
Abstract
Rat pluripotent stem cells, embryonic stem cells (ESCs), and induced pluripotent stem cells (iPSCs) as mouse and human ones have a great potential for studying mammalian early development, disease modeling, and evaluation of regenerative medicine approaches. However, data on pluripotency realization and self-renewal maintenance in rat cells are still very limited, and differentiation protocols of rat ESCs (rESCs) and iPSCs to study development and obtain specific cell types for biomedical applications are poorly developed. In this study, the RNA-Seq technique was first used for detailed transcriptome characterization in rat pluripotent cells. The rESC and iPSC transcriptomes demonstrated a high similarity and were significantly different from those in differentiated cells. Additionally, we have shown that reprogramming of rat somatic cells to a pluripotent state was accompanied by X-chromosome reactivation. There were two active X chromosomes in XX rESCs and iPSCs, which is one of the key attributes of the pluripotent state. Differentiation of both rESCs and iPSCs led to X-chromosome inactivation (XCI). The dynamics of XCI in differentiating rat cells was very similar to that in mice. Two types of facultative heterochromatin described in various mammalian species were revealed on the rat inactive X chromosome. To explore XCI dynamics, we established a new monolayer differentiation protocol for rESCs and iPSCs that may be applied to study different biological processes and optimized for directed derivation of specific cell types.
Introduction
E
We generated a set of ESCs and iPSCs of two rat strains—Brattleboro, which is a model of inherited hypothalamic diabetes insipidus, and WAG. The cell lines displayed a standard set of the properties attributable to pluripotent cells, including the ability to form chimeras in the heterologous mouse–rat system. The high-throughput RNA-seq approach was applied to better characterize the rESC and riPSC transcriptomes and to learn more about the core pluripotency network in rats. We compared the transcriptomes of rESCs, riPSCs, and rat embryonic fibroblasts (rEFs) to understand whether riPSCs that had become pluripotent during rEF reprogramming truly resembled rESCs at the molecular level. The riPSC transcriptome was found to be highly similar to that in rESC and significantly different from that in rEF. Expression patterns of rESC and riPSC lines corresponded to a ground state of pluripotency.
Both X chromosomes are known to be active in mouse pluripotent cell lines with the karyotype 40,XX while the developmental phenomenon of X-chromosome inactivation (XCI) occurs during their differentiation [7,8]. Reprogramming of mouse female differentiated cells to the pluripotent state leads to reactivation of the inactive X chromosome [9]. Typical human XX ESC and iPSC lines have an inactive X chromosome that can partially change its epigenetic and transcriptional statuses during extended cultivation [10]. Rare human pluripotent cell lines carry two active X chromosomes and appear to resemble mouse ESCs possessing the highest degree of pluripotency (the ground state of pluripotency). X-chromosome status monitoring in XX ESCs and iPSCs allows to trace pluripotency maintenance and epigenetic stability during cultivation and to control the completeness of the reprogramming process and specific epigenetic changes [11]. Differentiation of XX ESCs and iPSCs is used as an ex vivo model to study XCI molecular mechanisms, and differentiated derivatives of pluripotent cells with correct XCI are adequate tools for biomedical applications. However, X
In the study, X-chromosome status was examined in cultured rat pluripotent cells and during their differentiation. We established that XX rESCs and riPSCs were able to maintain two active X chromosomes, one of which underwent XCI in majority of cells during differentiation. To study XCI dynamics, we developed a new monolayer differentiation protocol. The protocol provided high cell viability and allowed rESCs and riPSCs to be differentiated into derivatives of three germ layers. We believe that the protocol will be valuable to model developmental processes using differentiation of rat pluripotent cells and to obtain specific cell types for biomedical applications.
Materials and Methods
The study was carried out according to The Guidelines for Manipulations with Experimental Animals and approved by the Ethics Committee of The Federal Research Center Institute of Cytology and Genetics, Novosibirsk, Permit Nos. 14.2 and 14.3. The study was conducted at the Center for Genetic Resources of Laboratory Animals ICiG SB RAS.
Cell cultures
rEFs were isolated from rat (the Brattleboro and WAG strains) embryos at day E12. Cells were maintained in 1:1 mixture of Dulbecco's modified Eagle's medium (DMEM) and Ham's F12 medium supplemented with 10% fetal bovine serum, GlutaMAX™, 5,000 U/mL penicillin/streptomycin, 1× nonessential amino acids, 1× sodium pyruvate, and 0.1 mM 2-mercaptoethanol.
rESCs and riPSCs were cultivated in N2B27 medium: a mixture of N2 (DMEM/F12 supplemented with 1× N2) and B27 (Neurobasal supplemented with 1× B27) media, GlutaMAX, 5,000 U/mL penicillin/streptomycin, 0.1 mM 2-mercaptoethanol, 1,000 U/mL mouse LIF (StemRD), 1 μM PD0325901 (StemRD), and 3 μM CHIR99021 (StemRD). All reagents were from Life Technologies unless otherwise stated.
All cells were grown at 37°C in an atmosphere containing 5% CO2.
rESC and riPSC derivation
rESC derivation was performed as described previously [5,6].
Viral preparation and infection were performed as described [12].
TetO-FUW-OSKM was a gift from Rudolf Jaenisch (Plasmid 20321) [12], pMDL-g/pRRE and pSRV-Rev were a gift from Didier Trono (Plasmid 12251, Plasmid 12253) [13], and pCI-VSVG was a gift from Garry Nolan (Plasmid 1733). Lipofectamine 2000 was used as a transfection reagent.
rEFs at the passage 3 were seeded at 104 cells/cm2 in a six-well plate. One hour before transduction, the growth medium was supplemented with 4 μg/mL hexadimethrine bromide (Polybrene, H9269; Sigma), and then virus particles diluted in culture medium were added to wells. After 18 h of incubation, the culture medium was replaced with a new medium containing doxycycline (44577; Sigma) at a final concentration of 2 μg/mL. Four days later, cells were plated on a 10-cm culture dish containing mitomycin-inactivated mouse EFs and cultured in the riPSC medium.
rESC and riPSC differentiation
Monolayer differentiation of rat pluripotent cells was performed for 14 days on tissue culture surfaces coated with different collagen types or gelatin. We used collagen III (C4407; Sigma) and collagen IV (C5533; Sigma). Collagen treatment was carried out according to the manufacturers' instructions. Pluripotent cells disaggregated with collagenase IV or TrypLE were seeded on a collagen or gelatin-treated surface at the density of 104 cells/cm2 in the medium containing 1:1 mixture of the N2B27 (without LIF, PD0325901, CHIR99021) and DMEM/F12 media supplemented with 10% fetal bovine serum, nonessential amino acids, GlutaMAX, 10 ng/mL basic fibroblast growth factor (bFGF), and 10 μM Y27632 (1254; R&D). The medium was changed every day. Small colonies of rat pluripotent cells (24 h after plating) were preferential to be used for monolayer differentiation. All reagents were from Life Technologies unless otherwise stated. All cells were grown at 37°C in an atmosphere containing 5% CO2.
Embryoid bodies (EBs) were formed by the hanging drop method in the medium for monolayer differentiation or in rat EB formation medium [14].
