
Abstract
Clonal endothelial progenitor cells (EPCs) have been implicated in the aberrant vascular growth that features infantile hemangioma (IH), the most common benign vascular tumor in childhood that may cause ulceration, bleeding, and/or permanent disfigurement. Endothelial colony-forming cells (ECFCs), truly endothelial EPCs endowed with clonal ability and capable of forming patent vessels in vivo, remodel their Ca2+ toolkit in tumor-derived patients to acquire an adaptive advantage. Particularly, they upregulate the proangiogenic store-operated Ca2+ entry (SOCE) pathway due to the overexpression of its underlying components, that is, stromal interaction molecule 1 (Stim1), Orai1, and transient receptor potential canonical 1 (TRPC1). The present work was undertaken to assess whether and how the Ca2+ signalosome is altered in IH-ECFCs by employing Ca2+ and nitric oxide (NO) imaging, real-time polymerase chain reaction, western blotting, and functional assays. IH-ECFCs display a lower intracellular Ca2+ release in response to either pharmacological (i.e., cyclopiazonic acid) or physiological (i.e., ATP and vascular endothelial growth factor) stimulation. Conversely, Stim1, Orai1, and TRPC1 transcripts and proteins are normally expressed in these cells and mediate a constitutive SOCE, which is sensitive to BTP-2, La3+, and Pyr6 and recharges the intracellular Ca2+ pool. The resting SOCE in IH-ECFCs is also associated to an increase in their proliferation rate and the basal production of NO compared to normal cells. Likewise, the pharmacological blockade of SOCE and NO synthesis block IH-ECFC growth. Collectively, these data indicate that the constitutive SOCE activation enhances IH-ECFC proliferation by augmenting basal NO production and sheds novel light on the molecular mechanisms of IH.
Introduction
I
Intracellular Ca2+ signals have long been known to play a key role in angiogenesis by stimulating endothelial cell proliferation, migration, and adhesion to substrate [11 –13]. More recently, both us [14 –17] and other research groups [18] have demonstrated that store-operated Ca2+ entry (SOCE) drives proliferation and in vitro tubulogenesis in healthy ECFCs. SOCE is activated upon depletion of the endoplasmic reticulum (ER) Ca2+ content in response to inositol-1,4,5-trisphosphate (InsP3) producing stimuli or to the pharmacological inhibition of sarcoendoplasmic reticulum Ca2+-ATPase (SERCA). The ensuing drop in intraluminal Ca2+ concentration is detected by the ER Ca2+ sensor stromal interaction molecule 1 (Stim1), which then oligomerizes and rapidly relocates toward ER-plasma membrane junctions, termed puncta; herein, Stim1 binds to and gates the Ca2+-permeable channels Orai1 and transient receptor potential canonical 1 (TRPC1) to mediate SOCE [18,19]. SOCE undergoes a dramatic remodeling in ECFCs derived from patients suffering from proliferative disorders. For instance, SOCE is upregulated due to the overexpression of Stim1, Orai1, and TRPC1 in renal cellular carcinoma (RCC) and primary myelofibrosis (PMF)-derived ECFCs (RCC-ECFCs and PMC-ECFCs, respectively) [19 –21]. SOCE controls the proangiogenic activity in the former [19], but not in the latter, which keeps on replicating despite the presence of specific SOCE inhibitors, such as N-(4-[3,5-bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl)-4-methyl-1,2,3-thiadiazole-5-carboxamide (BTP-2), La3+, and Gd3+ [20,22]. An additional hint at the plasticity of the Ca2+ toolkit endowed to ECFCs is provided by UCB-ECFCs. Unlike their peripheral counterparts, UCB-ECFCs express TRPC3, which acts in concert with SOCE to promote cell proliferation by triggering and sustaining vascular endothelial growth factor (VEGF)-induced Ca2+ oscillations [23]. Moreover, these cells lack type 1 InsP3 receptor (InsP3R1), which is present in circulating ECFCs, and contributes to mobilize intraluminally stored Ca2+ [15]. It is currently unknown whether the Ca2+ toolkit is altered and controls proliferation also in IH-derived ECFCs (IH-ECFCs). Likewise, there is no hint at the involvement of nitric oxide (NO) in IH-ECFC growth. NO is one of the most powerful stimulators of EPC egression from bone marrow and promotes EPC proliferation at neoangiogenic sites [24,25]. Moreover, SOCE has long been known as the most effective signal at inducing the endothelial NO synthase (eNOS) activity [26 –29]. NO could be a suitable link between the rearrangement of the Ca2+ toolkit, if any, and aberrant IH-ECFC proliferation.
Therefore, we endeavored the present investigation to evaluate whether and how the Ca2+ toolkit is remodeled in IH-ECFCs by paying particular attention to SOCE and its proangiogenic outcome. This was done by carrying out Ca2+ and NO measurements, quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR), western blot analysis, and proliferation assays. We found that, while Stim1, Orai1, and TRPC1 are normally expressed, SOCE is constitutively activated in IH-ECFCs most likely due to the partial depletion of their ER Ca2+ pool. This resting influx of Ca2+ enhances the basal production of NO, thereby accelerating IH-ECFC proliferation compared to normal cells. These data support the notion that tumorigenesis dramatically affects the Ca2+ toolkit in ECFCs by demonstrating for the first time that IH-derived ECFCs are different from healthy cells [22,30]. Furthermore, these results hint at SOCE as an alternative target to devise more efficient antiangiogenic treatments of IH.
Materials and Methods
Isolation and cultivation of ECFCs
Blood samples (40 mL) collected in ethylenediaminetetraacetic acid (EDTA)–containing tubes were obtained from healthy human volunteers, while 3–4 mL were obtained from pediatric patients affected by IH at the time of diagnosis (see Table 1 for demographic and clinical characteristics). All patients were out of cytoreductive therapy. The Institutional Review Board at “Istituto di Ricovero e Cura a Carattere Scientifico Policlinico San Matteo Foundation” in Pavia approved all protocols and specifically approved this study. Informed written consent was obtained according to the Declaration of Helsinki of 1975 as revised in 2008. We focused on the so-called ECFCs, a subgroup of EPCs, which are found in the CD34+ CD45− fraction of circulating mononuclear cells (MNCs), exhibit robust proliferative potential, and form capillary-like structures in vitro [31]. To isolate ECFCs, MNCs were separated from peripheral blood by density gradient centrifugation on a lymphocyte separation medium for 30 min at 400 g and washed twice in an endothelial basal medium (EBM)-2 with 2% fetal calf serum. A median of 36 × 106 MNCs (range 18–66) was plated on collagen-coated culture dishes (BD Biosciences) in the presence of the endothelial cell growth medium EGM-2 MV Bullet Kit (Lonza) containing EBM-2, 5% fetal bovine serum, recombinant human (rh) EGF, rhVEGF, rhFGF-B, rhIGF-1, ascorbic acid, and heparin and maintained at 37°C in 5% CO2 and humidified atmosphere. Nonadherent cells were discarded after 2 days, and thereafter, the medium was changed thrice a week. The outgrowth of ECs from adherent MNCs was characterized by the formation of a cluster of cobblestone-shaped cells. That ECFC-derived colonies belonged to endothelial lineage was confirmed as described in Sánchez-Hernández et al. [14] and Lodola et al. [19]. In more detail, EPC-derived colonies were characterized by staining them with anti-CD31, anti-CD105, anti-CD144, anti-CD146, anti-vWf, anti-CD45, and anti-CD14 monoclonal antibodies and by assessment of capillary-like network formation in the in vitro Matrigel assay.
F, female; M, male.
For our experiments, we have mainly used endothelial cells obtained from early passage ECFC (P1-3, which roughly encompasses a 15–18-day period) with the purpose to avoid (or maximally reduce) any potential bias due to cell differentiation. However, to make sure that the phenotype of the cells did not change throughout the experiments, in preliminary experiments, we tested the immunophenotype of ECFCs at different passages and found no differences [19]. We also tested whether functional differences occurred when early (P2) and late (P6) passage ECFCs were used by testing the in vitro capacity of capillary network formation in a Matrigel assay and found no differences between early and late passage ECFC-derived cells.
Solutions
Physiological salt solution (PSS) had the following composition (in mM): 150 NaCl, 6 KCl, 1.5 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES. In Ca2+-free solution (0Ca2+), Ca2+ was substituted with 2 mM NaCl, and 0.5 mM EGTA was added. Solutions were titrated to pH 7.4 with NaOH. In Mn2+-quenching experiments, 200 μM MnCl2 was added to the 0Ca2+ external solution. The osmolality of PSS as measured with an osmometer (Wescor 5500) was 338 mmol/kg.
