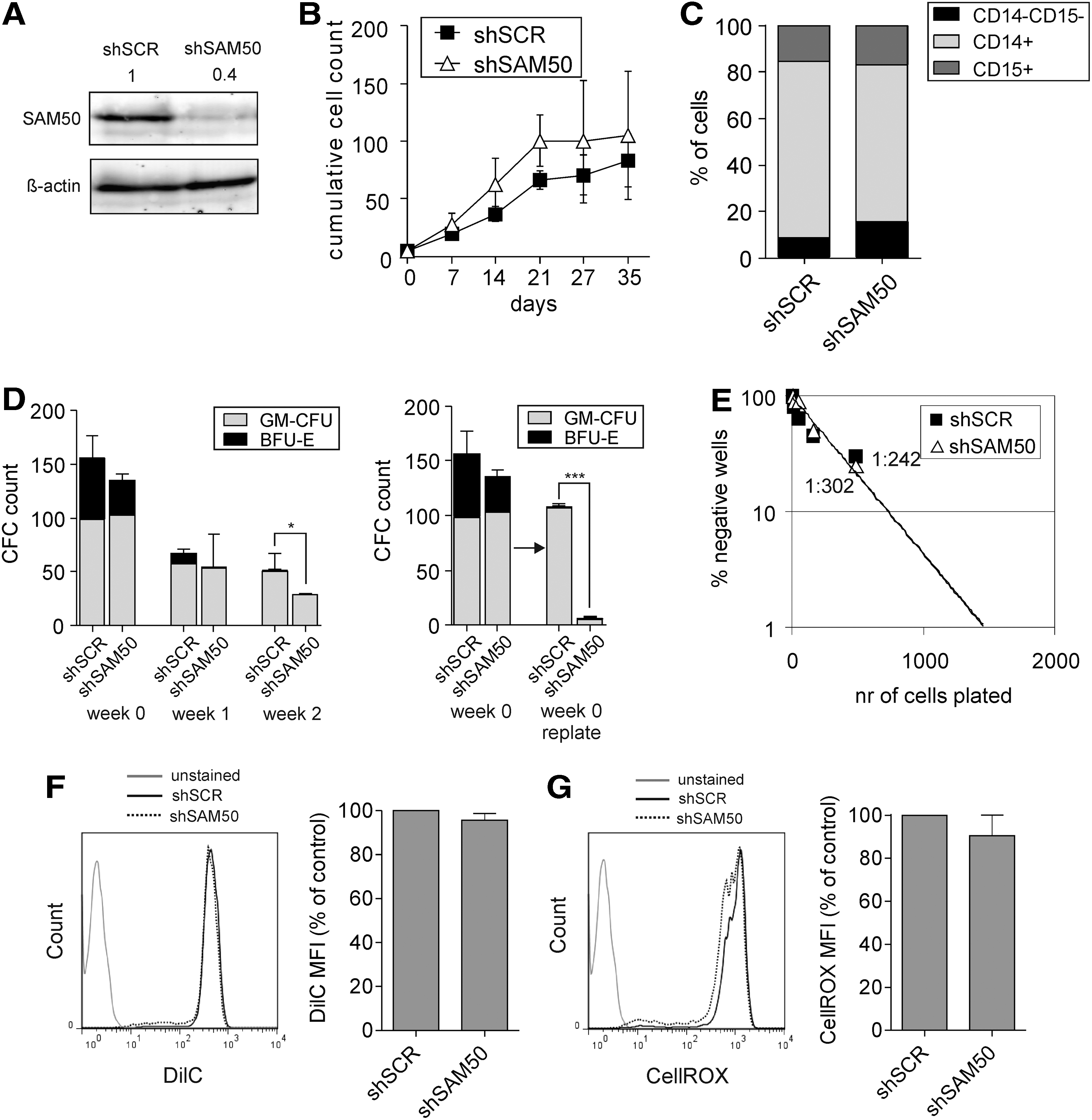
SAM50 downregulation affects replating capacity, but not proliferation or progenitor and stem cell frequencies of human HSPCs in stromal cocultures.
