
Abstract
A large number of cancer stem cells (CSCs) were identified and characterized; however, the origins and formation of CSCs remain elusive. In this study, we examined the origination of the newly identified CD34+ liver CSC (LCSC). We found that CD34+ LCSC coexpressed liver stem cell and myelomonocytic cell markers, showing a mixed phenotype, a combination of hepatobiliary stem/progenitor cells (HSPCs) and myelomonocytic cells. Moreover, human xenografts produced by CD34+ LCSCs and the parental cells, which CD34+ LCSC was isolated from, coexpressed liver cancer and myelomonocytic markers, also demonstrating mixed phenotypes. The xenografts and the parental cells secreted albumin demonstrating their hepatocyte origin and also expressed cytokines [interleukin (IL)-1b, IL-6, IL-12A, IL-18, tumor necrosis factor-alpha (TNF-α), and CSF1] and chemokines (IL-8, CCL2, and CCL5). Expression of these cytokines and chemokines responded to the stimuli [interferon-γ (INF-γ), IL-4, and lipopolysaccharide (LPS)]. Furthermore, human xenografts and the parental cells phagocytized Escherichia coli. CD34+ LCSC coexpressed CD45, demonstrating that its origin appears to be from a hematopoietic precursor. The percentage of cells positive for OV6, CD34, and CD31, presenting the markers of HSPC, hematopoietic, and myelomonocytic cells, increased under treatment of CD34+ LCSC with a drug. Cytogenetic analysis showed that CD34+ LCSC contained a greater number of chromosomes. HBV DNA integrations and mutations in CD34+ LCSC and the parental cells were identical to those in the literature or the database. Thus, these results demonstrated that CD34+ LCSCs were formed by fusion of HSPC with CD34+ hematopoietic precursor-derived myeloid intermediates; it appears that this is the first report that human CSCs have been formed by the fusion. Therefore, it represents a significant step toward better understanding of the formation of human CSC and the diverse origins of liver cancers.
Introduction
M
Human liver carcinoma (HLC) is the fifth most common cancer worldwide, with a median survival of 6–16 months [8]. Three types of cells that respond to liver tissue renewal or damage have been shown; they are mature hepatocytes, resident hepatobiliary stem/progenitor cells (HSPCs), and circulating bone marrow-derived cells (BMDCs) [9]. There are two major nonexclusive hypotheses of the cellular origin of liver cancers: that they derive from stem cells due to maturation arrest or from dedifferentiation of mature cells. A third possibility has also been suggested for rodent and human malignancy, although evidence in the liver has not yet been published; that circulating BMDCs may incorporate into benign or malignant tumors [10] or actually initiate tumor development [11]. Although a number of LCSCs have been apparently isolated and characterized based on putative CSC markers, such as CD90+ [12], CD133+ [13], CD44+ [13], or EpCAM+ [14], their origins are still unknown.
CD34+ stem cells play an important role during liver development and regeneration [15,16], thus we hypothesized that some HLCs might be derived from transformed CD34+ stem cells. Recently, we determined that a population of CD34+ stem cells functioned as LCSCs by forming three types of HLCs [combined cholangiohepatocellular carcinoma (CHC), hepatocellular carcinoma (HCC), and cholangiocarcinoma (CC)] [17], indicating that this LCSC exhibits bipotency, which can differentiate into hepatic and cholangiocytic lineages. In addition, CD34+OV6+ cells and their derivatives, OV6+ cells, produced HLC xenografts [17]; thus, it suggests that the origin of this LCSC may be derived from liver stem/progenitor cells. Importantly, we found that a large number of xenograft cells expressed CD31, which is normally present on endothelial cells, platelets, macrophages, granulocytes, and blood leukocytes [17]; moreover, CD34+CD31+ cells and their derivative CD31+ cells formed HLC xenografts [17], implying that the formation of this CSC may be also associated with CD31 lineage cells. These phenomena lead us to further investigate the origination of CD34+ LCSC. In this study, we attempted to determine its origin and to reveal how it was formed.
Materials and Methods
Cell lines and cell culture
Hepatoma cell lines, Hep G2 cells and PLC/PRF/5 cell (PLC), 293T cells, and human monocyte cell line, TIB-202/THP-1 cells, and mouse monocyte/macrophage cell line, TIB-71/RAW264.7 cells, were purchased from ATCC (Manassas, VA). The cell culture conditions for growing and expanding these line cells were as per the manufacturer's instructions.
Cultures of CD34+ LCSCs and the xenograft cells
CD34+ LCSC was clonogenically cultured, expanded, and maintained on mouse embryonic fibroblast (MEF; GlobalStem, Gaithersburg, MD) feeder cells as previously described [18]; briefly, PLC cells were stained with antibody against human CD34, and CD34+ LCSCs were sorted from PLC cells and seeded on MEF feeder cells with defined medium, which consists of epidermal growth factor (10 ng/mL), basic fibroblast growth factor (4 ng/mL), 1× ITS, 1× antibiotic/antimycotic (all from Invitrogen, Grand Island, NY),
Immunohistochemistry assay
CD34+ LCSCs grown on mouse feeder cells, the cultured xenograft cells, and hepatoma cell lines were fixed with 4% PFA and stained as previously described [19], with different primary and secondary antibodies. All antibodies used are listed in Supplementary Table S1 (Supplementary Data are available online at
The generation of cDNA and genomic DNA
RNA was extracted from the cultured xenograft cells, hepatoma cell lines, CD34+ LCSCs, human and mouse monocyte/macrophage cell lines, and 293T cells using the Qiagen mini RNA kit, and cDNA was generated and quantitative polymerase chain reaction (qPCR) was performed as previously described [19]. Genomic DNA was extracted from CD34+ LCSCs, the xenograft cells, PLC, and Hep G2 cells using the Qiagen Genomic Isolation Kit. DNA extraction was performed as per the manufacturer's instructions. Sequencing was done at the University of California at Davis Sequencing Facility. All primers/probes used are listed in Supplementary Table S2.