The FITC Annexin V-PI double staining kit (640914; Bio Legend) was used to evaluate apoptosis and necrosis levels of rat cells differentiated in the monolayer and EBs according to the manufacturer's instructions. Single-cell suspension was prepared using enzyme-free cell dissociation buffer (13151-014; Life Technologies). Stained cells were assessed by flow cytometry on BD FACS Canto II, FACS Diva software. Life imaging of the first 3 days of differentiation was performed on a Cell-IQ MLF (CM Technologies) imaging system in the Interinstitutional Shared Center of Cell technologies SB RAS.
Slide preparation, immunofluorescence staining, and RNA and DNA FISH
Nuclei and metaphase spread preparation, immunofluorescence staining (IF), IF-RNA-DNA FISH, IF-DNA FISH, as well as BrdU incorporation, and detection were performed as described [15,16]. Xist RNA was detected using BAC PR23 31G17 as a probe. Rat X chromosome was detected with BAC CH230 367L11, which contained a fragment of XCI center. IF and FISH slides were analyzed on a Nikon Ni-E microscope using NIS-Elements software. Our filter set allowed distinguishing Alexa 488, Alexa 594, and Cy3. The first two fluorochromes were used for IF, while the last one for DNA FISH. About 50 metaphase spreads or ∼150–200 nuclei were analyzed in each case. Protocol for immunofluorescent detection of germ layer markers was essentially the same as published in ref. [2]. Primary and secondary antibodies are listed in Supplementary Table S1 (Supplementary materials are available online at
RNA and DNA isolation, reverse transcription with subsequent PCR, and real-time PCR
Total RNA and genomic DNA were isolated from 5 × 106 cells using TRI Reagent (Ambion). Polymerase chain reaction (PCR) was carried out in a 50 μL volume using 5 U of Taq polymerase per reaction. Real-time PCR was performed on a LightCycler480 (Roche) in three replicates. cDNA samples were used as templates for reactions in a dilution of 1/100. Amplification was performed using the fluorescent SYBR Green I intercalator and Real-time PCR reagent kit (PCR-mix, Syntol). The primer sequences used are listed in Supplementary Table S2.
Bisulfite sequencing
Bisulfite conversion was performed in a thermocycler using the EZ DNA Methylation-Gold Kit (Zymo Research). Converted DNA was used as a template for PCR with primers that were previously described [17]. Sequences were analyzed using the QUMA Quantification tool for methylation analysis [18].
Karyotyping
Karyotyping was performing as described (19). Briefly, cell lines in logarithmic growth phase were incubated for 1 hour with 5 mg/ml ethidium bromide, then for 1.5 hours with 0.5 mg/ml colchicine. Cells were next incubated in 0.075M KCl solution for 30 min at 37°C. For chromosome counting and identification DAPI (200 ng/ml in 2×SSC) was used for 5 min. DAPI-stained chromosomes were identified according to the international classification using the AxioPlan2 Imaging (Carl Zeiss) equipped with CCD camera (CV M300, JAI Corporation), CHROMA filter sets.
Teratoma formation assay
Teratoma formation assay was performed according to standard protocol [20]. rESCs and riPSCs (at passages from 10 to 40) were injected subcutaneously to dorsal flank of nu/nu mice. Paraffin-embedded tissue slices (7–10 μm) were stained with ready-to-use reagent kits (Bio-Optica): Picro-Mallory trichrome (04-021822), Masson's trichrome (04-011802), P.T.A.H.-hematoxylin (04-060802), Luxol fast blue Krever Barrera (04-200812), Azan trichrome (04-001802), Picrofucsin Van Gizon (04-030802), Mucicarmine (04-190812), WVG long method (04-051802), and hematoxylin and eosin. Images were captured and analyzed using Axioscop2+ supplied with CCD camera (AxioCam HRc) and software, AxioVision, in the Interinstitutional Shared Center for Microscopic Analysis of Biological Objects SB RAS (
Chimera generation assay
For chimeric blastocyst and embryo (E10–E12) generation, embryos of the mouse BALB/cJLacY, ICR, and OG2 strains were used as hosts. Pseudopregnant F1: (♀C57BL/6J × ♂CBA/Lac) and F1: (♀CBA/Lac × ♂C57BL/6J) females were used as recipients. Chimeras were generated according to standard protocol [20] through microinjection of 10–15 cells in 3.5-day C57BL/6 and ICR mouse blastocysts. Micromanipulations with embryos were performed using Olympus SZX-7 and IX-71 (Olympus) microscopes and Narishige Group, VacuTip, and TransferTip-ES (Eppendorf) micromanipulators. Embryos were dissected at 8–9 and 10–11 days of development (E-9 and E10-11) in DPBS (pH 7.4–7.6) using an Olympus SZX-7 (Olympus) microscope.
mRNA-Seq
rEFs (RNFF1, RNFM1), rESCs (RES27, dB50), and riPSCs (NF13, NF21, SU3, and QV8) were chosen for mRNA-Seq analysis; 107 cells were lysed in TRIzol reagent and total RNA was extracted according to the manufacturer's instructions. RNA quality was analyzed by Agilent Bioanalyzer and RIN was 9.2–9.6 for all samples. Ten micrograms of total RNA was used to obtain poly(A) RNA by the Dynabeads mRNA Purification Kit. Poly(A) RNA was used to prepare the library for Illumina GAIIx sequencer using the NEBNext® mRNA Library Prep Reagent Set for Illumina® according to the protocol. For each library, more than 40 million single reads were generated. The raw data were submitted to the SRA database under accession number ID SRP019984.
Results
Generation and characterization of iPSCs and ESCs of Brattleboro and WAG rats
Considering further application of rat pluripotent cells for disease modeling, we have generated rESCs and riPSCs not only of the WAG rat strain but also of the Brattleboro rats suffering from hereditary diabetes insipidus. The Brattleboro rat strain carries an autosomal recessive mutation (diabetes insipidus, di/di) in the gene encoding arginine vasopressin. The mutation results in an inability to synthesize this hormone due to a single guanine deletion in exon 2 and in the development of diabetes-like phenotype [21 –23] (Supplementary Fig. S1). ESCs of inbred WAG and Brattleboro rats were derived from the blastocysts using the 2i protocol in the N2B27 medium [5,6]. riPSCs were generated from female and male EFs by transduction with two lentivirus types. One carried cDNA of the mouse OSKM genes (Oct4, Sox2, Klf4, and Myc) under control of a doxycycline regulatory element and the other did a reverse tetracycline transactivator gene. After derivation, rat pluripotent stem cells (rPSCs), both rESCs and riPSCs, were not additionally subcloned for further analyses. Then, standard tests confirming pluripotency of the derived cell lines were performed. Table 1 lists the rESC and riPSC lines obtained. It should be noted that no difference was found between pluripotent cell lines generated from the Brattleboro and WAG rats.
EF, embryonic fibroblast; ESC, embryonic stem cell; iPSC, induced pluripotent stem cell.