To investigate whether mitochondrial oxidative phosphorylation was affected by SAM50 depletion, we used the XFe Analyzer of extracellular flux to measure the OCR and ECAR of the BCR-ABL-expressing leukemic K562 cell line. OCR levels of shSAM50-transduced K562 cells were markedly reduced compared to control shSCR-transduced cells, indicative of decreased mitochondrial respiration. At the same time, ECAR levels were not significantly changed, indicating that glycolysis was not affected by SAM50 downregulation (Fig. 2C). Subsequently, we measured OCR and ECAR in BCR-ABL and shSAM50 double-transduced CD34+ CB cells. Also in this case, the OCR levels were significantly downregulated upon SAM50 depletion, while the decrease in ECAR levels did not reach significance (Fig. 2D). Finally, the two additional independent shRNA SAM50 vectors were also tested in K562 cells and both reduced OCR, while the reduction in ECAR was less pronounced and reached significance only with shSAM50 #2 (Fig. 2E). Overall, these results showed that SAM50 downregulation negatively affected mitochondrial membrane potential and resulted in decreased mitochondrial respiration in BCR-ABL-expressing cells.
Downregulation of SAM50 in normal HSPCs does not affect proliferation, progenitor and stem cell frequencies, or mitochondrial membrane potential, but decreases the replating capacity
Leukemic cells have been proven to be more sensitive to therapeutics targeting mitochondrial metabolism than normal hematopoietic cells [23]. Therefore, we investigated whether downregulation of SAM50 could also be used as a strategy to specifically target leukemic, but not normal hematopoietic cells. To study the long-term effects of SAM50 downregulation in human HSPCs, shRNA-transduced CD34+ CB cells were sorted and plated on a bone marrow stromal feeder layer for 5 weeks. The efficiency of SAM50 downregulation in shRNA-transduced cells was assessed by western blot (Fig. 3A). During the coculture, SAM50-depleted cells showed no proliferative disadvantage compared to control shSCR-transduced cells (Fig. 3B).
Differentiation along the myeloid lineage was also unaffected (Fig. 3C). In addition, SAM50 downregulation did not result in a significant decrease in progenitor frequency as assessed by CFC assay using cells directly after transduction and sorting (Fig. 3D, week 0) or by using cells taken at week 1 from MS5 cocultures shown in Fig. 3B. Only a slight reduction in CFCs was noted using cells taken at week 2 from MS5 cocultures (Fig. 3D). Similarly, no differences were seen in the most primitive stem cell fraction upon SAM50 knockdown as determined by LTC-IC assays (1:302 vs. 1:224, P = 0.4, Fig. 3E). Also, the mitochondrial functions as assessed by the measurement of the mitochondrial membrane potential and ROS levels were not different between SAM50-depleted and control HSPCs (Fig. 3F, G). However, the replating capacity of colony-forming cells was significantly reduced upon SAM50 downregulation, suggesting that under stress conditions SAM50 also becomes important in normal cells (Fig. 3D).
BCR-ABL-transduced human HSPCs show increased mitochondrial activity
We speculated that the high sensitivity of BCR-ABL-expressing cells to SAM50 downregulation could originate from their increased mitochondrial activity. To this end, we directly compared mitochondrial membrane potential and ROS levels in normal and BCR-ABL-expressing human HSPCs. CD34+ cells isolated from a single batch of CB were used to eliminate interbatch variability, and either cotransduced with BCR-ABL and shRNA constructs, or transduced with shRNA only. Sorted cells were plated in stromal cocultures and FACS analyses were performed at the same time for normal and BCR-ABL-transduced cells. FACS analysis of DilC dye showed that the mitochondrial membrane potential significantly increased in BCR-ABL-expressing HSPCs, compared to controls (Fig. 4A). ROS levels in BCR-ABL HSPCs were also increased, although to levels that were not statistically significant (Fig. 4B). Overall, these data indicate that the mitochondrial activity of leukemic cells is altered in comparison to normal CD34+ cells.