The evaluation of partial expression of CD45
Six pairs of primer sets (CD45 A to CD45 F) were designed to amplify the entire CD45 cDNA from the C-terminal to N-terminal from CD34+ LCSCs, the xenograft tumor cells, and PLC. Their sequences, locations, and amplicon sizes of these six primer sets are listed in Supplementary Table S2. The amplicons were sequenced and compared with the sequence of CD45 in NCBI GenBank. To confirm the partial expression of CD45, many antibodies recognizing CD45 leukocyte common antigen, N-terminal, central, and C-terminal areas were used to perform immunostaining using CD34+ LCSCs and their derivatives. These antibodies are listed in Supplementary Table S1.
The determination of HBV DNA integration in CD34+ LCSC and PLC
Three primer sets (HBV S, HBV C/P, and HB-Hu) were designed based on the literature [20 –22] to amplify HBV surface antigen gene, core gene, polymerase gene, and HBV-human DNA junctions from genomic DNAs of CD34+ LCSC and PLC. Fragments of HBV-human DNA junctions were sequenced and compared with those in the literature [22] and in NCBI GenBank. The sequences, locations, and amplicon sizes of these three primer sets are listed in Supplementary Table S2.
The determination of mutations in CD34+ LCSC and PLC
Three genes (CDKNA2, STK11, and TP3) were sequenced in CD34+ LCSCs and PLC from their genomic DNAs, three DNA fragments containing the mutations in three genes were amplified by three primers of CDKNA2, SKT11, and TP3 and further sequenced and compared with those in each gene from the Sanger COSMIC database (
Stimulation by cytokines and lipopolysaccharide
PLC cells, the xenograft cells, Hep G2 cells, THP-1 cells, and 293T cells were treated with interferon-γ (INF-γ; 100 ng/mL), interleukin-4 (IL-4, 10 ng/mL; R&D Systems, Minneapolis, MN), and lipopolysaccharide (LPS, 1 ng/mL; Sigma-Aldrich) for 24 h, then the cells were harvested for cDNA generation, and expression changes of cytokines and chemokines were determined by qPCR in these cells after stimulation. All primers/probes used are listed in Supplementary Table S2.
Phagocytosis assay
PLC cells, the xenograft cells, RAW264.7 cells, and 293T cells were used to assess phagocytosis employing the Escherichia coli Phagocytosis Assay Kit (Cell Biolabs, Inc., San Diego, CA). Phagocytosis was performed as per the manufacturer's instructions.
Enzyme-linked immunosorbent assay analysis
Enzyme-linked immunosorbent assay (ELISA) analysis was performed as previously described [19], human albumin values secreted into the medium by PLC and the xenograft cells were measured using the Human Albumin ELISA Quantitation Kit (Bethyl, Montgomery, TX) and normalized to total cell number cultured.
Cytogenetic analysis
Metaphase chromosomes and banding were prepared by the standard trypsin-Giemsa method [23]. Briefly, the cells were exposed to 100 ng/mL KaryoMax colcemid solution (Invitrogen) for 4 h, lysed in 0.075 M potassium chloride for 15 min, followed by fixation in methanol and glacial acetic acid (3:1 v/v) for 15 min. Giemsa bandings were prepared according to the manufacturer's staining protocol.
Drug resistance
CD34+ LCSCs were removed from culture with MEF feeder cells and seeded, expanded, and maintained on a commercial extracellular matrix (ECM) plate, which is for culturing LCSCs (Celprogen, Torrance, CA) under our defined medium to remove feeder cells. The CD34+ LCSCs were treated with cisplantin at 2 μg/mL at day 8 for 6 days, then cells were harvested and stained with antibodies against seven markers, then the percentages of cells positive for CD34, CD31, EpCAM, CD44, CD90, CD133, and OV6 were measured by flow cytometry.
Statistical analysis
All data are summarized as mean±standard error of the mean from at least three independent measurements. An unpaired Student's t-test was used to analyze the data. P<0.05 was considered statistically significant.