[Ca2+]i and NO measurements
ECFCs were loaded with 4 μM fura-2 acetoxymethyl ester (fura-2/AM; 1 mM stock in dimethyl sulfoxide) in PSS for 1 h at room temperature. After washing in PSS, the coverslip was fixed to the bottom of a Petri dish and the cells observed by an upright epifluorescence Axiolab microscope (Carl Zeiss), usually equipped with a Zeiss ×40 Achroplan objective (water-immersion, 2.0 mm working distance, 0.9 numerical aperture). ECFCs were excited alternately at 340 and 380 nm, and the emitted light was detected at 510 nm. A first neutral density filter (1 or 0.3 optical density) reduced the overall intensity of the excitation light and a second neutral density filter (optical density = 0.3) was coupled to the 380-nm filter to approach the intensity of the 340 nm light. A round diaphragm was used to increase the contrast. The excitation filters were mounted on a filter wheel (Lambda 10; Sutter Instrument). Custom software, working in the LINUX environment, was used to drive the camera (Extended-ISIS Camera; Photonic Science) and the filter wheel and to measure and plot online the fluorescence from 10 up to100 rectangular “regions of interest” (ROI). Each ROI was identified by a number. Since cell borders were not clearly identifiable, an ROI may not include the whole cell or may include part of an adjacent cell. Adjacent ROIs never superimposed. [Ca2+]i was monitored by measuring, for each ROI, the ratio of the mean fluorescence emitted at 510 nm when exciting alternatively at 340 and 380 nm (shortly termed “ratio”). An increase in [Ca2+]i causes an increase in the ratio [14,19]. Ratio measurements were performed and plotted online every 3 s. The experiments were performed at room temperature (22°C).
Mn2+ has been shown to quench Fura-2 fluorescence. Since Mn2+ and Ca2+ share common entry pathways in the plasmalemma, Fura-2 quenching by Mn2+ is regarded as an index of divalent cation influx [32]. Experiments were carried out at the 360 nm wavelength, the isosbestic wavelength for Fura-2, and in the Ca2+-free medium supplemented with 0.5 mM EGTA, as previously described [33]. This avoids Ca2+ competition for Mn2+ entry and therefore enhances Mn2+ quenching.
NO was measured as described in Berra-Romani et al. [29]. Briefly, ECFCs were loaded with the membrane-permeable NO-sensitive dye 4-Amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) diacetate (10 μM) for 60 min at room temperature and washed in PSS for further 1 hour. DAF-FM fluorescence was measured by using the same equipment described for Ca2+ recordings, but with a different filter set, that is, excitation at 480 nm and emission at 535 nm wavelength (emission intensity was shortly termed “NOi”). NO measurements were performed and plotted online every 5 s. Again, offline analysis was performed by using custom-made macros developed by Microsoft Office Excel software. The experiments were performed at room temperature. DAF-FM is essentially nonfluorescent until it irreversibly reacts with the nitrosonium cation produced by spontaneous oxidation of newly synthesized NO. The resulting fluorescent compound is trapped in the cytoplasm, so that DAF-FM fluorescence summates with continual NO production. Basal NO levels were measured in N- and IH-ECFCs bathed in PSS by averaging the value of DAF-FM fluorescence at 300 ± 50 s from the beginning of the recording. The impact of SOCE on resting NO production was evaluated in IH-ECFCs preincubated in the absence of external Ca2+ (0Ca2+) and presence of NG-nitro-l-arginine methyl ester (L-NAME) (75 min, 100 μM), BTP-2 (20 min, 20 μM), or 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) (2 h, 30 μM).
RNA isolation and real-time qRT-PCR
Total RNA was extracted from the ECFCs using the QIAzol Lysis Reagent (Qiagen). Single cDNA was synthesized from RNA (1 μg) using random hexamers and M-MLV reverse transcriptase (Promega). Reverse transcription was always performed in the presence or absence (negative control) of the reverse transcriptase enzyme. qRT-PCR was performed in triplicate using 1 μg cDNA and specific primers (intron-spanning primers), as previously described [14,19] (Table 2). Briefly, GoTaq qPCR Mastermix (Promega) was used according to the manufacturer's instruction and qRT-PCR performed using Rotor Gene 6000 (Corbett). The conditions were as follows: initial denaturation at 95°C for 5 min; 40 cycles of denaturation at 95°C for 30 s; annealing at 58°C for 30 s; and elongation at 72°C for 40 s. The qRT-PCRs were normalized using β-actin as the housekeeping gene. Melting curves were generated to detect the melting temperatures of specific products immediately after the PCR run. The triplicate threshold cycle (Ct) values for each sample were averaged resulting in mean Ct values for both the gene of interest and the housekeeping gene β-actin. The gene Ct values were then normalized to the housekeeping gene by taking the difference: ΔCt = Ct[gene] − Ct[β-actin], with high ΔCt values reflecting low mRNA expression levels. The sequences of the bands were checked by using the Big dye terminator cycle sequencing kit (Applied Biosystem). PCR products were also separated with agarose gel electrophoresis, stained with ethidium bromide, and acquired with the Image Master VDS (Amersham Biosciences Europe). The molecular weight of the PCR products was compared to the DNA molecular weight marker VIII (Roche Molecular Biochemicals).
Sample preparation and immunoblotting
ECFCs from normal subjects and IH patients were homogenized by using a Dounce homogenizer in a solution containing the following: 250 mM sucrose, 1 mM EDTA, 10 mM Tris-HCl, pH 7.6, 0.1 mg/mL phenylmethylsulfonyl fluoride (PMSF), 100 mM β-mercaptoethanol, and Protease Inhibitor Cocktail (P8340; Sigma). The homogenates were solubilized in the Laemmli buffer [14] and 30 μg proteins were separated on 10% sodium dodecyl sulfate– polyacrylamide gel electrophoresis and transferred to the Hybond-P PVDF Membrane (GE Healthcare) by electroelution. After 1 h blocking with Tris-buffered saline (TBS) containing 3% bovine serum albumin (BSA) and 0.1% Tween (blocking solution), the membranes were incubated for 3 h at room temperature with affinity-purified antibodies diluted 1:200 in the TBS and 0.1% Tween: anti-Stim1 (sc-166840), anti-Orai1 (sc-68895), anti-TRPC1 (E-6; sc-133076), anti-TRPC4 (sc-15063), and anti-InsP3R-I/II/III (sc-377518) from Santa Cruz Biotechnology and anti-Stim2 (PRS4123) and anti-Orai3 (HPA015022) from Sigma-Aldrich. The membranes were washed and incubated for 1 h with peroxidase-conjugated mouse, rabbit, or goat IgG (1:120,000 in blocking solution), from Dakocytomation (P0260), Chemicon (AP132P), and Santa Cruz (sc-2354), respectively. The bands were detected with the ECL™ Select western blotting detection system (GE Healthcare Europe GmbH). Control experiments were performed as described in Sánchez-Hernández et al. [14]. Prestained molecular weight markers Prism Ultra Protein Ladder (10–245 kDa) (ab116028) from Abcam were used to estimate the molecular weight of the bands. Blots were stripped [34] and reprobed with anti β-actin rabbit antibody as the loading control (Rockland Immunochemicals for Research; code, 600–401-886). The antibody was diluted 1:2,000 in the TBS and 0.1% Tween. Bands were acquired, densitometric analysis of the bands was performed by the Total Lab V 1.11 computer program (Amersham), and the results were expressed as a percentage of the gene/β-actin densitometric ratio.
Protein content
Protein contents of all the samples were determined by the Bradford's method using BSA as standard [35].
Proliferation assays
As described elsewhere [14,19], growth kinetics were evaluated by plating a total of 1 × 104 IH-ECFC-derived cells (first passage) in 30-mm collagen-treated dishes in the EGM-2 MV medium or, where appropriate, in the presence of EBM-2 medium. Cultures were incubated at 37°C (in 5% CO2 and humidified atmosphere) and cell growth assessed every day until confluence was reached in both cultures. At this point, cells were recovered by trypsinization from all dishes and the cell number assessed by counting in a hemocytometer. To assess the impact of constitutive SOCE and NO on IH-ECFC proliferation, cells were seeded in the presence or absence of 100 μM L-NAME, 20 μM BTP-2, 10 μM Pyr6, or 30 μM BAPTA. Preliminary experiments showed no unspecific or toxic effect for each agent when used at these concentrations. The percentage of growth inhibition by the drugs was calculated by dividing the total number of cells obtained in the presence of either L-NAME or BTP-2 or Pyr6 or BAPTA by the number of cells in control experiments and multiplying the ratio by 100.
Statistics
All the Ca2+ data have been collected from ECFCs isolated from peripheral blood of at least three healthy volunteers or IH patients. Pooled data are given as mean ± SE and statistical significance (P < 0.05) was evaluated by the Student's t-test for unpaired observations. The amplitude of Ca2+ release in response to either cyclopiazonic acid (CPA) or ATP was measured as the difference between the ratio at the peak of intracellular Ca2+ mobilization and the mean ratio of 1-min baseline before the peak. The magnitude of SOCE evoked by either CPA or ATP upon Ca2+ restoration to the bath was measured as the difference between the ration at the peak of extracellular Ca2+ entry and the mean ration of 1 in baseline before Ca2+ readdition. The rate of Mn2+ influx was evaluated by measuring the slope of the fluorescence intensity curve at 400 s after Mn2+ addition [36].