All the rESC and riPSC lines grew as round-shaped colonies of small-sized cells with a high nucleus–cytoplasm ratio. The riPSCs and rESCs expressed endogenous alkaline phosphatase and pluripotency markers—the OCT4 and NANOG transcription factors and SSEA1 surface antigen (Fig. 1A, B). RT-PCR assay demonstrated that the lines expressed the genes responsible for pluripotent state maintenance, such as Oct4, Nanog, Sox2, Eras, as well as Rex1, and Dppa3 (Supplementary Fig. S2A). According to bisulfite sequencing data, the promoter region DNA of Oct4 and Nanog was demethylated in the riPSC and rESC lines and was highly methylated in the initial rEFs, where these genes were repressed (Supplementary Fig. S3). Since the riPSC lines were derived using the lentivirus construct that was able to integrate into the genome, the cells were assayed for whether the exogenous reprogramming factors were expressed in the absence of doxycycline. Using specific primers, the exogenous Oct4, Sox2, Klf4, and Myc were demonstrated not to be expressed in all the riPSC lines (except for SU18 and NF7) (Supplementary Fig. S2B, C).
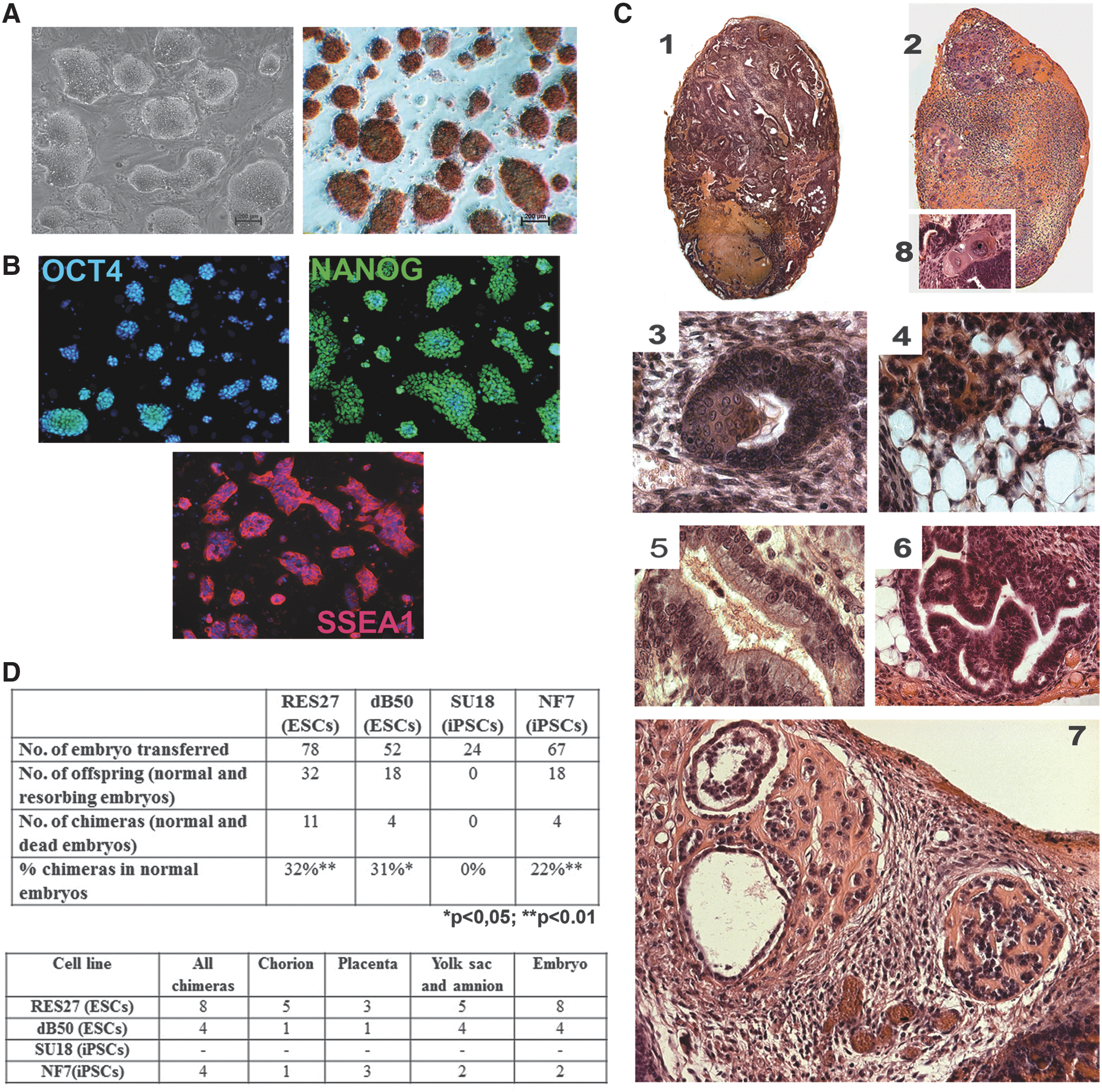
Characterization of pluripotent stem cell lines of Brattleboro and WAG rats. Representative images demonstrating
The riPSCs and rESCs were able to be differentiated into derivatives of all three germ layers as was shown by the teratoma test (Fig. 1C). A subcutaneous injection of the riPSCs and rESCs to nu/nu mice induced tumor development. Histological analysis detected the regions of fold-like and rosette-like primitive neuroepithelium as well as cell morphotypes characteristic of a premature glial-like tissue. The loose connective and fatty tissues were abundant (Fig. 1C4). The cartilage and primary osseous trabeculae were detectable only in the tumors derived from the NF7 cell line (Fig. 1C7). Hematopoietic (erythropoietic) foci were observed in the tumors obtained from the dB50 and NF7 lines. The epithelial tubular structures lined with ciliated (Fig. 1C5), enzyme-secreting, and mucoid-secreting gut-like epithelium were rather abundant (Fig. 1C6). The regions with squamous epithelium were also observed both within complex epithelial tubular structures and (much less frequently) as a developing autonomous keratin lens (Fig. 1C3). A specific feature of all the tumors was a stable presence of various syncytial trophoblast- and cytotrophoblast-like derivatives (Fig. 1C1,2,8).
The rESCs and riPSCs were assayed for their ability to contribute to the tissue development of chimeric organisms, which is the strongest evidence of their pluripotency. A xenogenic model, rat ↔ mouse, was used. Ten to fifteen cells were injected into 3.5-day blastocysts of C57BL/6 and ICR mice. On days 8–9 and 10–11 of mouse embryonic development (E8–9 and E10–11), the embryos were dissected and tested for chimerism (colonization) using primers for microsatellite regions. The rates of chimeras for the dB50, SU18, RES27, and NF7 lines have been shown to be 11%, 0%, 27%, and 22%, respectively (Fig. 1D). Thus, no chimeras have been obtained for the SU18 cell line. Such a situation is rather likely, assuming that the injected stem cells interfere with normal morphogenesis of the chimeric blastocyst and/or specialization of transient cell types.