Results
CD34+ LCSCs coexpressed liver stem cell and myelomonocytic cell markers
Besides CSC markers [17], CD34+ LCSCs cultured on the feeder cells also coexpressed liver stem cell markers with monocyte/macrophage markers, such as OV6 and CD14, OV6, and CD68, alpha-fetoprotein (AFP) and microphage antigen, AFP and CD68, and OV6 and AFP (Fig. 1). In adult liver, HSPCs are the transit amplifying population derived from facultative stem cells, which are activated during progenitor-dependent regeneration [15]. In mice and rats, these are commonly referred to as oval cells, and it has been shown that some HCCs and CCs in these models are of oval cell origin [24]. HSPCs express OV6 as well as the bile duct marker, CK19 [15,16,25]. Liver sinusoidal endothelial cells and BM-derived Kupffer cells (KCs) express CD31 and they are implicated in the pathophysiological process of liver injury [26]; however, other endothelial markers such as CD105, von Willebrand factor, and factor VIII were not detected in our CD34+ LCSCs and their derivatives. Normally, HSPCs do not express myelomonocytic genes, but CD34+ LCSCs coexpressed CD68, CD14, CD31, and macrophage antigen, thus demonstrating that both liver stem/progenitor markers and myelomonocytic markers were expressed in the same cells and suggesting that CD34+ LCSCs showed mixed phenotypes from two different cell types, a combination of HSPCs and myelomonocytic cells (monocyte/macrophage). CD34+ LCSCs expressed another myelomonocytic cell marker, lysozyme, and alpha-1-antitrypsin (α1-AT) (Fig. 1). α1-AT can be expressed by hepatocytes and macrophage, but CD34+ LCSCs do not express more mature hepatocyte markers, such as albumin and Hep Par1, thus α1-AT was expressed by CD34+ LCSCs as a macrophage marker. However, EMR1, a mature human KC marker, was not expressed in CD34+ LCSCs, suggesting that the monocyte/macrophage characteristics of CD34+ LCSCs might be derived from a CD34+ precursor-derived myeloid intermediate rather than a local KC.

Characterization of CD34+ LCSCs. CD34+ LCSCs cultured on the feeder cells were costained and merged with antibodies against markers of liver stem cells (AFP, OV6) and myelomonocytic cells [CD14, CD68, macrophage antigen (Macro Ag)], and also stained with antibodies against α1-AT or lysozyme. Scale bar: 100 μm. LCSC, liver cancer stem cell. α1-AT, alpha-1-antitrypsin; AFP, alpha-fetoprotein. Color images available online at