As to mRNA and protein analysis, all data are expressed as mean ± SE. The significance of the differences of the means was evaluated with Student's t-test. In the proliferation assays, results are expressed as percentage (±SD) of growth compared to controls (given as 100% growth), obtained from three different sets of experiments, each performed in duplicate. Differences were assessed by the Student's t-test for unpaired values. All statistical tests were carried out with GraphPad Prism 4.
Chemicals
EBM and EGM-2 were purchased from Clonetics (Cell System). Fura-2/AM was obtained from Molecular Probes (Molecular Probes Europe BV). BTP-2 and Pyr6 were purchased from Calbiochem. All other chemicals were obtained from Sigma Chemical Co.
Results
Intracellular Ca2+ release is lower, while SOCE is higher in IH-derived ECFCs
The resting intracellular Ca2+ levels in N- and IH-ECFCs were evaluated after digital subtraction of the fluorescence background and were not statistically different (P < 0.05), the average values of the Fura-2 ratio being 0.897 ± 0.006, n = 585 and 0.899 ± 0.0042, n = 1,380, respectively. Similar to N-ECFCs [15], we could not detect any spontaneous oscillation in intracellular Ca2+ concentration ([Ca2+]i) over long-time (up to 1 h) recordings (not shown). Intracellular Ca2+ mobilization and SOCE activation were analyzed by exploiting the so-called “Ca2+ add-back” protocol [14,19,37]. Briefly, the cells were first bathed in the absence of extracellular Ca2+ (0Ca2+) and then exposed to CPA (10 μM) to selectively block the SERCA activity. CPA prevents Ca2+ sequestration into ER lumen and leads to a progressive Ca2+ efflux through yet unidentified leakage channels, thereby depleting the ER Ca2+ pool. The ensuing increase in [Ca2+]i is indicative of the amount of intraluminally stored Ca2+ [19,20]. After recovery of the initial elevation in [Ca2+]i to the baseline, extracellular Ca2+ was restituted to the perfusate to monitor SOCE amplitude. As shown in Fig. 1A and B, both components, that is, intracellular Ca2+ release and SOCE, of CPA-induced Ca2+ signal were significantly (P < 0.05) lower in IH-ECFCs compared to their healthy counterparts. In agreement with these results, the rate of CPA-induced Ca2+ inflow was significantly (P < 0.05) slower in IH-ECFCs as respect to normal cells (Fig. 1C). Moreover, the percentage of responding IH-ECFCs was significantly (P < 0.05) lower related to N-ECFCs (Fig. 1D), which is consistent with a reduction in ER Ca2+ concentration ([Ca2+]ER). Next, the Ca2+ add-back protocol was conducted by challenging the cells with the physiological autacoid ATP (100 μM), which binds to P2Y receptors to initiate InsP3 synthesis, deplete the ER Ca2+ store, and activate SOCE [14,19,20]. Similar to CPA, ATP-induced Ca2+ release and store-dependent Ca2+ inflow were significantly (P < 0.05) lower in IH-ECFCs relative to control cells (Fig. 1E, F). Again, ATP-evoked Ca2+ influx was significantly (P < 0.05) slower in IH-derived ECFCs (Fig. 1G). Extracellular Ca2+ was replenished after ATP withdrawal from the bath to prevent the activation of P2X receptors or second messenger-operated channels [14,37], such as TRPV4 [16]. Similar to CPA, the percentage of IH-ECFCs, which produced a Ca2+ signal in response to ATP, was significantly (P < 0.05) lower compared to control cells (Fig. 1H). Control experiments were also conducted by applying the Ca2+ add-back protocol in absence of either CPA or ATP and confirmed that no Ca2+ entry occurs when the agonist is omitted from the bathing medium (not shown). We have recently demonstrated that VEGF triggers proangiogenic Ca2+ oscillations in N-ECFCs by triggering a concerted interplay between InsP3-dependent Ca2+ mobilization and SOCE [15,38]. Nevertheless, VEGF (10 ng/mL) failed to induce any detectable elevation in [Ca2+]i in most of IH-ECFCs, while it triggered a prolonged train of Ca2+ spikes in ∼0% of N-ECFCs (Fig. 1I, J). However, the reduction in ATP and VEGF sensitivity was not due to the downregulation of InsP3Rs as evident by the qRT-PCR analysis of ECFC transcripts carried out using specific primers for all the three known InsP3R isoforms (Fig. 1K) [19,20]. InsP3R expression was further examined by immunoblotting, which unveiled a large band, deriving from the sum of single 313/260/250 kDa bands that correspond, respectively, to InsP3R1/2/3 (Supplementary Fig. S1; Supplementary Data are available online at

SOCE is expressed in IH-derived ECFCs.
The molecular components of SOCE are normally expressed in IH-ECFCs
To assess whether SOCE machinery was downregulated in IH-ECFCs, thereby leading to a decrease in CPA- and ATP-evoked Ca2+ inflow, we examined the expression of Stim1, Orai1, and TRPC1 by carrying out a qRT-PCR examination of mRNA extracts from IH-ECFCs. We also evaluated their paralogues [13,39], namely Stim2, Orai2-3, and TRPC3-7, by using the specific primers illustrated in Table 2, as shown elsewhere [14,19,20]. Negative controls were obtained by omitting the reverse transcriptase in the cDNA synthesis step (not shown). We could not find any difference in transcript levels between N- and IH-ECFCs: mRNAs encoding for Stim1, Orai1, and TRPC1 were expressed at the same level in both cell types (Fig. 2). Similar to N-ECFCs, IH-ECFCs were also endowed with Stim2, Orai2-3, and TRPC4 transcripts (Fig. 2) and lacked TRPC3, TRPC5, TRPC6, and TRPC7 (not shown). Therefore, the analysis of the Ca2+ toolkit suggests that IH-ECFCs are strikingly different from UCB-ECFCs, which present TRPC3 and are devoid of InsP3R1 (see above) [23]. A western blot investigation of Stim1, Orai1, and TRPC1 expression was then conducted by exploiting affinity-purified antibodies [14,19,20]. Immunodetection disclosed a doublet of ∼77 and 100 kDa for Stim1, while Orai1 and TRPC1 showed major bands of ∼33 and ∼110 kDa, respectively (Figs. 3 and 4). We also assessed the protein expression of Stim2, which displayed another doublet of about 77 and 100 kDa, Orai3 and TRPC4, which showed major bands of the expected size, that is, 33 and 110 kDa, respectively (Figs. 3 and 4). We did not focus further on Orai2, which plays a key role in Ca2+ inflow only in brain neurons [40]. Densitometry of the bands could not detect any difference in the expression of Stim1-2, Orai1, Orai3, TRPC1, and TRPC4 proteins between N- and IH-ECFCs (Figs. 3 and 4). In conclusion, these data demonstrate that the molecular players of SOCE are normally expressed in IH-ECFCs despite the fact that store-dependent Ca2+ influx upon extracellular stimulation is significantly dampened in these cells.