It has been earlier demonstrated that reprogramming may involve various alterations in the cell genome, ranging from single-nucleotide substitutions to considerable variations in the copy numbers of extended genomic elements and large chromosome rearrangements [24,25]. To estimate the effects of reprogramming on karyotype stability, the initial rEFs and riPSCs were analyzed (Supplementary Table S3 and Supplementary Fig. S4). The modal chromosome number in the rEFs was 42. RNFF1 and RNFM1 contained 11% and 6% of polyploid metaphases, as well as 26% and 28% of aneuploid metaphases, respectively. No structural chromosome rearrangements were observed. The modal class in all the riPSC lines was 42 chromosomes and varied from 34% in SU3 to 55.5% in the NF7 and NF29 lines. No loss of sex chromosomes was detected. However, chromosome rearrangements appeared as compared with the rEF lines. In particular, the NF29 line was likely to consist of two subclones, one of which (21.9%) carried metaphases with a Robertsonian translocation, Rb (9.9), while 11% of the metaphases contained circular chromosomes and solitary Robertsonian translocations, Rb (3.3) and Rb (3.4). It is noteworthy that the rESCs also displayed chromosome rearrangements. All the rESC lines were polymorphic in the length of the chromosome 18 p-arm; 17.1% of the metaphases of the dB50 line carried various Robertsonian translocations—Rb (4.3), Rb (2.7), Rb (3.12), and Rb (1.9), none of which was prevalent. Thus, the rESC and riPSC lines obtained were characterized by genomic instability, such as structural and numerical chromosome aberrations. However, the fact that no rearrangements have been detected in the rEFs suggests that this may be due to cultivation conditions and a more stable karyotype may be obtained through optimization of the conditions.
Both X chromosomes are active in mouse female pluripotent cell lines, whereas one of the two X chromosomes is inactive in differentiated female cells [7,8]. We studied X-chromosome status in rat differentiated and pluripotent cells using immunofluorescent staining. In RNFF1 (female EF line), one of the two X chromosomes was inactive and demonstrated exclusion of H3K4me3, a marker of active chromatin, and accumulation of H3K27me3, a marker of inactive chromatin (Fig. 2A). As for the riPSCs and rESCs, both X chromosomes were enriched with H3K4me3 and no enrichment of H3K27me3 was observed (Fig. 2B). Thus, both X chromosomes were active in the rat pluripotent cells. This implies that the inactive X chromosome was reactivated during reprogramming of rEFs to the pluripotent state. It is worth noting that the mouse X chromosome is also reactivated at the final stages of reprogramming, which is an evidence of successful attainment and maintenance of the pluripotent state [26].
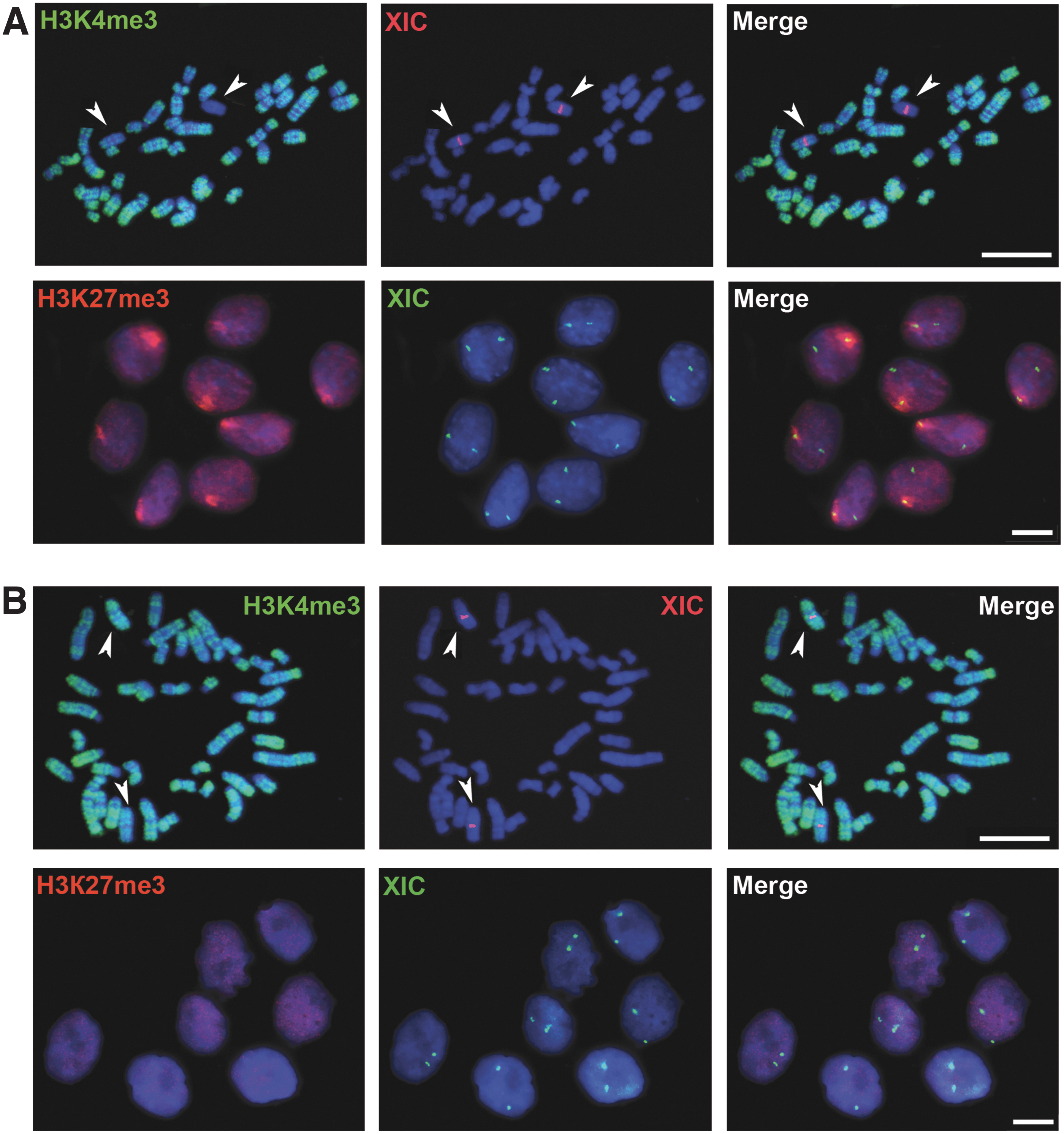
X chromosome is reactivated during rEF reprogramming to the pluripotent state.
Thus, we have obtained a set of pluripotent cell lines (iPSCs and ESCs) of the WAG and Brattleboro rats. Our next step was massively parallel mRNA sequencing (RNA-Seq) on the Illumina GAIIx platform for a more detailed analysis of the cell lines. rEF (RNFF1 and RNFM1), rESC (RES27 and dB50), and riPSC (NF13, NF21, SU3, and QV8) lines were selected for transcriptome analysis.
Transcriptome analysis
A total of 547 million raw reads were obtained from sequencing of 10 libraries with an average length of 45 nt. Sequenced reads were aligned to the rat rn4 genome using TopHat [27]; on the average, 55% of reads were aligned. The counts of mapped reads for each gene were calculated and used in subsequent analyses. The normalization of read counts between samples was performed using the DESeq method [28] (Supplementary Table S4). The raw data were submitted to the SRA database under accession number ID SRP019984. Expression values from the RNA-Seq data were calculated by quantifying the number of sequence reads for each gene with standardized RPKM values (reads per kilobase of exon model per million mapped sequence reads).