The xenografts coexpressed liver cancer cell and myelomonocytic cell markers
Using immunohistochemistry (IHC), the xenograft cells produced by the transplantation of CD34+ LCSCs into mice coexpressed Hep Par1 and CD68, CK19 and CD68, AFP and CD68, α1-AT and CD31, as well as α1-AT and CD14 (Fig. 2). These results also demonstrated that both liver cancer markers and myelomonocytic cell markers were expressed in the same cells as the hybrid cells exhibited both parental cell phenotypes. This phenomenon strongly suggested that CD34+ LCSCs might be formed by the fusion of HSPCs with myelomonocytic cells.
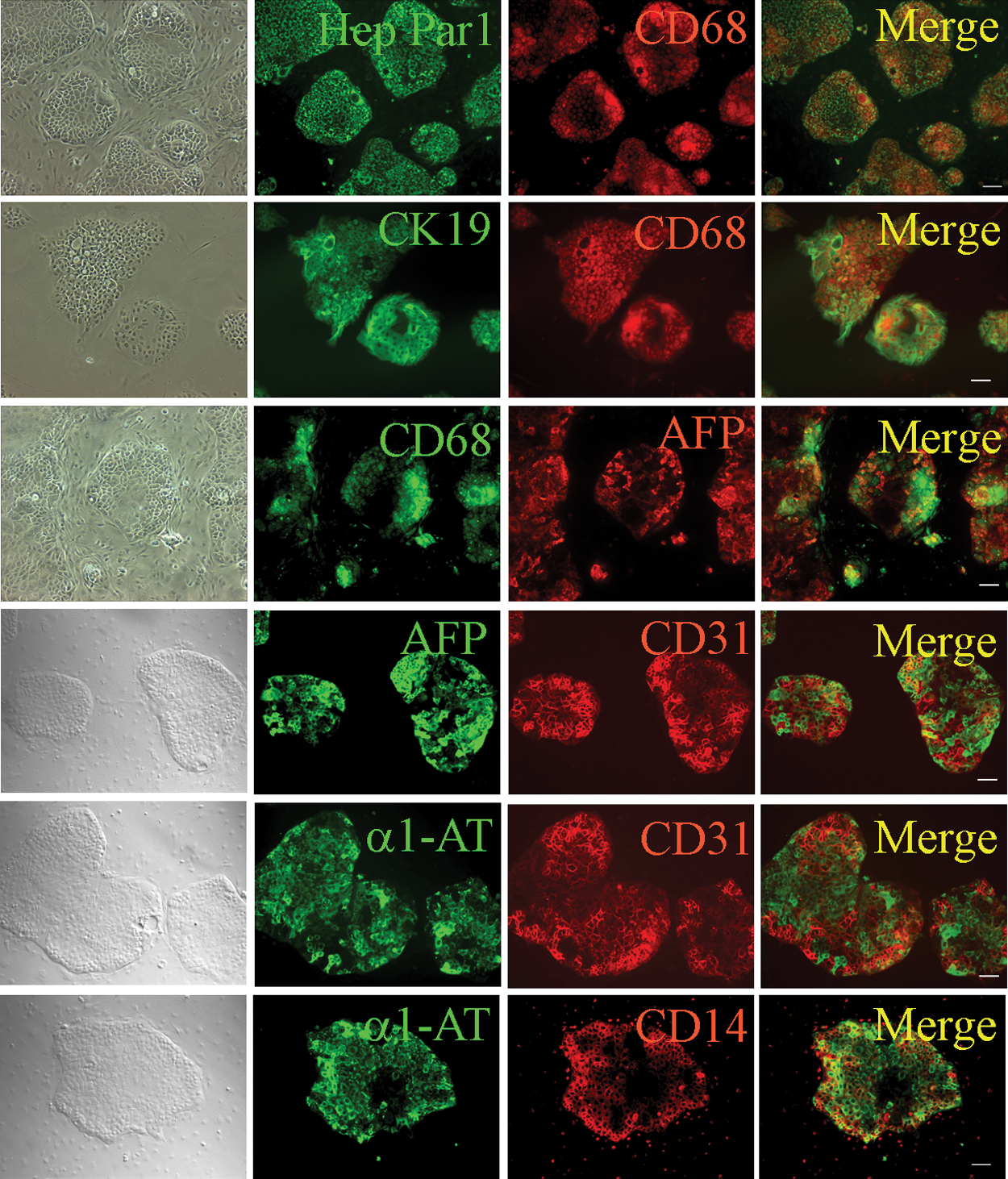
Characterization of HLC xenografts. Cultured HLC xenograft cells produced by the transplantation of CD34+ LCSCs were costained and merged with antibodies against markers of liver cancer cells (Hep Par 1, CK19, AFP, and α1-AT) and myelomonocytic cells (CD68, CD31, and CD14). Scale bar: 100 μm. HLC, human liver carcinoma. Color images available online at
CD34+ LCSC and the xenografts expressed CD45
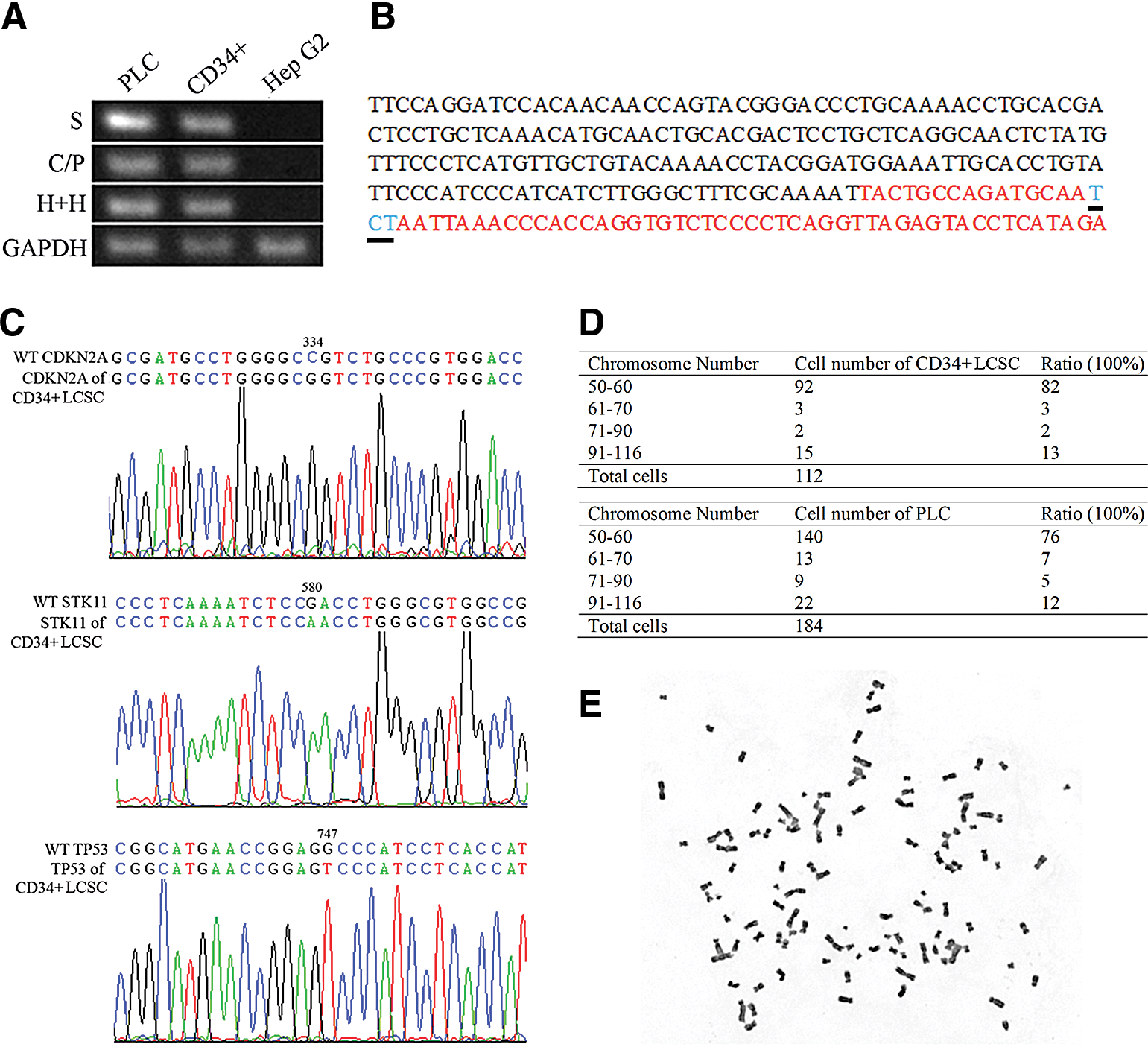
Normally HSPCs and liver cancer cells do not express hematopoietic genes; however, CD34+ LCSCs coexpressed OV6 and CD45, AFP and CD45, CD45 and macrophage antigen; HLC xenograft cells coexpressed Hep Par1 and CD45 and CK19 and CD45 (Fig. 3A), suggesting that CD34+ LCSCs might be derived from a CD34+ hematopoietic precursor. Interestingly, the CD45 gene was only partially expressed in CD34+ LCSCs, the xenograft cells, and PLC from which CD34+ LCSC was isolated [17]. Using six pairs of primer sets, which amplify the entire CD45 cDNA from the C-terminal to N-terminal, all of six fragments could be amplified in human primary hepatocytes; however, only two fragments near the C-terminal were amplified in CD34+ LCSCs, the xenograft cells, and PLC, and none was amplified in Hep G2 cells (Fig. 3B). The sequences of these two fragments were identical to those in NCBI GenBank (Fig. 3C and Supplementary Fig. S1). This phenomenon was also confirmed by IHC. Many antibodies recognizing CD45 leukocyte common antigen, N-terminal, central, and C-terminal areas were used in this study (Supplementary Table S1); however, interestingly, only antibodies against CD45 C-terminal and nearby regions produced positive staining, demonstrating that the entire CD45 gene is not expressed in these cells.