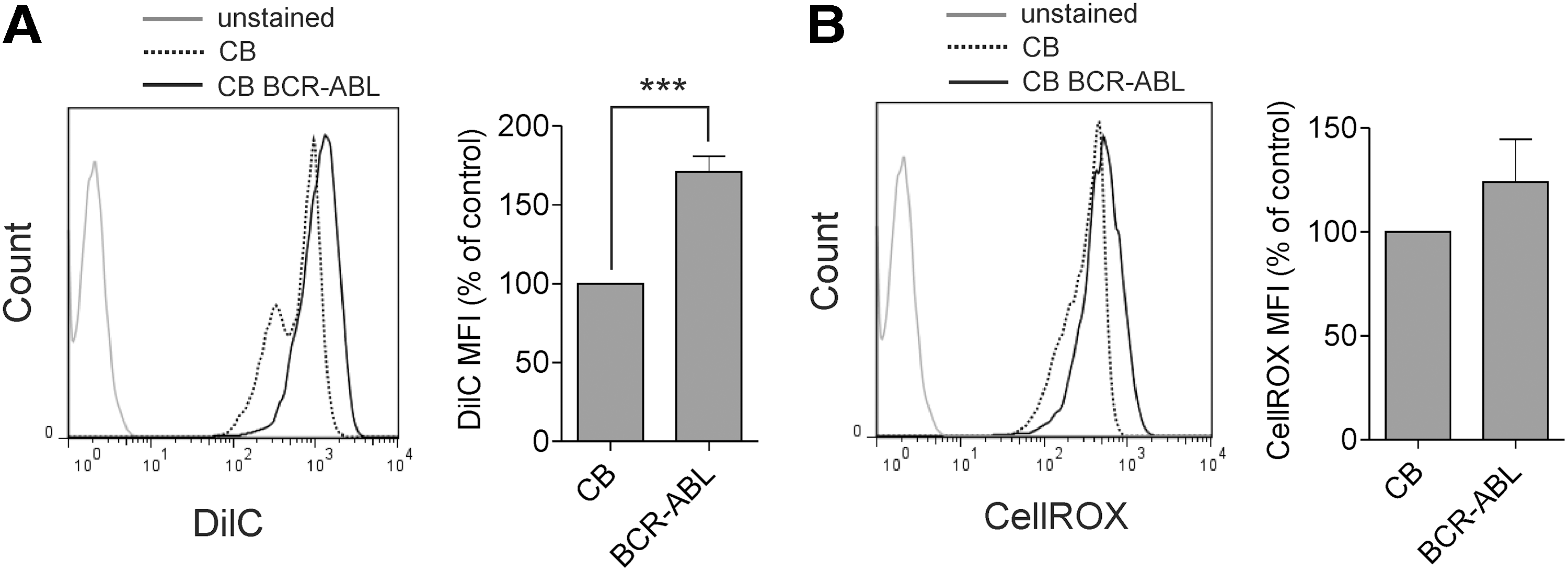
Increased mitochondrial activity of BCR-ABL-transduced human HSPCs.
Cytokine-driven proliferation of normal HSPCs results in increased mitochondrial activity and increased sensitivity to SAM50 downregulation
The BCR-ABL oncogene is a potent driver of proliferation of transformed CB cells [36,39]. Therefore, we wondered whether inducing a high proliferation rate on CB CD34+ cells by culturing them in a cytokine-rich medium would increase their mitochondrial activity and render them more sensitive to SAM50 depletion. CD34+ CB cells were cultured in a coculture setting with or without the addition of cytokines (20 ng/mL SCF and IL-3). As expected, cells cultured with cytokines showed an increased proliferation rate (compare shSCR control data in Fig. 3B without cytokines with data in Fig. 5D with cytokines). FACS analysis using DilC staining showed a significant increase in mitochondrial membrane potential in cells cultured with the addition of cytokines (Fig. 5A). Also the ROS levels produced within these cells were increased, although the results did not reach statistical significance (Fig. 5B). OCR levels were significantly increased upon the addition of cytokines (Fig. 5C). Together, this showed that the cytokine-induced proliferation is paralleled by increased mitochondrial activity in CB cells.