The RNA-Seq data were used in cluster analysis of the cell lines obtained. The riPSC lines grouped into a single cluster, displaying a high similarity of their gene expression patterns (the lowest correlation coefficient was 0.86) (Fig. 3A). It is worth noting that the correlation coefficient of gene expression between the riPSC (QV8) and rESC (dB50) lines was as high as 0.96. This suggests that the pluripotent state attained through rEF reprogramming is almost indistinguishable from that in the rESCs. The RES27 (rESCs) and NF21 (riPSCs) lines showed the lowest similarity (the correlation coefficient was 0.86), demonstrating a variation in the expression profiles between pluripotent cells. This variation is likely to be cell line specific and not to be associated with the reprogramming per se. Two rEF lines displayed considerable similarity (the correlation coefficient was 0.89) in their gene expression profiles, formed a separate cluster, and significantly differed from the pluripotent cells (the highest correlation coefficient was 0.34). Gene expression heat map for genes involved in self-renewal and pluripotency maintenance in mice and humans [29] was constructed for the rat cell lines (Fig. 3B). The results evidently demonstrated a high transcription level of the pluripotency markers and similarity within the rESC and riPSC lines, as well as a low transcription level of the pluripotency genes and similarity within the rEF lines (Fig. 3B, C).
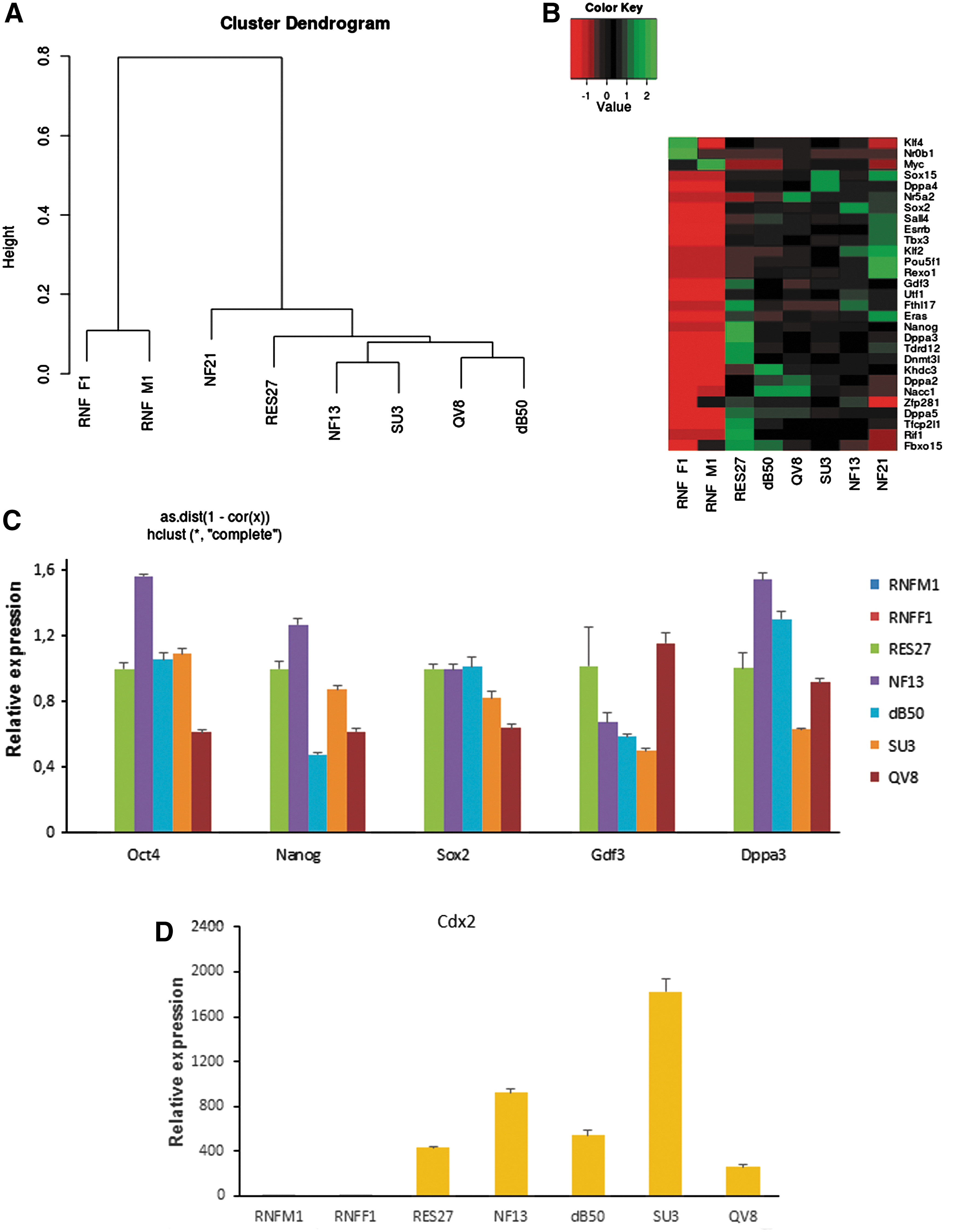
Transcriptome characterization of pluripotent stem cell lines of Brattleboro and WAG rats.
Several states of pluripotency differing in a number of characteristics are distinguished [30,31]. It is currently postulated that mouse ESCs derived in a serum-free medium under 2i conditions display a ground state of pluripotency. Their properties are much closer to pluripotent cells derived from preimplantation blastocysts compared with the so-called conventional ESCs generated in a serum-containing medium [30]. The ground state and conventional ESCs are, in turn, matched against mouse epiblast stem cells (EpiSCs) derived from postimplantation embryos (E5.5–E7.5) [32]. Based on the RNA-Seq data, we decided to find out which of the pluripotency states was acquired by the rESC and riPSC lines and maintained there. A heat map for several genes differing in their transcription levels in mouse ESCs and EpiSCs (mESCs and mEpiSCs) [32] was constructed for the rESC and riPSC lines (Supplementary Fig. S5). The transcription level of the genes in riPSCs and rESCs showed a higher similarity to that in mESCs rather than to that in mEpiSCs. Moreover, XX mEpiSCs are characterized by the presence of one inactive X chromosome, whereas XX mESCs carry two active X chromosomes [31]. We have shown that both X chromosomes have active chromatin marks in the XX rESCs and riPSCs. When examining the transcriptomes of the XX riPSC lines using SNP data (Supplementary Table S5), the transcripts of both X chromosomes were detectable at approximately the same rates. Each riPSC line is a clone derived from a single EF, which suggests that one X chromosome in the XX riPSCs was reactivated during reprogramming and completely restored not only modifications of active chromatin but also the corresponding level of gene transcription. Thus, both riPSCs and rESCs do not match the criteria for EpiSCs. Next, we assayed the rESCs and riPSCs for the mRNA levels of genes displaying different transcription levels in mESCs of ground state and conventional pluripotencies [32]. A high transcription level of Prdm14 and a low transcription level of Myc, Mycn, Id1, and Id2 suggest that the riPSCs and rESCs possess the ground state of pluripotency.