Characterization of CD45 expression.
HLC xenograft cells and PLC expressed cytokines and chemokines
HLC xenograft cells and PLC expressed cytokines IL-1b, IL-6, IL-12A, IL-18, tumor necrosis factor-alpha (TNF-α), and CSF1, and chemokines IL-8, CCL2, and CCL5, as well as myelomonocytic cell markers (CD68 and CD14), as determined by qPCR (Fig. 4A). These cytokines and chemokines are secreted by many different cell types; however, the macrophage is the primary cell, which produces all of them. Moreover, expression of these cytokines and chemokines could be enhanced by the treatment with INF-γ, IL-4, or LPS (Table 1). Response to stimulation by these three factors is a unique macrophage characteristic.
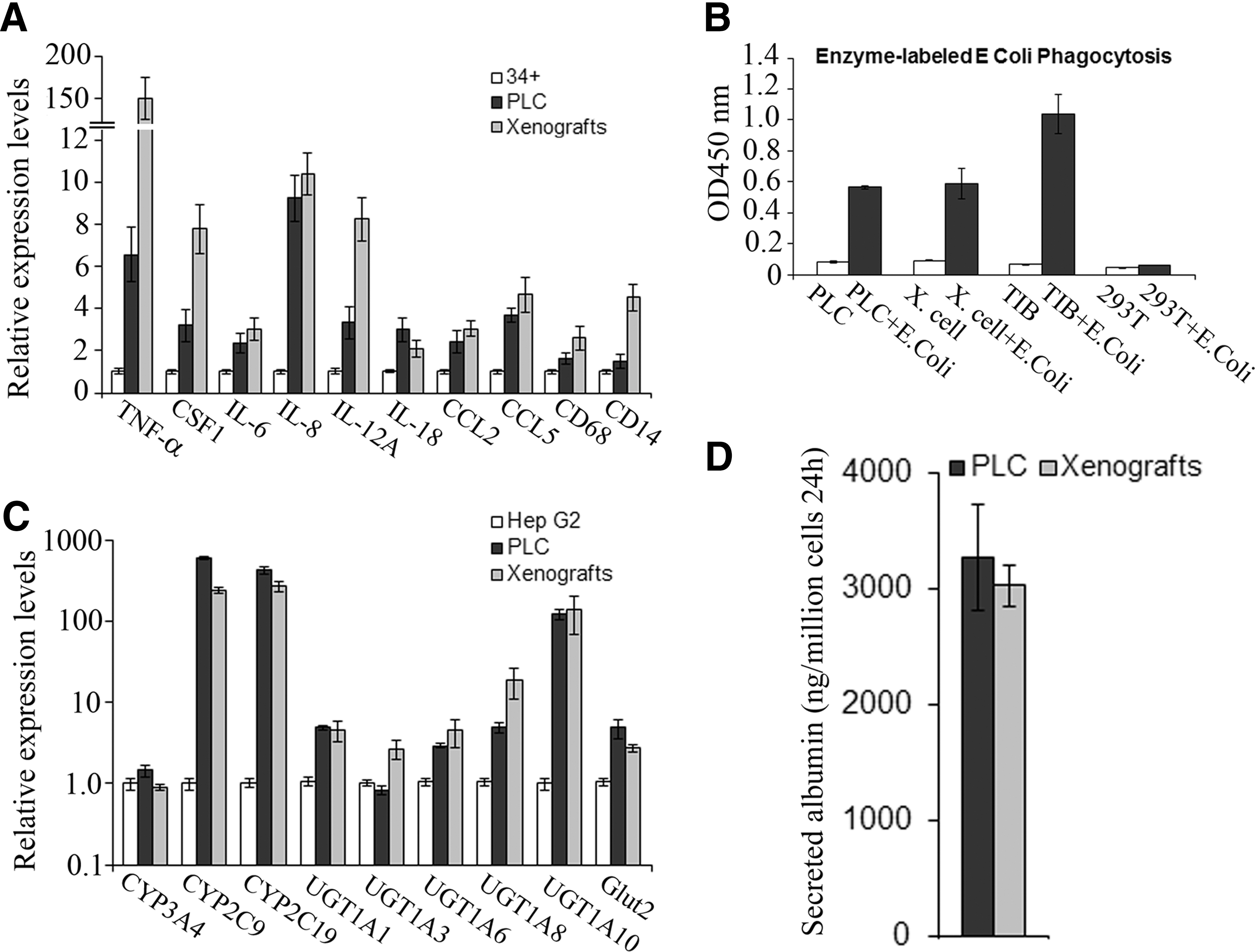
Analysis of gene expression and functions.