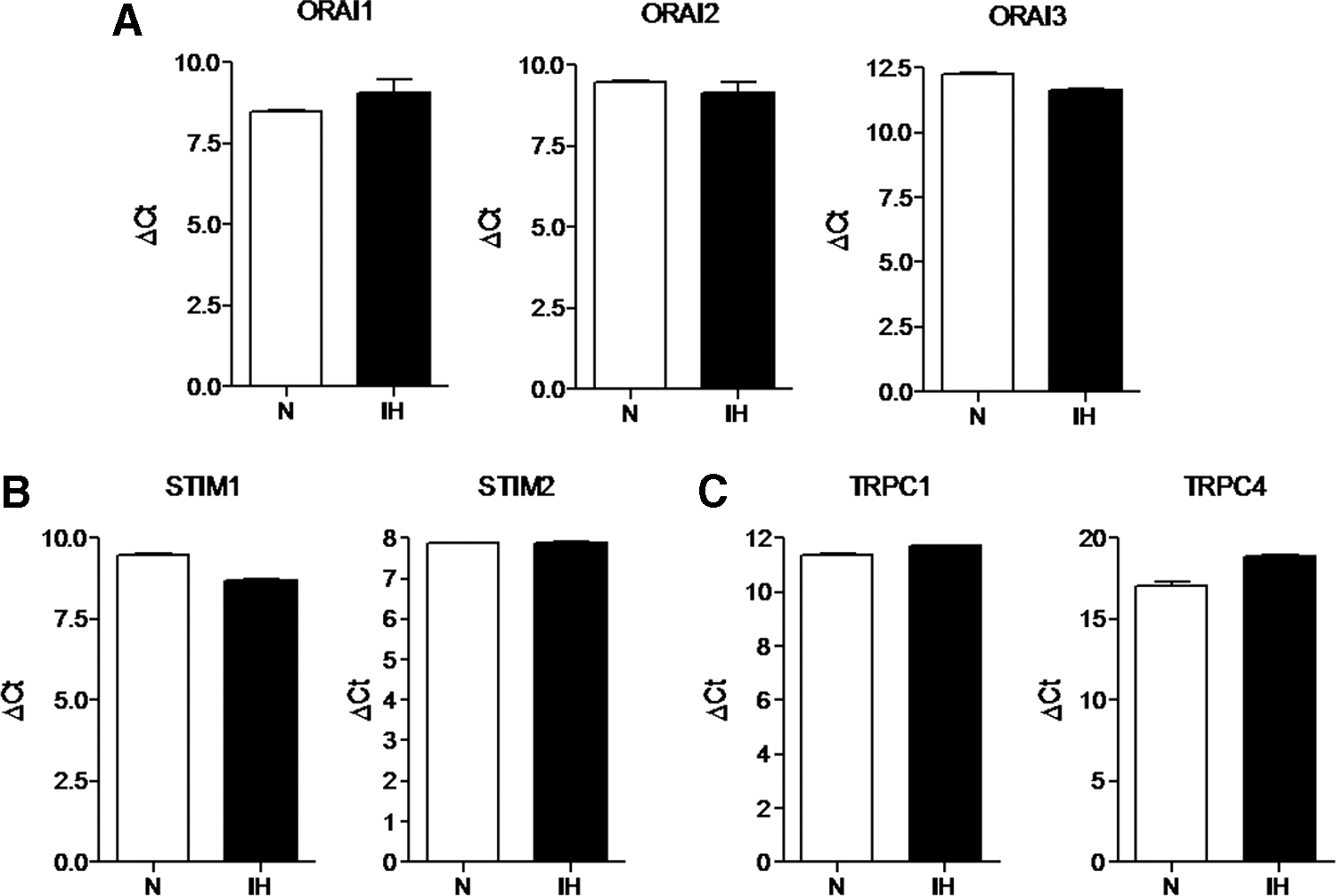
Expression of Orai1-3, Stim1-2, TRPC1, and TRPC4 transcripts in IH-derived ECFCs. The transcript levels of Orai1-3
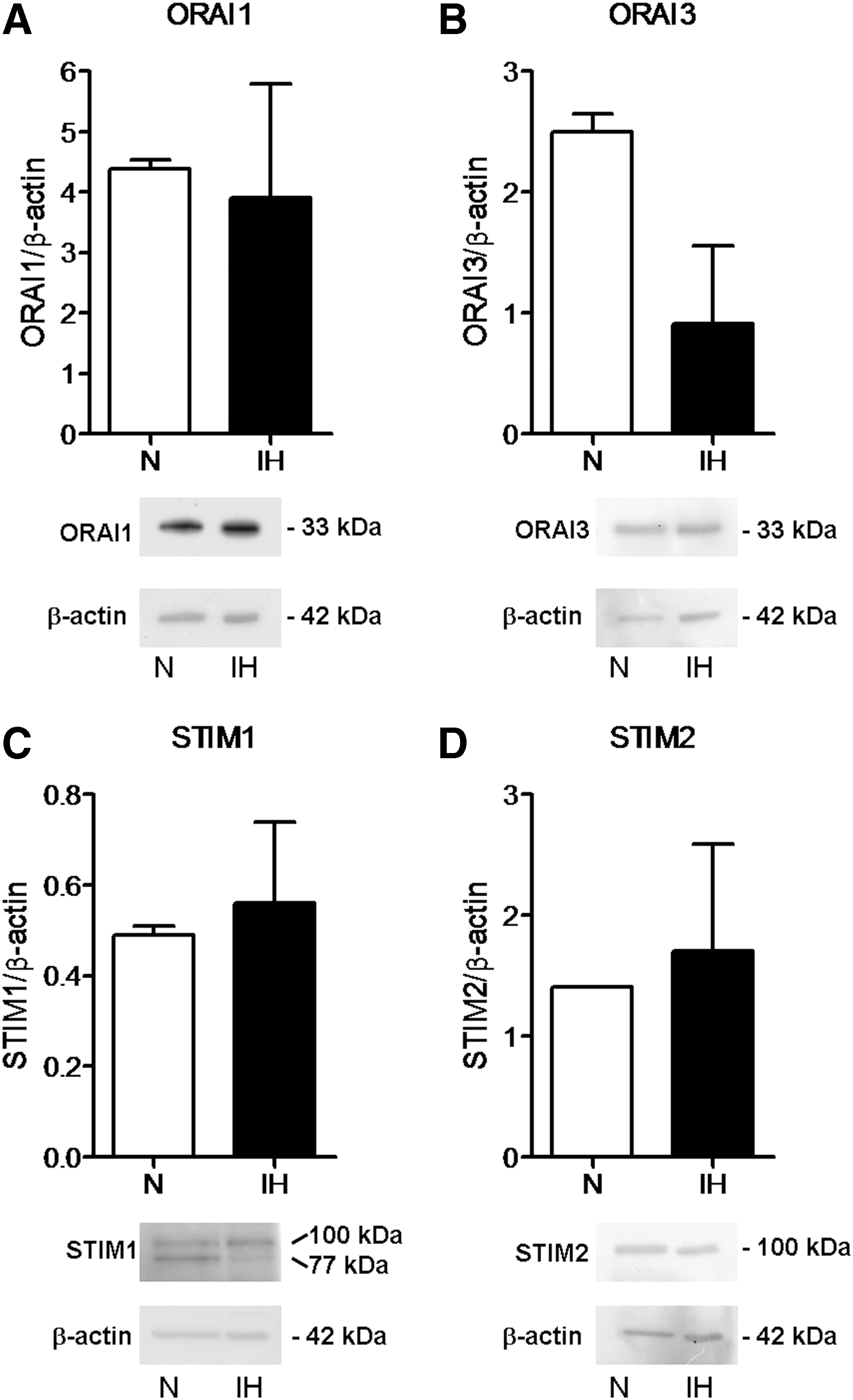
Expression of Orai1, Orai3, Stim1, and Stim2 proteins in IH-derived ECFCs. The protein levels of Orai1
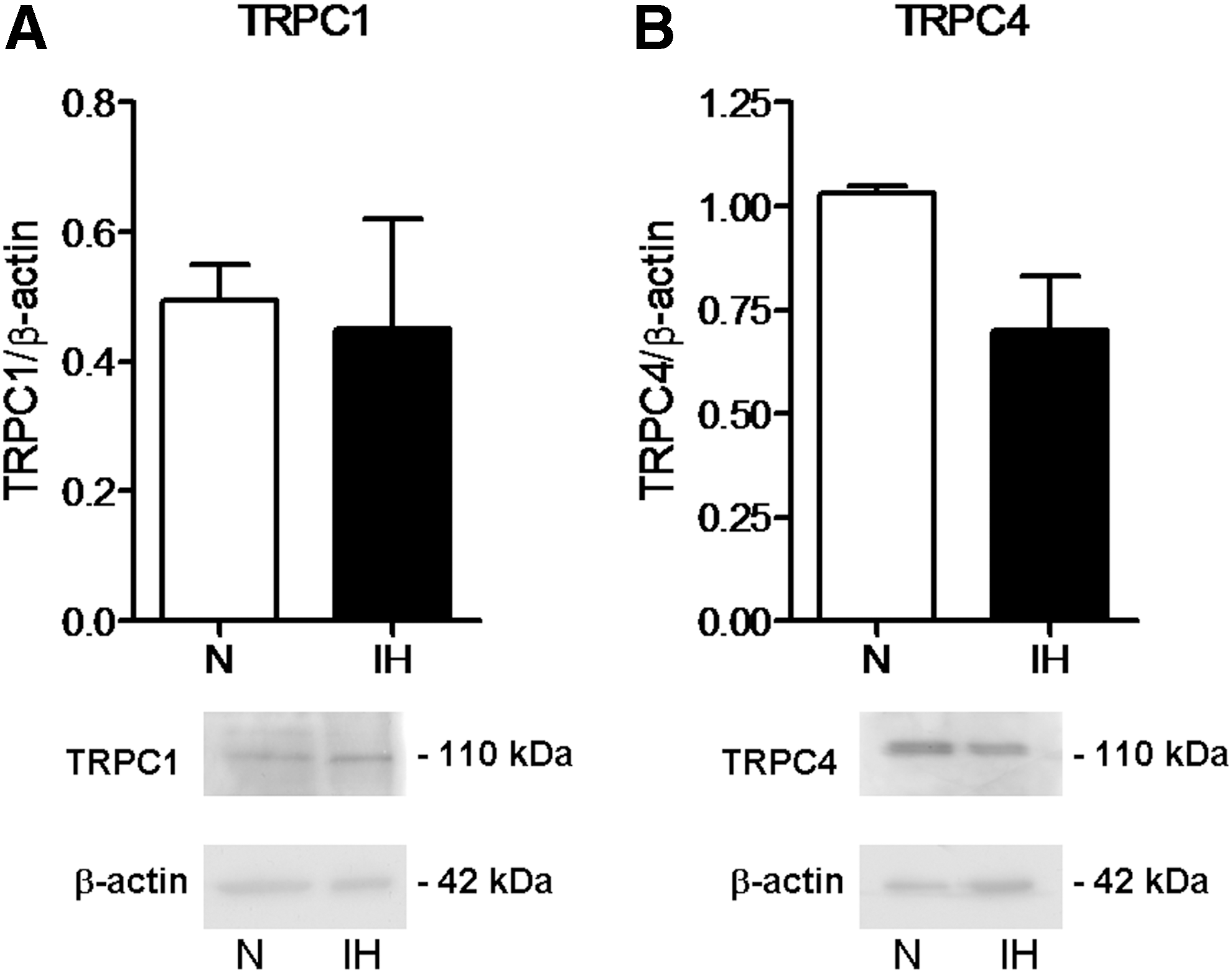
Expression of TRPC1 and TRPC4 proteins in IH-derived ECFCs. The transcript levels of TRPC1
The pharmacological blockade of SOCE affects intracellular Ca2+ release in IH-ECFCs
We have previously shown that the pharmacological inhibition of SOCE with YM-58483/BTP-2 and lanthanides selectively interferes with CPA- and ATP-induced Ca2+ entry without affecting intracellular Ca2+ mobilization in N-ECFCs [14,19,20,22,41]. Conversely, preincubating IH-ECFCs with BTP-2 (20 min, 20 μM) inhibited both components, that is, Ca2+ release and SOCE, of the Ca2+ signal elicited by either CPA (10 μM) (Fig. 5A, B) or ATP (100 μM) (Fig. 5C, D). The same effect was achieved when IH-ECFCs were pretreated with La3+ (20 min, 10 μM) (Fig. 6A–D). Please note that the inhibitory effect exerted by La3+ on both phases of the Ca2+ response to CPA was greater compared to BTP-2. These results were further confirmed by using the novel pyrazole derivative Pyr6 (10 μM), which specifically blocks Orai1-dependent Ca2+ inflow [42]. Similar to BTP-2 and La3+ [14,19], Pyr 6 (10 μM, 20 min) blocked CPA-evoked SOCE, but not intracellular Ca2+ release, in N-ECFCs (Supplementary Fig. S2A, B). Conversely, it repressed both components of the Ca2+ response to CPA in IH-ECFCs (Supplementary Fig. S2C, D). Thus, Pyr6 may be regarded as a specific blocker of SOCE also in ECFCs. Moreover, these data suggest that SOCE is constitutively active to refill the endogenous Ca2+ stores in IH-ECFCs. If this hypothesis was true, then, depleting extracellular Ca2+ for at least 20 min would