In the teratoma test, we noticed that the rESC and riPSC lines gave high yields of syncytial trophoblast and cytotrophoblast derivatives. It was described that use of a GSK3 inhibitor, CHIR99021, in cultivation of rat and mouse ESCs caused ectopic expression of Cdx2 [33 –35] encoding a well-known trophoectodermal transcription factor [36]. According to RT-PCR and transcriptome analysis, Cdx2 was not expressed in the rEF lines and was highly expressed (on the average, 1,192 normalized read counts) in the rat pluripotent cells (Fig. 3D and Supplementary Table S4). The mRNA transcription levels of major genes (Eomes, Elf5, Ets2, Gata3, Tcfap2c, Esrrb, and Hand1) involved in establishment and maintenance of trophoblast stem cells were also assessed [37]. We observed a high level of the mRNAs for Eomes, Ets2, Tcfap2c, and Esrrb, of which at least Eomes and Esrrb (as well as Cdx2) are the target genes in activation of the Wnt-β-catenin signaling with CHIR99021. Thus, the formation of trophoblast derivatives in the teratoma test may be explained by the fact that CHIR99021 induces ectopic expression of several trophoblast genes.
The RNA-Seq data also allowed us to estimate the change in transcription profile of the genes involved in formation of cell structural and functional elements. The changes in expression of genes encoding components of extracellular matrix and intercellular contacts were analyzed. In the rESCs and riPSCs, a decrease in the transcription level was observed for all genes encoding various collagen types (except for Col13a1) as well as elastin (Eln) and fibronectin (Fn1) precursor genes. Col13a1 was the only gene with 10- to 145-fold increased expression in the riPSCs compared with the rEFs.
Various cell types utilize different intercellular contacts. The major function of occluding junctions is tight joining of adjacent cells. The major proteins providing for the junctions are claudins and occludin. We observed that the mRNA levels for claudin (Cldn4, -7, -6, and -9) and occludin genes were elevated in the rESCs and riPSCs. The anchoring junctions are divided into two subtypes—adherens junctions and desmosomes. They hold cells together and are formed of transmembrane adhesion proteins that belong to the cadherin family. Transcriptome analysis demonstrated that the gene encoding E-cadherin increased its expression level (351- to 1,906-fold) in the rESCs and riPSCs, whereas the genes encoding the other cadherins/protocadherins were repressed. The second subtype comprises focal adhesions and hemidesmosomes that bind cells to the extracellular matrix and are formed of transmembrane adhesion proteins of the integrin family. According to the transcriptome data, Itgb1 and Itgb5 were repressed, whereas Itgb2 and Itgb4 were activated in the rESCs and riPSCs.
Thus, the transcriptome analysis has demonstrated a high correlation between the rESC and riPSC lines in their transcription profiles that corresponded to the ground state of pluripotency. The rESC and riPSC lines were characterized by a low transcription level of the genes encoding components of extracellular matrix and a high mRNA level of the genes encoding proteins forming tight junctions.
XCI dynamics during rESC and riPSC monolayer differentiation
XCI is known to occur during differentiation of mouse pluripotent cells [38]. However, rESCs and riPSCs are extremely difficult to differentiate in vitro. We first used an in vitro differentiation protocol proposed by Cao et al. [14]. The protocol allows efficient differentiation of rat pluripotent cells through EB formation in the presence of conditioned medium collected from mouse EFs and a quarter of PD0325901 and CHIR99021 concentration used for cultivation of pluripotent cells. To improve cell survival during differentiation, a Rho-associated kinase inhibitor, Y27632, was also used in the EB-based protocol. Under the differentiation conditions, XCI was initiated in the XX rESCs and riPSCs, but this occurred very asynchronously, so it was very difficult to trace XCI dynamics (data not shown).
To study XCI dynamics, we developed a new differentiation protocol of rat pluripotent cells. We noticed that rat pluripotent cells successfully survived and proliferated on collagen III-coated culture dishes. We assumed that collagen III provided specific environment required for cell viability and carried out differentiation of the rat pluripotent cells in the monolayer on a surface coated with collagen III. The 1:1 mixture of the N2B27 medium and DMEM/F12 medium was used. The mixture was supplemented with 10% fetal bovine serum, nonessential amino acids, GlutaMAX, 10 μM Y27632, and 10 ng/mL bFGF. Importantly, PD0325901, CHIR99021, LIF, and conditioned medium that are used for pluripotency maintenance were not added to the differentiation medium. In the medium, cells of different rESC and riPSC lines successfully attached to the collagen III-treated surface even when they were in a single-cell suspension and actively proliferated (Supplementary Movie S1). From the first differentiation day, the cells changed their morphology (Fig. 4A). After 2 days of the monolayer differentiation, we quantified cell viability by FACS using double staining with Anexin V and propidium iodide (Fig. 4B). The percentage of living cells in monolayer differentiation was about 82%. This is ∼30% higher than viability of the cells differentiated in EBs even in the same medium. On differentiation day 12, we observed a monolayer of cells with different morphologies (Fig. 4A). Immunofluorescent analysis of the differentiated cells revealed derivatives of three embryonic germ layers (Fig. 4C). Moreover, as evident from RT-PCR, nearly the same differentiated derivatives were obtained both under the monolayer and EB differentiation conditions (Supplementary Fig. S6). Furthermore, we asked whether collagen III was the only substrate for successful differentiation of rat pluripotent cells in the monolayer or it could be replaced with other collagen types or even gelatin. We established that the rat pluripotent cells were also be able to successfully survive and be differentiated on surfaces coated with collagen IV (data not shown) or gelatin (Fig. 4B). As for untreated surfaces, rESCs and riPSCs attached inefficiently and had only ∼30% viability (Fig. 4B and Supplementary Fig. S7).