Human monocyte cell line, THP-1 cells (TIB), PLC, the xenograft cells (Xeno), Hep G2 cells (G2), and 293T cells were treated with INF-γ, IL-4, and LPS for 24 h, then expression changes of cytokines and chemokines in treated cells were measured by qPCR when compared with those in corresponding untreated cells. Data represent mean±SEM.
IL-4, interleukin-4; INF-γ, interferon-γ; LPS, lipopolysaccharide; PLC, PLC/PRF/5 cells; qPCR, quantitative polymerase chain reaction; SEM, standard error of the mean; TIB, TIB-202/THP-1 cells; TNF-α, tumor necrosis factor-alpha.
HLC xenograft cells and PLC had phagocytotic activity
HLC xenograft cells and PLC had the capacity to phagocytose E. coli (Fig. 4B); E. coli size is 2 μm in this assay kit. Phagocytosis is the process by which the cells bind and internalize the particles larger than 0.75 μm in diameter, and it can be performed only by phagocytes (neutrophils, monocytes, macrophages, dendritic cells, and mast cells). This process is different from endocytosis, which is mediated by small vesicles (∼100 nm in diameter) and used by all cells of the body.
Human liver phenotype and function by HLC xenograft cells and PLC
HLC xenograft cells and PLC not only expressed Hep Par 1, ALB, AFP, and CK19 (Figs. 2 and 3A) but also expressed metabolizing phase I and II enzymes, CYP3A4, CYP2C9, CYP2C19, UGT1A1, UGT1A3, UGT1A6, UGT1A8, UGT1A10, and phase III transporter protein, glucose transporter protein 2 (Glut2) (Fig. 4C). In the assay of hepatocyte function, HLC xenograft cells and PLC cells secreted albumin into the medium (Fig. 4D).
The determination of the original of PLC
Because the original PLC showed HBV DNA integration in its genome, employing PCR and sequencing, we found that integration of the HBV surface antigen gene [20], core gene, and polymerase gene [21] and HBV-human DNA junctions [22] were identical in the parental PLC and CD34+ LCSCs and were negative in Hep G2 cells (Fig. 5A). Of note, the sequences from these DNA fragments that spanned HBV-human DNA junctions were almost exactly the same as in those published almost three decades ago [22] (Fig. 5B and Supplementary Fig. S2) (there were three base pair differences in the human DNA sequence, but ours are identical to those in NCBI GenBank). The Sanger COSMIC database showed that there were three single mutations that occurred in three genes, CDKNA2 at c.334C>G, STK11 at c.580G>A, and TP53 at c.747G>T (
The original of PLC and CD34+ LCSCs.
Cytogenetic analysis
Fusion between two different cell types produces a heterokaryotic hybrid cell that initially contains the genetic elements and organelles of both cell types. A change in the chromosome number in CD34+ LCSCs may indicate whether there was any possibility that the nucleus was extruded from the heterokaryon. Cytogenetic analysis showed that the mean modal number (50–60) of the chromosomes was 82% in CD34+ LCSCs and 76% in PLC cells, matching the number from the data published three decades ago [27]; we also found that 13% of CD34+ LCSCs and 12% of PLC cells had a greater chromosome number with a range from 90 to 116 (Fig. 5D, E, and Supplementary Fig. S4). Our speculation is that the cells with a chromosome number ranging from 50 to 60 are the progeny of LCSCs and their derivatives that resulted from the extrusion of additional nuclei during proliferation or differentiation. This could explain the partial expression of CD45 in CD34+ LCSCs, the xenograft cells, and PLC line. The entire CD45 gene contains many exons, which locate at many different chromosomes; a possible explanation for this phenomenon is that the fusion event caused partial exclusion of the CD45 gene from the hematopoietic precursor of the heterokaryon. On the other hand, our belief is that the cells with a greater number of chromosomes (aneuploid) are the cells that kept the major chromosomes of two cells after fusion. BM-derived hepatocytes generated by fusion also showed some aneuploidy after transplantation in mice, although malignant hepatic transformation was not observed in any of the mice in the study [28,29]; however, the observation time may not have been long enough to observe any potential tumorigenicity associated with aneuploidy.