selectively reduce CPA-induced Ca2+ mobilization in IH-ECFCs, but not in N-ECFCs, which still display intracellular Ca2+ release after 20 min in the presence of three structurally different SOCE inhibitors. Our subsequent experiments revealed that this is indeed the case. In more detail, CPA was added after 100 s (Fig. 7A) and after 20 min (Fig. 7B) in the continuous absence of external Ca2+ to assess whether Ca2+ inflow was required to refill endogenous stores and ensure Ca2+ mobilization upon stimulation [43]. This protocol revealed that CPA-induced intracellular Ca2+ release did not decrease with the time when N-ECFCs were maintained in 0Ca2+ before stimulation (Fig. 7A, C). Conversely, the Ca2+ response to CPA was dramatically reduced in IH-ECFCs continuously bathed in 0Ca2+ for 20 min (Fig. 7A, C). Taken together, these data suggest that a basal influx of Ca2+, presumably mediated by an SOCE pathway, ensures replenishment of the ER Ca2+ stores in IH-ECFCs, while it is not indispensable for refilling the ER Ca2+ pool in N-ECFCs (at least over a period of 20 min). This hypothesis would explain why BTP-2, La3+, Pyr6, and the depletion of external Ca2+ block the intracellular Ca2+ release, which cannot occur in the absence of ER store recharging, in IH-ECFCs, but not in N-ECFCs. Moreover, we speculate that the basal SOCE is remarkably larger in IH-ECFCs as related to N-ECFCs. This surmise would explain why challenging the cells with store-depleting agents, such as CPA and ATP, does not fully activate Ca2+ influx in IH-ECFCs. Indeed, a large fraction of store-dependent channels, that is, Orai1 and TRPC1, would already be open under resting conditions and could not be further recruited by another incoming stimulus.
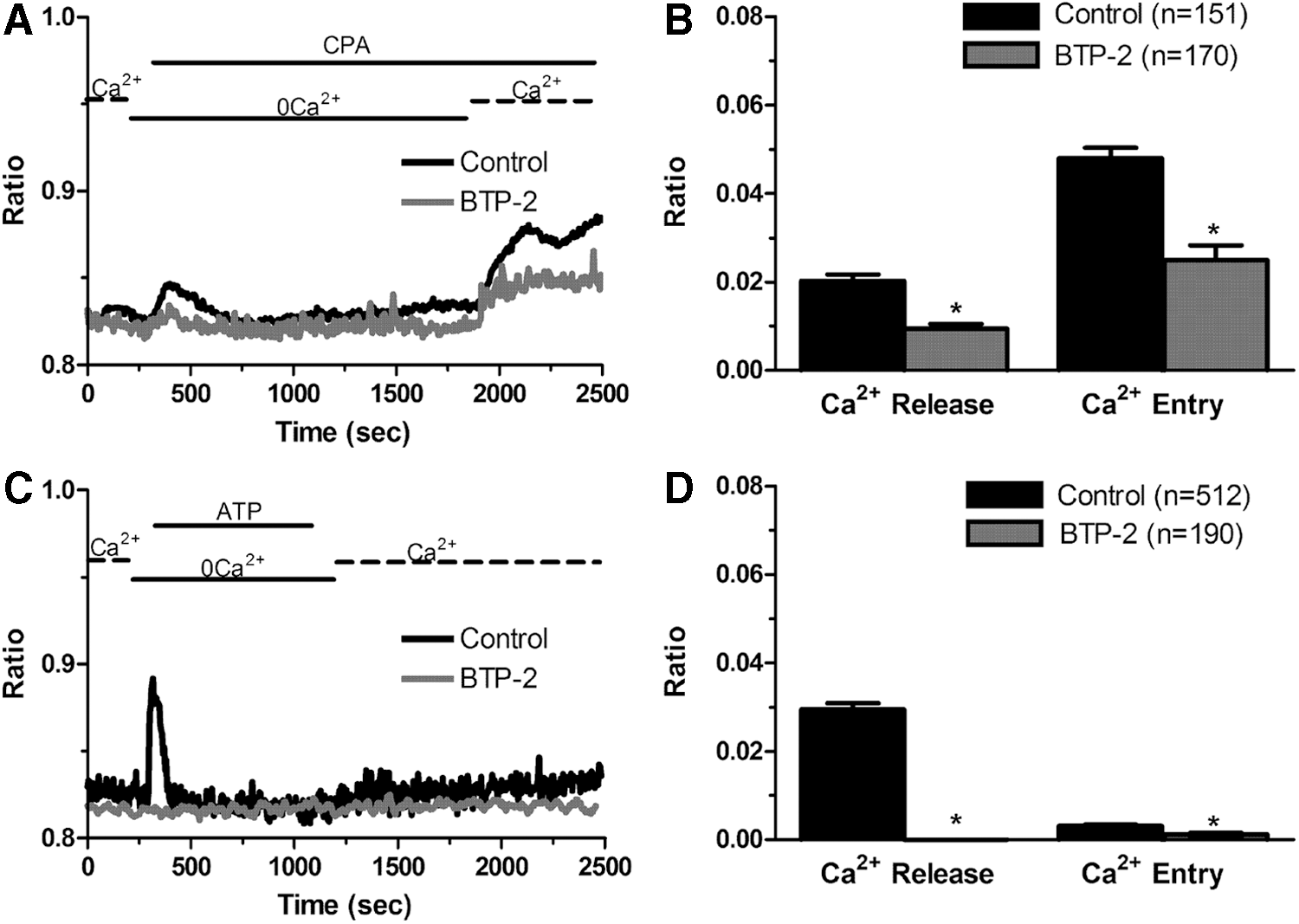
BTP-2 inhibits Ca2+ release and SOCE in IH-derived ECFCs.

La3+ inhibits Ca2+ release and SOCE in IH-derived ECFCs.