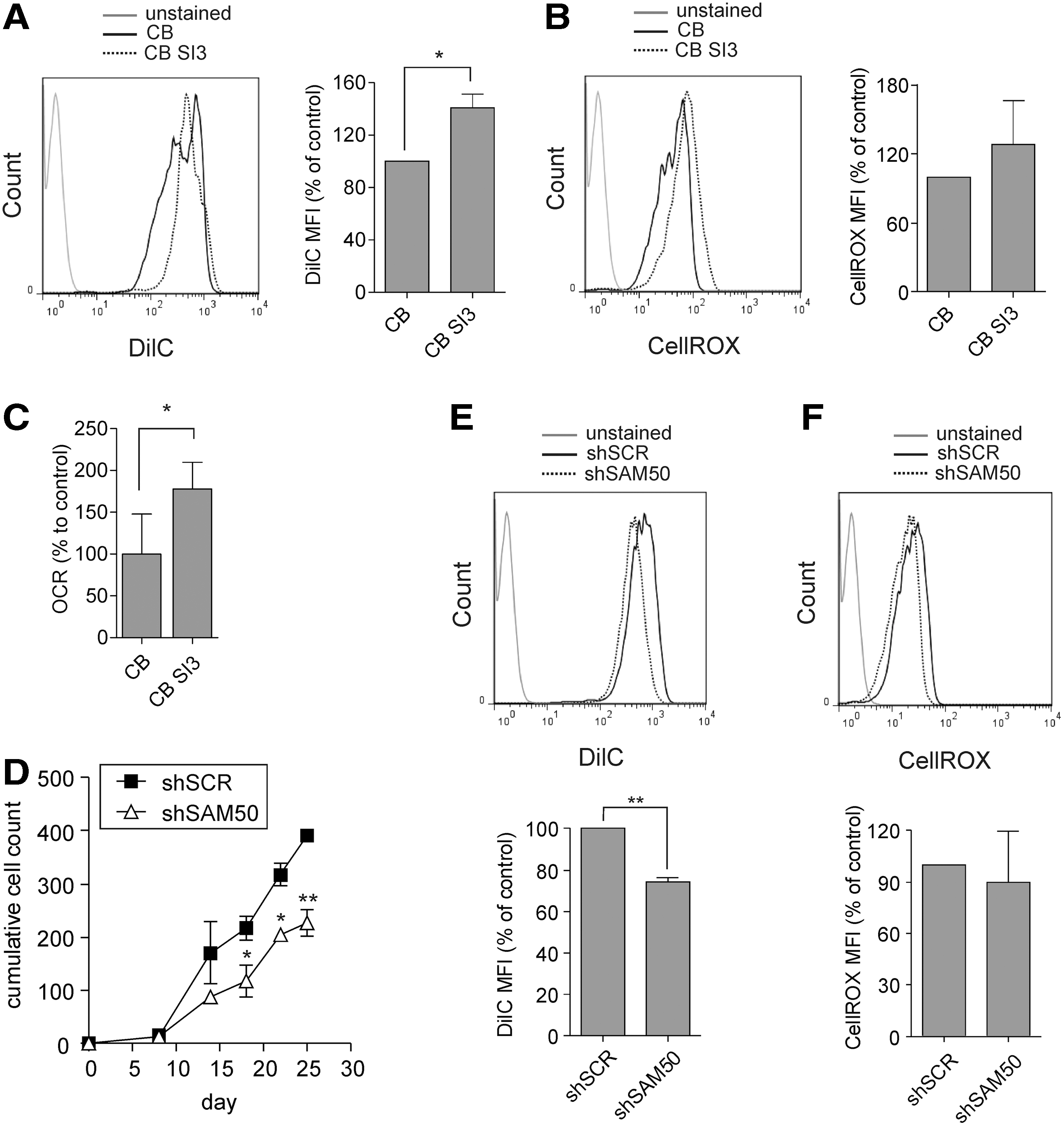
Increased mitochondrial activity and increased sensitivity to SAM50 downregulation in normal HSPCs upon cytokine stimulation.
Next, we transduced CD34+ CB cells with the SAM50-targeting or shSCR control shRNA construct, and transduced cells were sorted and plated on stroma in the same cytokine-rich medium. Under the proliferation-driven culture conditions, shSAM50-transduced CB cells showed a significant proliferative disadvantage, which was not seen in the regular CB cocultures without the extra addition of cytokines (Figs. 5D and 3B). Also the mitochondrial membrane potential was significantly decreased and ROS levels were slightly, but not significantly, reduced (Fig. 5E, F). Overall, these data suggest that a high proliferation rate induced by cytokines increased the mitochondrial activity in CB cells, which correlated with an increased sensitivity to SAM50 downregulation.