Drug resistance
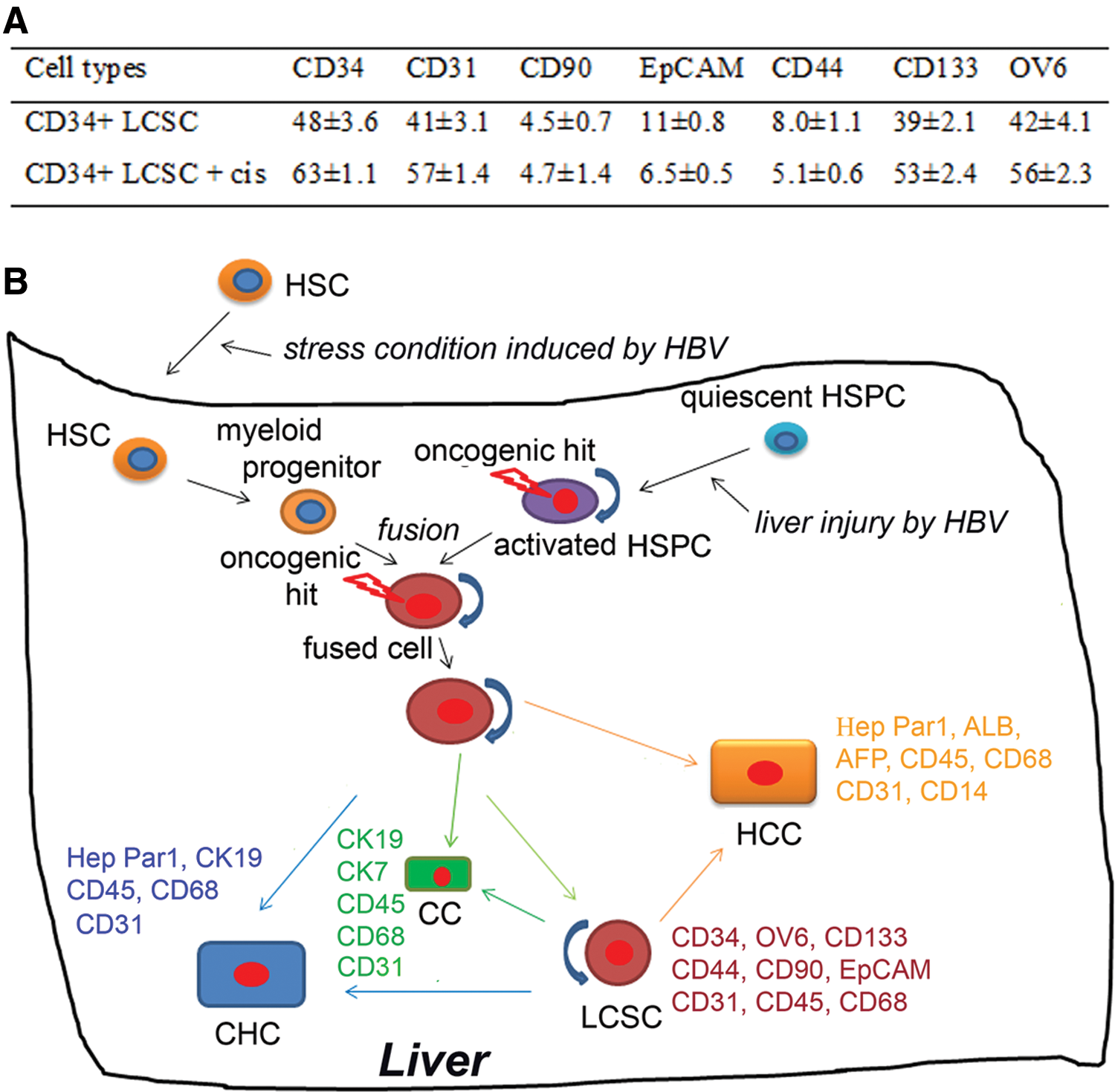
CD34+ LCSCs exhibited the resistance to the drug, cisplatin, CD34+ LCSCs were removed from the feeder cells to the ECM plate and treated with cisplatin at day 8 for 6 days. Flow cytometry showed that the percentages of cells positive for CD34, CD133, CD31, and OV6 were increased, indicating that surface markers presenting hematopoietic cells (CD34), myelomonocytic cells (CD31), and HSPCs (OV6) were drug resistant (Fig. 6A). Thus, this result further demonstrated that CD34+ LCSC was fused by hematopoietic precursor-derived myelomonocytic cells with HSPCs. CD133 has been shown as a hematopoietic marker [30] and LCSC marker [13], however, its role in this study remains unknown. Interestingly, except CD133, the populations of other putative CSC markers, CD90, CD44, and EpCAM, were not changed or even decreased under the treatment with cisplatin.
Drug resistance and illustration of the origination of CD34+ LCSCs.
Discussion
The stem cells are candidates for targets of transformation because of their self-renewal and longevity, which would allow the sequential accumulation of genetic or epigenetic mutations required for oncogenesis [31]. Although a large number of CSCs, including LCSCs [12 –14], have been apparently isolated and characterized, the origins of these CSCs remain unknown. Due to their important role in liver development and regeneration [15,16], CD34+ stem cells might represent candidates to be targeted and transformed as LCSCs. We recently determined that a population of CD34+ stem cells functioned as LCSCs [17]; the results in this study showed that both CD34+ LCSCs and their derivatives exhibited mixed phenotypes. PLC is depicted as a well-differentiated hepatoma cell line [32]; however, we found that the xenografts and PLC also expressed cytokines and chemokines and their expression could be enhanced by stimulus. Moreover, the xenografts and PLC could phagocytose E. coli. Thus, they appeared to express characteristics of the two parental cell types as hybrid cells do. This phenomenon strongly suggests that CD34+ LCSCs might be formed by fusion. Because of lacking EMR1 and coexpression of CD45, these results indicated that CD34+ LCSCs were formed with CD34+ hematopoietic-derived myeloid intermediates rather than local KCs.