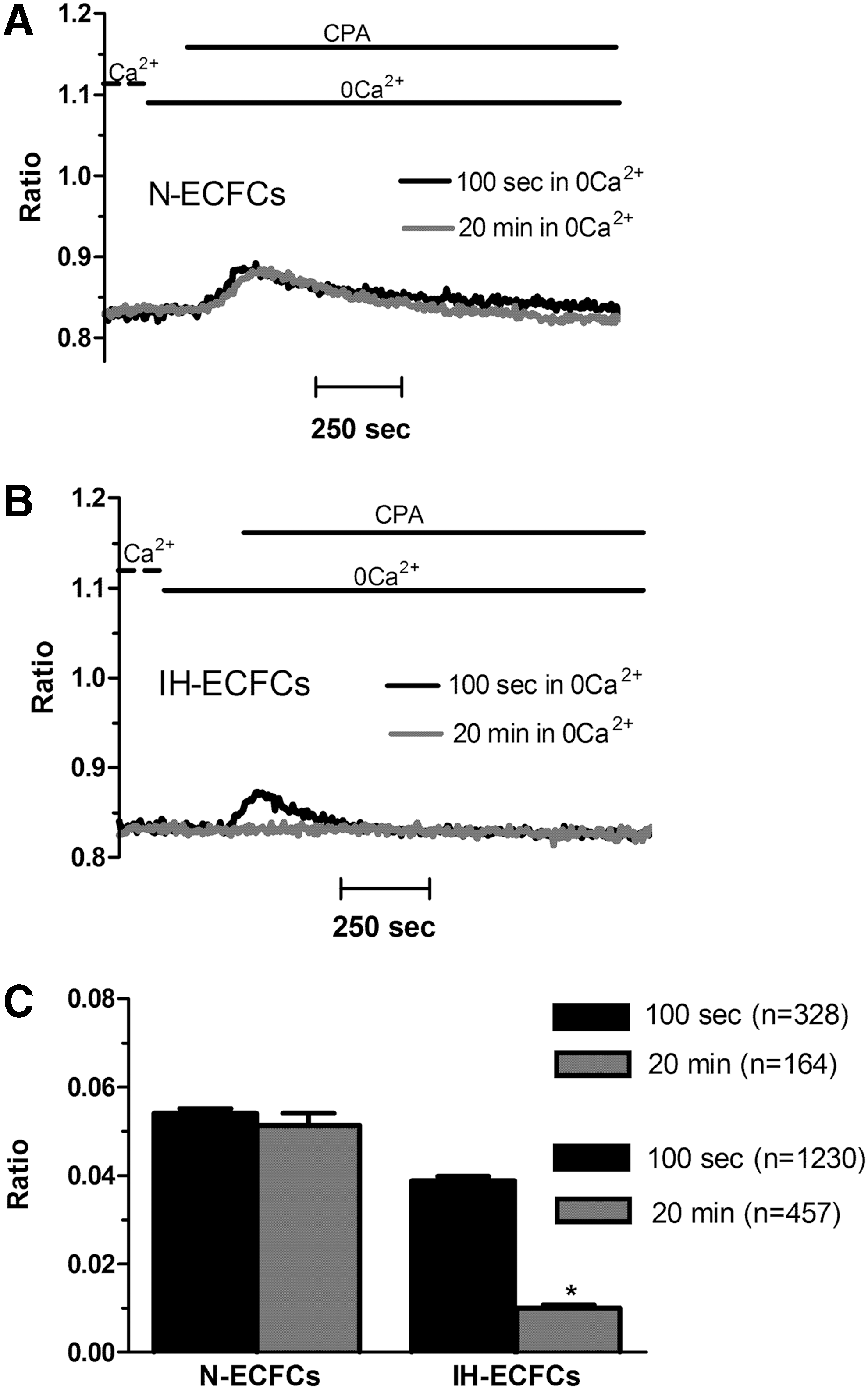
Ca2+ entry is necessary to refill the endoplasmic reticulum Ca2+ stores in IH-derived ECFCs, but not in their healthy counterparts.
A question then arises as to why resting Ca2+ entry is not evident by simply changing the extracellular Ca2+ concentration as observed in other cell types [44]. The basal entry of Ca2+ could be underestimated by Ca2+ clearance through Ca2+-transporting systems, such as SERCA and Na+/Ca2+ exchanger (NCX). Therefore, we repeated the Ca2+ add-back protocol in the absence of any extracellular agonist and by replacing external Ca2+ with an equimolar amount of Ba2+, which is not captured by SERCA and NCX. This maneuver revealed that Ba2+ entry occurs in both type of ECFCs (Supplementary Fig. S3A), although it is significantly (P < 0.05) more frequent in IH-ECFCs (Supplementary Fig. S3B); in addition, both the amplitude and rate of Ba2+ inflow were significantly (P < 0.05) greater in tumor ECFCs compared to healthy cells (Supplementary Fig. S2A, C, D). Collectively, these data support the notion that a basal influx of Ca2+, presumably mediated by an SOCE pathway, is present in both types of ECFCs, but is dramatically enhanced in IH-ECFCs.
Constitutive SOCE is present and enhanced in IH-ECFCs
To confirm whether SOCE is constitutively activated both in N- and in IH-ECFCs, we took advantage of the Mn2+-quenching technique. Extracellular Mn2+ quenches Fura-2/AM fluorescence by flowing through Ca2+-permeable conductances [32,33,45], such as store-dependent channels [36,45]: this is more evident when exciting the cells at 360 nm, which is the isoemissive wavelength for Fura-2 and is independent on [Ca2+]i. The Mn2+-quenching technique has widely been used to monitor resting Ca2+ entry in multiple cell types, including endothelial cells [32,33,36,45]. Figure 8A and B show that most of N-ECFCs lack a clear decay in Fura-2 fluorescence signal upon Mn2+ addition to the perfusate. Conversely, a clear basal influx of Mn2+ was evident in majority of IH-ECFCs, which showed a rather linear quenching of Fura-2 fluorescence (Fig. 8B, C). As described elsewhere [46], the slope of the first 400 s of the quenching trace can be used to measure the rate of Mn2+ entry into the cells. In agreement with previous data, the rate of fluorescence decay was significantly higher in IH-ECFCs compared to the few N-ECFCs displaying a basal Mn2+ entry (Fig. 8C). Subsequently, resting Mn2+ inflow in IH-ECFCs was assessed upon the pharmacological inhibition of SOCE: BTP-2 (20 min, 20 μM) and La3+ (20 min, 10 μM) did not affect the fraction of cells exhibiting constitutive Mn2+ inflow, but significantly (P < 0.05) reduced its slope (Fig. 9). As for the Ca2+ response to CPA (see above), La3+ was more effective at blocking resting Ca2+ inflow compared to BTP-2. Likewise, Pyr 6 (10 μM, 20 min) did not affect the percentage of IH-ECFCs displaying basal Mn2+ entry, but significantly (P < 0.05) decreased its rate (Supplementary Fig. S4). These data do support the hypothesis that constitutive SOCE is present and enhanced in IH-ECFCs compared to N-ECFCs. Basal Ca2+ influx in human umbilical vein endothelial cells might be driven by Stim2 [47], a lower affinity ER Ca2+ sensor as relative to Stim1 [48]. To assess whether Stim2 contributes to resting SOCE in IH-ECFCs, we pretreated the cells with the aminoglycoside antibiotic G418 (500 μg/mL), an established Stim2 inhibitor [49,50]. G418, however, did not interfere with basal Mn2+ influx, thereby ruling out Stim2 involvement in such a process (Fig. 10). Overall, these data supported the notion that Stim1, Orai1, and TRPC1 mediate constitutive SOCE and determine ER Ca2+ loading in IH-ECFCs. If this hypothesis was true, CPA-evoked Mn2+ entry would be predicted to be significantly higher in N-ECFCs, in which SOCE is silent in the absence of external stimulation, compared to IH-ECFCs, in which store-operated channels (SOCs) are already open under basal conditions. Accordingly, CPA induced a large Mn2+ influx in N-ECFCs (Fig. 11A, C), while CPA-elicited Mn2+ entry was significantly (P < 0.05) lower than basal Mn2+ entry in IH-ECFCs (Fig. 11B, C). It is, therefore, possible to conclude that resting SOCE is significantly greater in IH-ECFCs compared to their healthy counterparts.

Basal Ca2+ entry is enhanced in IH-derived ECFCs.

The pharmacological blockade of store-operated Ca2+ channels blocks the constitutive Ca2+ influx in IH-derived ECFCs.

G418 does not inhibit constitutive SOCE in IH-derived ECFCs.