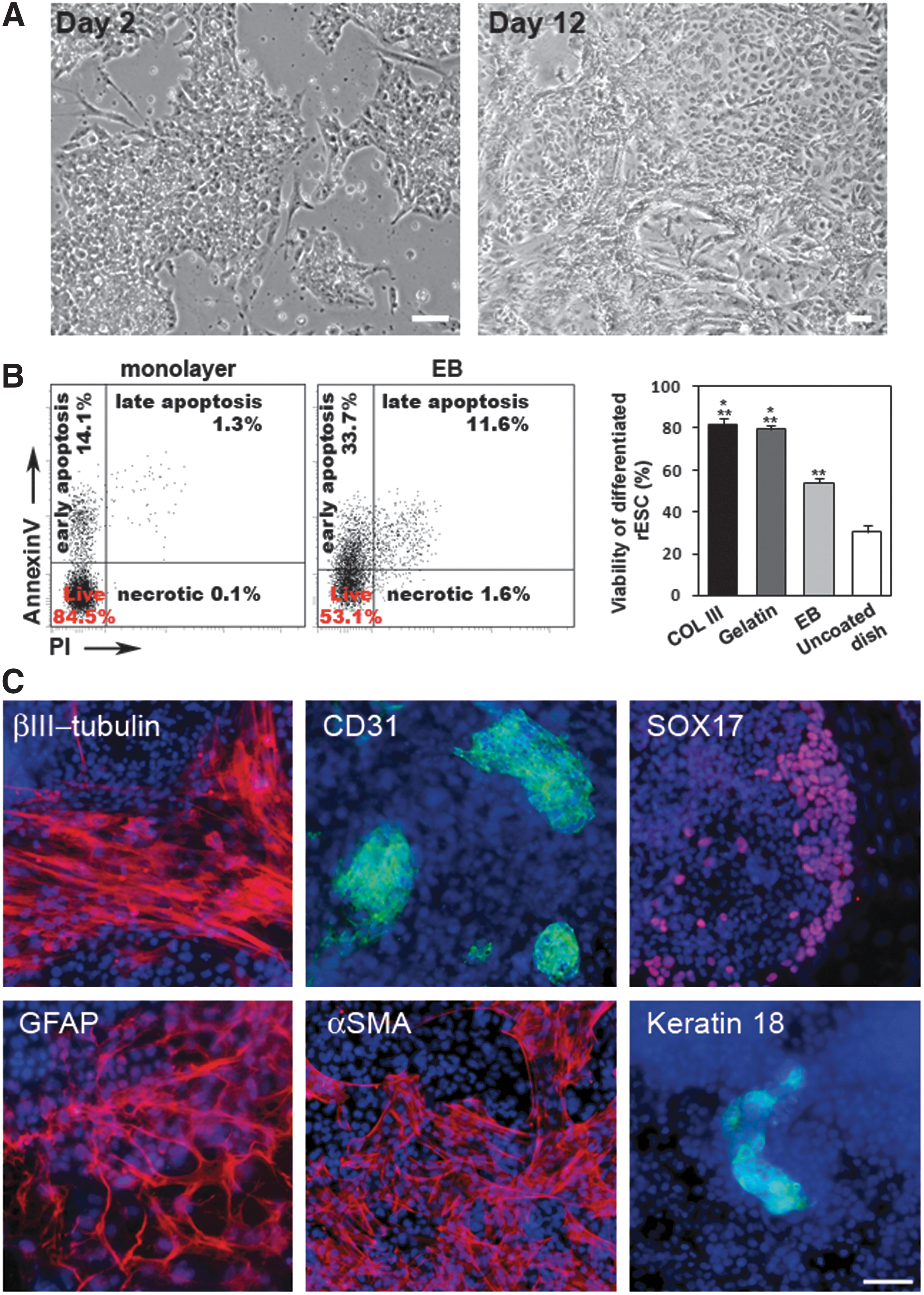
Monolayer differentiation of rat pluripotent cells on collagen III-coated surfaces.
Next, we used the monolayer differentiation protocol to study XCI dynamics in differentiating XX rESCs (RES6, RES27) and riPSCs (NF13). After the rat pluripotent cells were plated on collagen III in the differentiation medium, daily cell aliquots were taken for cytocentrifugation and subsequent XCI assays up to differentiation day 14. We started with assessment of Xist RNA accumulation, which is a key event for XCI initiation. In undifferentiated XX rat pluripotent cells, no Xist RNA signals were detected by RNA FISH (Fig. 5A). Xist RNA clouds associated with one of the two X chromosomes appeared on differentiation day 3 for the RES6 and NF13 lines and only on day 5 for the RES27 line. One day later, Xist RNA clouds were revealed in the majority of the cells (Fig. 5A). The Xist RNA dynamics has been shown to be reproducible for each of the cell lines studied.
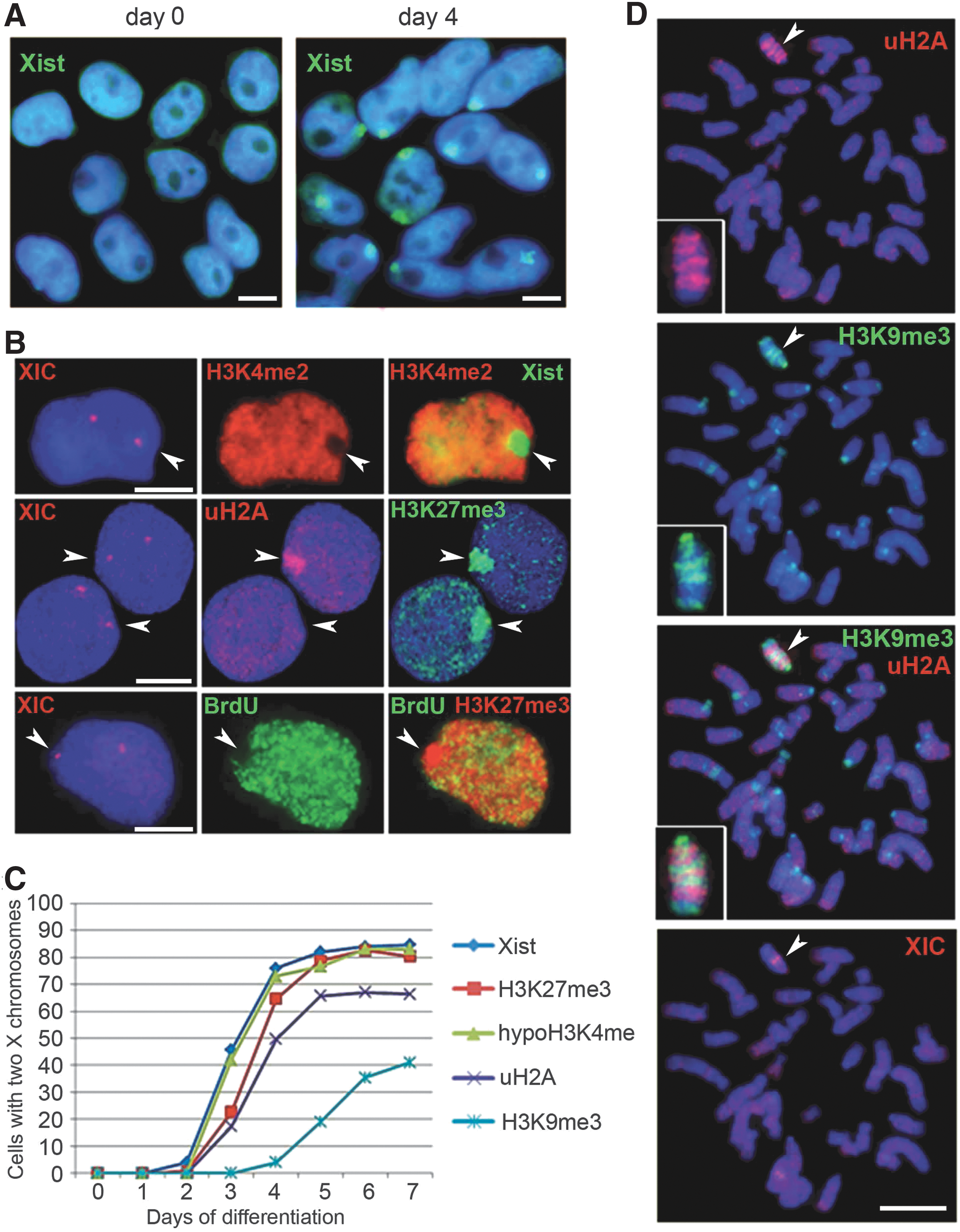
XCI in differentiating female rat pluripotent stem cells.