Our conclusion about the origin and the formation of CD34+ LCSCs is based on evidence of similar occurrences in the literature. Although BM-derived stem cells have been shown to directly differentiate into hepatocytes in vivo [33,34], many other investigations have shown that BMDCs can be transformed to a hepatocyte phenotype by fusion with hepatocytes in adult livers of rodents and humans [28,29,35 –38]. BM-derived HSCs can migrate to and engraft into injured adult livers [39]. PLC line was derived from the liver carcinoma of a patient who had long-term HBV infection [40]. Liver injury caused by hepatitis viral infection dramatically increases the number of BM-derived HSCs and attracts HSCs to home to the liver [41]. Thus, it is certainly plausible that a cell from the myeloid lineage fused with a cell from the hepatocyte lineage and became an LCSC candidate. In the aforementioned studies, the evidence for fusing employed BM transplantation from sex-mismatched donors. The very recent report showed that the fusion of BMDCs with the tumor cells occurred in human cancer from a case of a patient with a brain metastasis produced from the transplant of bone marrow received from the patient's brother for the treatment of lymphoma, the fusion between BMDCs and the tumor cells was determined by donor–patient hybrid genome following bone marrow transplantation (42). In our study, we were not able to provide the chromosome identification from a sex-mismatched donor and hybrid genome analysis. Instead, we provide evidence that cell function from two distinct cell types coexisted in this LCSC, an alternative method to demonstrate that this hybrid LCSC was formed by fusion.
The greatest challenge is to understand the pathophysiology of the tumorigenesis. Cancer cell fusing with migratory BMDCs is a source of metastasis in some cancers [42,43] and such fusion may represent a potential mechanism for the origin and formation of CSCs [44,45]. Tumorigenesis caused by a mutation from the integration of HVB DNA is widely accepted, and in this case, there are three mutations in three genes, including p53, a tumor suppressor gene. The consequence of these mutations is thought to cause normal liver cells to become cancer cells during long-term HBV infection. Hepatocytes are the host of HBV, and if the patient's liver carcinoma was caused by the direct accumulation of hepatocyte-based cancer cells generated by HBV-induced mutation, the phenotype of this liver carcinoma should be HCC. However, PLC line is quite different from other hepatocyte-based HCC lines, such as Hep G2, Hep 3B, and Huh7. PLC and the xenograft cells produced by the injection of parental PLC into the mice expressed high levels of both hepatocyte and cholangiocyte markers, showing that they were of a CHC phenotype in our previous study [17].
Our hypothesis is that after migrating to the liver attracted by the stress signals due to HBV infection, CD34+ HSCs were differentiated to myeloid intermediates under pathological conditions. HSPCs were activated by the liver damage caused by HBV infection. Myeloid intermediates were then fused with activated HPSCs, and the hybrid cells continued their differentiation along liver lineages, resulting in both hepatocyte cancer cells and cholangiocyte cancer cells with additional myelomonocytic cell markers [17] (Fig. 6B). The results in this study explained why CD34+OV6+ cells and CD34+CD31+ cells and their derivatives, OV6+ cells and CD31+ cells, formed HLCs and their xenografts contained a large number of CD31+ cells in our previous study [17]. The oncogenic hit might occur in activated HSPCs caused by the mutation before fusion or in hybrid cells caused by the mutation coupled with the consequence of fusion such as forming aneuploidy (Fig. 6B). This remains unknown. Theoretically, oncogenic HPSCs could form three types of HLCs; however, HPSCs do not express hematopoietic and myelomonocytic cell markers, neither do hepatocytes and cholangiocytes differentiated from HPSCs. Taken together, these results demonstrate that CD34+ LCSCs are not derived from HPSCs alone; rather, they are formed by fusion of activated HPSCs with the CD34+ hematopoietic-derived myeloid intermediate (myelomonocytic cells).
In summary, BM-derived HSCs and their progenitors can be transformed into hepatocytes by fusion [28,29,35 –38]. Similar situations also occur in other tissues and organs such as neurons and cardiomyocytes through such transformation [46 –48] under physiological conditions. Moreover, macrophage-tumor cell fusion or fused hybrids have been reported in melanoma patients [49], renal carcinoma patients [50], colorectal cancer patient and pancreatic cancer patient [51], and lymphoma patient [43]. Thus, the fusion might clinically be an approach of the formation of human CSCs. In this study, our results further indicate that the CD34+ LCSC was certainly formed through fusion; it appears that this is the first report that human CSCs have been formed by the fusion, and it is also the first report that an HLC appears to be initiated and developed from the interaction of HSPCs with CD34+ hematopoietic precursors, thus revealing a diversity of origins for HLCs. Therefore, our results represent a significant step toward better understanding of the formation of human CSCs and the diverse origins of human cancers. Our results also suggest a novel mechanism for the development of tumors: circulating BM-derived precursors can become the origins of tumorigenesis and carcinogenesis through fusion or transdifferentiation or reprogramming under pathological conditions after homing to injured organs.
Footnotes
Acknowledgments
This work was supported, in part, by the NIH grant, DK075415 (to M.A.Z.), and, in part, by the GlaxoSmithKline Research Fund of the Korean Association for the Study of the Liver (to S.C.P. and J.R.E.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.