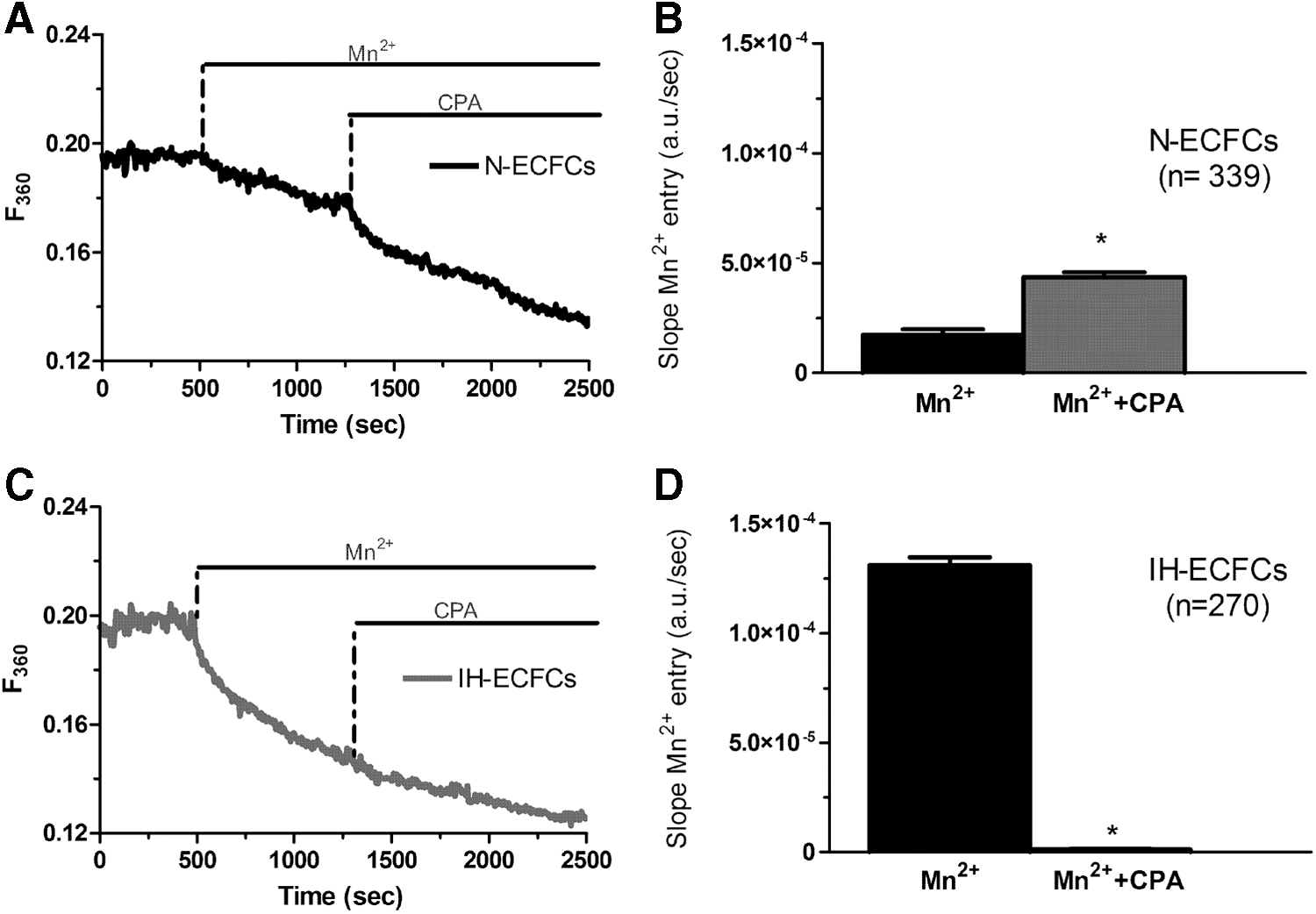
CPA induces Mn2+ entry in normal ECFCs, but not in those isolated from IH patients.
Constitutive SOCE leads to IH-ECFC proliferation by enhancing basal NO production
The aforementioned results clearly indicate that basal SOCE controls ER Ca2+ content in IH-ECFCs, but not in healthy cells. In addition to refilling the ER Ca2+ reservoir, the resting SOCE could control a number of Ca2+-dependent processes relevant to IH neovascularisation [12,13,51], including ECFC proliferation. We first found that IH-ECFCs proliferated at a greater rate compared to N-ECFCs (Fig. 12A). To assess whether IH-ECFC growth is controlled by SOCE, we adopted the experimental approach we already used to address this issue in N-ECFCs [19], primary myelofibrosis (PMF)-ECFCs [20], and RCC-derived tumor cell lines [52]. We previously found that both BAPTA (30 μM), a membrane-permeable buffer of intracellular Ca2+ levels, and BTP-2 (20 μM) block N- and RCC-ECFC proliferation [14,19], while BTP-2 is barely effective in PMF-ECFCs [20] and primary cultures of human metastatic RCC [52]. Notably, both N- and RCC-ECFCs were grown in the presence of EGM-2, a commercially available (see Materials and Methods section) endothelial differentiation medium, which is largely employed to favor ECFC expansion and survival [30,31]. Under these conditions, BAPTA and BTP-2 inhibit ECFC growth by interfering with the proangiogenic SOCE evoked by VEGF [15], the most effective growth factor contained in EGM-2. Herein, BAPTA and BTP-2 also significantly (P < 005) reduced IH-ECFC growth (Fig. 12B). However, SOCE is only barely activated following depletion of the InsP3-sensitive Ca2+ pool (Fig. 1A–H) and VEGF does not recruit SOCE in these cells (Fig. 1I–K). Therefore, BAPTA and BTP-2 cannot act on VEGF-induced SOCE in this preparation. Conversely, IH-ECFCs exhibit a robust SOCE, which is constitutively activated and is sensitive to BTP-2. Collectively, these data indicate that (1) BTP-2 targets the basal store-dependent Ca2+ influx in IH-ECFCs and (2) the hyperfunctioning of this pathway results in their enhanced proliferation rate compared to N-ECFCs. These results were further confirmed by using Pyr6 (10 μM), which dramatically decreased cell growth both in N- (Fig. 12C) and IH-ECFCs (Fig. 12B). Importantly, Pyr6 curtailed the proangiogenic intracellular Ca2+ oscillations initiated by VEGF (10 ng/mL) in N-ECFCs (Supplementary Fig. S5). This last result corroborates the notion that, while Pyr6 suppresses N-ECFC growth by impairing VEGF-evoked SOCE, it reduces IH-ECFC proliferation by impairing the resting SOCE.
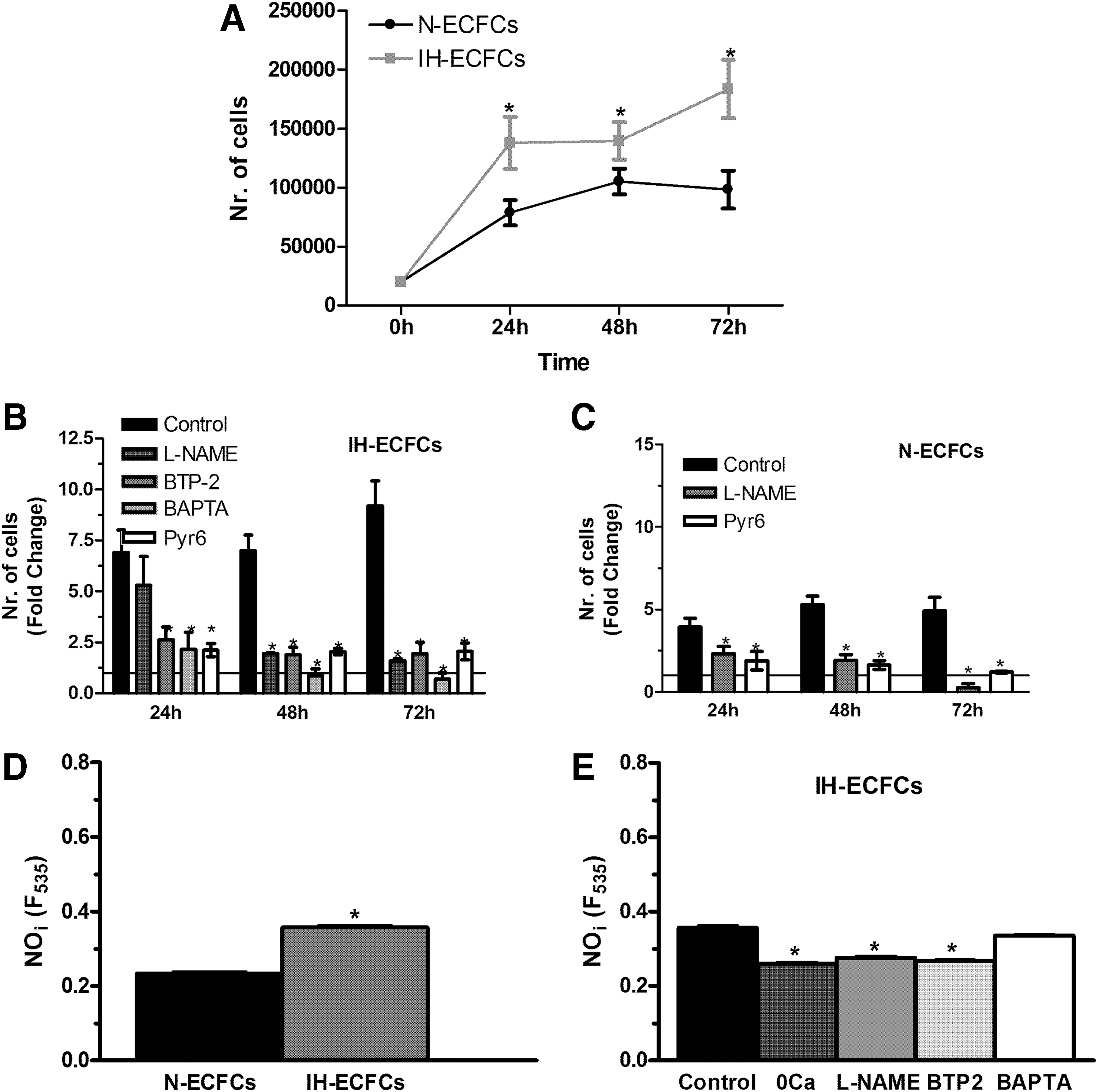
SOCE leads to higher cell proliferation rate and NO production in IH-derived ECFCs.