In parallel, we examined changes in histone modifications during XCI in differentiating rat pluripotent cells (Fig. 5B–D and Supplementary Fig.S8). Using a combination of IF, RNA, and DNA FISH, we tested X chromosomes for loss of active chromatin marks and enrichment of repressive histone modifications that had been found on the inactive X chromosome in other mammals [15,16,38 –41]. In all the three cell lines, shortly after Xist RNA coating, X chromosome undergoing XCI was depleted in H3K4me2, an active chromatin marker, and enriched with H3K27me3 and ubiquitylated H2A (uH2A), repressive chromatin modifications. H3K9me3 appeared on the inactive X chromosome only after H2A ubiquitylation and was the latest among the histone modifications tested. The accumulation of Xist RNA and H3K27me3 was seen in the majority of differentiating cells. The percentage of the cells with uH2A and H3K9me3 enrichment on the inactive X chromosome was significantly lower and varied between cell lines. Using BrdU incorporation followed by IF, we showed that the inactive X chromosome started replicating later in S-phase than the active X chromosome in some cells since day 5. However, the proportion of cells with one late replicating X chromosome did not exceed 5% even on day 14. It should be noted that up to 16% of cells with two X chromosomes, but without any early signs of XCI, namely Xist RNA clouds, H3K27me3 enrichment, and H3K4 hypomethylation, could be found in all the three cell lines even on differentiation day 14.
Differentiated derivatives of all the three rPSC lines, including NF13, H3K27me3, and uH2A, colocalized on the inactive X chromosome as discrete bands that alternated with H3K9me3 bands (Fig. 5D and Supplementary Fig. S8). This alternating pattern was previously found on the inactive X chromosome of other placental mammals and seems to reflect two different types of facultative heterochromatin acting in XCI [15,16,38 –41]. However, no H3K9me3 enrichment (excluding the centromeric region) was detected on the inactive X chromosome in the female rEFs used for the NF13 line generation. Taking into account that H3K9me3 was recruited on the inactive X chromosome only in about a half of cells during rESC and riPSC differentiation, we suggest that participation of H3K9me3 in XCI is not necessary for all cells.
Discussion
Pluripotent cells are among the most promising in vitro models for the research into molecular pathogenesis of various diseases and for development of methods and approaches to disease therapies. Two types of pluripotent cells—ESCs and iPSCs—are currently being used in research [42]. The laboratory rat is one of the oldest objects used in modeling several significant diseases, such as various types of hypertension, Alzheimer's disease, and cardiovascular diseases. However, rat stem cells have been successfully derived only recently [5,6,19], and insufficient knowledge of their properties considerably interferes with their application in this area.
In this work, we generated rESC and riPSC lines from rEFs and performed a large-scale analysis of the lines using a standard set of PSC assays and genome-wide transcriptome analysis by RNA-Seq and studied X-chromosome status in the rat pluripotent cells and their differentiated derivatives. We have demonstrated that the cell lines derived express the genes characteristic of the pluripotent state, are able to give derivatives of all embryonic germ layers during differentiation, and contribute to tissues of chimeric organisms. The RNA-Seq assay has shown that all the rESC and riPSC lines are highly similar in their transcriptome profiles and considerably differ from rEFs. In addition, we have not found any difference in the transcription profiles between the pluripotent cell lines derived from Brattleboro and WAG rats. The transcription profiles in the rat pluripotent cells corresponded to the ground state of pluripotency in several characteristics.
Interestingly, the rPSC lines gave rise to syncytial trophoblast and cytotrophoblast derivatives. We supposed that this phenomenon was due to use of the GSK3 inhibitor, CHIR99021, during rPSC cultivation. This is in agreement with previous studies [33 –35] that demonstrated that GSK3 inhibition led to activation of some differentiation-associated genes, including several trophoblast-specific genes. Similar to rPSCs, mouse ESCs and iPSCs were recently discovered to give rise to trophoblast cells due to Cdx2 overexpression [43], indicating the existence of common transcriptional mechanisms that regulate lineage-specific differentiation in rats and mice.
We have shown that maintenance of two active X chromosomes is a feature of the rESCs and riPSCs. Establishing the pluripotent state during the rEF reprogramming is coupled with reactivation of the inactive X chromosome. The reactivation leads to loss of heterochromatic marks from the inactive X chromosome and to restoration of transcriptional activity of its genes. The pluripotency network is known to be able to directly repress initiation of XCI in mouse ESCs and iPSCs [44]. This may be also the case in the rESCs and riPSCs. Loss of the pluripotency in the XX rESCs and riPSCs during differentiation is accompanied by XCI. The rESC and riPSC lines differ in time of XCI initiation. However, XCI dynamics is very similar in differentiating cells of different rESC and riPSC lines and resembles that during differentiation of pluripotent cells and embryogenesis in mice [7,8,38]. Two types of facultative heterochromatin previously described in other mammals have been identified on the rat inactive X chromosome. The two types have been shown to be established on the inactive X chromosome during differentiation of rat pluripotent cells. We believe that the rESCs and riPSCs can be further used as an ex vivo model to study XCI mechanisms and will help clarify what enzymes are responsible for H3K9me3 recruitment to the inactive X chromosome. In almost all the cells demonstrating XCI, the inactive X chromosome is associated with Xist RNA accumulation and H3K27me3 enrichment. Recruitment of uH2A and H3K9me3 on the inactive X chromosome occurs only in a part of the cells. Whether the phenomenon is cell-type specific or a disruption in XCI needs to be elucidated. It should be noted that XCI does not take place in all differentiating cells. The cells without signs of XCI can be observed up to differentiation day 14. The cells that did not initiate XCI seem to die during subsequent cultivation. Inability of cells to trigger XCI and karyotype abnormalities may cause cell death during rat pluripotent cell differentiation.
We have developed a new monolayer differentiation protocol for rESCs and riPSCs to study XCI. Existing protocols allow differentiating rat pluripotent cells only through EB formation. In the EB-based protocols, a gradual removal of PD0325901 and CHIR99021 that are necessary for rat pluripotent cell cultivation is used [14,19]. Cell viability in an optimized EB-based protocol is about 75%. However, differentiation of pluripotent cells in the monolayer is more preferable in some cases. For example, addition of soluble morphogenes and extracellular microenvironment can be more precisely controlled in monolayer culture than in EBs [45]. In addition, the EB-based protocol with gradual removal of PD0325901 and CHIR99021 is not suitable to study dynamics of biological processes. It was impossible to predict when XCI initiated in differentiating rESCs and riPSCs using the EB-based protocol. Even if we found first signs of XCI in some EBs, we could detect no XCI markers in a new pool of EBs the next day. In the study, we first suggested a new method of rat pluripotent cell differentiation in the monolayer on the surface coated with collagen III, IV, or gelatin. The substrates were found to provide good cell viability during differentiation, which allowed removal of PD0325901 and CHIR99021 from the differentiation medium on day 0. Due to the complete removal of PD0325901 and CHIR99021, cells may overcome faster inhibition of differentiation signaling pathways. This results in synchronous differentiation of rat pluripotent cells, as was evident from the XCI dynamics study. The protocol appears to be useful to study dynamics of different biological processes associated with cell differentiation. Using the protocol, derivatives of three germ layers can be obtained. This gives us the opportunity to optimize the protocol for directed differentiation into specialized cell types.
Footnotes
Acknowledgments
The authors thank Elena V. Dementyeva for critical comments and suggestions on the article and Vladimir V. Sherstyuk for technical assistance. Rat cell isolation and analysis, RNA-Seq experiments, and analysis of transcriptomes were supported by the Russian Science Foundation (14-14-00271); XCI dynamics study was supported by RFBR (research project 15-04-03947 A).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.