Discussion
Interfering with mitochondrial metabolism has previously been shown to be an effective strategy to specifically target leukemic cells. In AML cells, inhibition of mitochondrial translation with the drug tigecycline or by shRNA-mediated knockdown of the EF-Tu mitochondrial translation factor showed a selective antileukemic effect [23]. In chronic lymphocytic leukemia, interrupting the MRC by a benzodiazepine derivative PK11195 specifically induced cell death in leukemic, but not normal T and B cells [40]. Also, in BCR-ABL-expressing leukemic cells pharmacological inhibition or decreased expression of small GTPase RAC2 selectively targeted leukemic and not normal HSPCs by decreasing their mitochondrial activity and ROS production [30,31,41,42].
In the current study we show that shRNA-mediated downregulation of a mitochondrial protein SAM50 resulted in a strongly reduced proliferation of leukemic but not normal human HSPCs, using multiple independent shRNAs against SAM50. The replating capacity of leukemic cells both in coculture and CFC assay was strongly decreased, suggesting that also the more primitive, LSC-containing population was affected. Also the mitochondrial functions such as OCR were affected by SAM50 downregulation in BCR-ABL expressing, but not normal CB cells.
SAM50, as a part of the MIB complex, is necessary for the assembly of the MRC and formation of cristae [35]. In agreement with this, our experiments showed that downregulation of SAM50 resulted not only in a decreased mitochondrial membrane potential in BCR-ABL-expressing human HSPCs, but also lower OCR, indicative of decreased oxidative phosphorylation. SAM50 expression was not increased in AML CD34+ cells as compared to normal bone marrow CD34+ cells (unpublished observation) and, therefore, could not account for the dependence of leukemic cells on SAM50. When we directly compared normal and BCR-ABL-transduced CB CD34+ cells, we observed an increased mitochondrial membrane potential and (although not statistically significant) increased ROS levels. This was in accordance with previous findings, where increased mitochondrial biogenesis and increased respiratory rates were shown to be characteristic for leukemic cells [23,40]. Thus, we concluded that an increase in mitochondrial activity of leukemic cells makes them more vulnerable to strategies disrupting mitochondrial functions.
Although, in the steady state, the proliferation and progenitor frequency of normal CB cells in coculture was not affected by SAM50 downregulation, the replating capacity of progenitor cells was strongly reduced. This suggests that upon stress, possibly due to increased energy requirements, cells rely more strongly upon intact mitochondrial functions. Malignant transformation, with its increased proliferation and metabolic rate, also creates stress within the cells. Moreover, accumulation of ROS results in the so-called cancer-associated oxidative stress [20]. Although elevated ROS levels can be advantageous for malignant cells by inducing prosurvival signaling pathways, too high ROS cause detrimental DNA and protein damage [43]. Therefore, disruption of the redox balance in either direction can negatively affect growth and survival of transformed cells.
Addition of cytokines to the coculture medium not only increased the proliferation rate of normal CB cells, but also rendered them more sensitive to SAM50 downregulation. This effect could not only simply be a result of a higher proliferation rate that requires more energy produced by the mitochondria, but also a direct effect of cytokine signaling. Several growth factors and cytokines, including IL-3, were found to stimulate ROS production [44,45]. In accordance with that, in our study we found not only increased mitochondrial membrane potential, but also increased, although not statistically significant, ROS levels in cytokine-stimulated CB cells. Although the effect of SAM50 downregulation on the growth of CB cells in cytokine-rich cultures was clear, it was less dramatic than in BCR-ABL-transduced CB cells. This suggests that driving cell proliferation is not the only aspect of the oncogenic kinase activity that is responsible for the increased sensitivity to SAM50 downregulation. Elucidating these additional mechanisms that make leukemic cells highly dependent on mitochondrial functions could contribute to more efficient and selective targeting of leukemia.
Footnotes
Acknowledgments
The authors would like to thank Dr. Vera Kozjak-Pavlovic from the University of Würzburg, Germany for kindly providing anti-SAM50 antibody, as well as all the members of the Experimental Hematology Laboratory for helpful discussions. The authors thank Jeanet Dales for help with CB isolations. This work was supported by a grant from The Netherlands Organization for Scientific Research (NWO-VIDI 91796312) to J.J.S.
Author Disclosure Statement
No competing financial interests exist.