To further confirm that it is the constitutive SOCE that accelerates IH-ECFC replication, we plated both types of ECFCs in the EBM-2, which has the same composition as EGM-2, but is not supplemented with growth factors. Even in the absence of extracellular mitogens, IH-ECFCs still displayed a higher growth rate compared to N-ECFCs (Supplementary Fig. S6), although they could not reach full confluence after 5 days of observation. Therefore, the greater proliferation rate of IH-ECFCs is attributable to an intrinsic factor that is lacking in N-ECFCs. The pharmacological blockade of SOCE with BTP-2 (20 μM) and Pyr6 (10 μM) blocked IH-ECFC replication (Supplementary Fig. S6), while it only marginally affected N-ECFC growth (Supplementary Fig. S6). Of course, under these culture conditions, BTP-2 and Pyr6 could not interfere with VEGF-evoked SOCE, but only with the basal one. Collectively, these findings support the hypothesis that the hyperactivation of the constitutive SOCE leads to higher replication rate of IH-ECFCs. Furthermore, they show that the main targets of BTP-2 and Pyr6 are the basal and the VEGF-evoked SOCE in IH- and N-ECFCs, respectively.
NO has long been known to stimulate EPC mobilization from bone marrow and proliferation at the neoangiogenic sites [53,54]. Furthermore, SOCE is among the most powerful stimulators of the Ca2+/calmodulin-dependent eNOS [26 –28,55]. Therefore, we speculated that constitutive SOCE could lead to higher basal NO synthesis and stimulate IH-ECFC growth. We measured NO production in ECFCs by exploiting the NO-sensitive fluorophore DAF-FM as previously shown both by others [28,56,57] and us [29] in mature endothelial cells. In agreement with our hypothesis, basal NO levels were significantly (P < 0.05) higher in IH-ECFCs compared to N-ECFCs (Fig. 12D). In addition, resting NO levels in IH-ECFCs were significantly (P < 0.05) reduced by L-NAME (75 min, 100 μM), a selective eNOS inhibitor, and BTP-2 (20 min, 20 μM), as well as by removal of external Ca2+ (0Ca2+) (Fig. 12E). Conversely, NO production was unaffected by BAPTA (2 h, 30 μM) (Fig. 12D). Nevertheless, this result could be explained by the tight coupling between SOCs, that is, Orai1 and TRPC1, and eNOS, which could prevent BAPTA from reaching the inner mouth of the channel pore during such a short preincubation time [58]. Indeed, the blockade of NO synthesis with L-NAME (100 μM) prevents N- and IH-ECFC proliferation (Fig. 12B, C). Therefore, these results collectively support the notion that constitutive SOCE leads to higher NO production and cell proliferation in IH-ECFCs.
Discussion
The cellular and molecular mechanisms that drive IH neovascularisation are still unclear, yet, many evidences hint at EPCs as the most likely precursors of the endothelial cells lining tumor neovessels [1,3,6,8]. In this context, it has been proposed that IH arises from the clonal expansion of tissue resident or bone marrow-derived EPCs [1,3,4]. Unlike CFU-ECs and CACs, ECFCs are truly endothelial precursors that may either circulate in peripheral blood or reside in the arterial vascular wall [9,10]. Therefore, although their role in IH growth and development remains to be clearly established, they represent a suitable tool to study the signaling pathways involved in the higher proliferation rate of EPCs in IH. We and others have recently demonstrated that SOCE is the most important proangiogenic Ca2+ entry pathway in N-ECFCs [14 –16,18]. Furthermore, we showed that the overall Ca2+ signalosome, and SOCE in particular, is remodeled in RCC-ECFCs [19], thereby conferring an adaptive advantage to tumor-derived ECFCs [22,59]. Consequently, the present investigation was undertaken to evaluate the rearrangement, if any, of the Ca2+ machinery in IH-ECFCs and to examine its functional impact on IH-ECFC proliferation. Our data strongly suggest that the constitutive activation of store-dependent channels enhances the growth rate of IH-ECFCs in an NO-dependent manner.
SOCE in N-ECFCs is mediated by the interaction between the ER Ca2+-sensor, that is, Stim1, and two Ca2+-permeable channels on the plasma membrane, that is, Orai1 and TRPC1 [18,19]. The depletion of the InsP3-dependent Ca2+ pool likely might cause these proteins to associate into a ternary super-molecular complex, as described in human platelets [60] and megakaryocytes [61]. Alternatively, Orai1-mediated Ca2+ inflow may recruit TRPC1 to the plasma membrane, where it is subsequently gated by Stim1 [62]. This issue remains to be addressed both in normal and tumoral ECFCs. SOCE, in turns, stimulates N-ECFC proliferation by promoting the nuclear translocation of the Ca2+-sensitive transcription factor, NF-κB [15]. We exploited the Ca2+ add-back protocol to monitor both the ER Ca2+ content, that is, the primum movens of Orai1 and TRPC1 activation, and SOCE amplitude. Our results clearly showed that both CPA, a selective SERCA inhibitor, and ATP, a physiological InsP3-producing autacoid, caused a lower intracellular Ca2+ release in IH-ECFCs compared to N-ECFCs. These data indicate that ER Ca2+ levels are significantly reduced in IH-ECFCs, as also shown in RCC-ECFCs [19], while ER Ca2+ content is dramatically higher in PMF-ECFCs [20]. Consistently, qRT-PCR and western blotting analysis revealed that InsP3Rs are normally expressed in IH-ECFCs, which supports the hypothesis that InsP3 impinges on a lower amount of releasable ER Ca2+ to trigger intracellular Ca2+ signals. In agreement with these data, VEGF did not mobilize intraluminally stored Ca2+, as also reported in RCC-ECFCs [19]. Whatever the underlying mechanism, which remains to be unveiled by future studies (but see below), the steady-state reduction in [Ca2+]ER is associated to the reduction of SOCE in these cells. This result differs from those obtained in RCC- and PMF-ECFCs [19,20] and confirms that ECFCs exhibit distinct Ca2+ signatures depending on the source [23,59]. Pharmacological characterization revealed that SOCE was inhibited by BTP-2 and low micromolar doses of La3+ and Pyr6, which may inhibit both Orai1 and TRPC1 [22,41,48]. Nevertheless, two unexpected findings hinted at a key clue to understand SOCE downregulation in IH-ECFCs. First, Stim1, Orai1, and TRPC1 were equally expressed at both mRNA and protein level in N- and IH-ECFCs, which indicated that the decrease in SOCE amplitude was not due to the downregulation of its molecular players. Second, in addition to Ca2+ inflow, BTP-2, La3+, and Pyr6 prevented CPA- and ATP-induced Ca2+ mobilization, which was not affected in N-ECFCs [14] as well as in RCC- [19] and PMF-ECFCs [20]. In an attempt to reconcile these conflicting results, we hypothesized that a constitutive SOCE recharges the ER Ca2+ pool in IH-ECFCs, but not N-ECFCs. We further speculated that such basal SOCE is remarkably larger in IH-ECFCs compared to healthy cells. This would explain the following: (1) why inhibiting SOCs suppresses intracellular Ca2+ release (no more Ca2+ within the ER) and (2) why CPA and ATP fail to fully activate Ca2+ inflow in IH-ECFCs (if most SOCs are already operative under resting conditions, the fraction of recruitable channels is lower than in healthy cells, where they are silent unless stimulated). Consistent with the first hypothesis, CPA-induced intracellular Ca2+ release did not reduce with the time (up to 20 min) when the cells remained in 0Ca2+ in N-ECFCs, while it was dramatically decreased in N-ECFCs. These data indicate that Ca2+ entry in unstimulated ECFCs is required to refill the ER only in IH-ECFCs during the period under observation. This pathway is likely to be provided by SOCE as it is sensitive to three distinct selective SOC inhibitors (i.e., BTP-2, La3+ and Pyr6). Caution is of course warranted in the interpretation of pharmacological data as some of the drugs employed in the present study may not be specific to SOCE, as discussed in Moccia et al. [22,41]. However, La3+ has widely been shown to selectively affect Orai1 in the low micromolar range, while it does not reliably block store-independent channels at this concentration [63]. Likewise, Pyr6 has recently been introduced as a specific SOCE blocker and a powerful tool to discriminate this pathway from receptor-operated Ca2+ inflow [42]. Finally, BTP-2 may also affect both TRPC3 and TRPC5 [64], which are however absent in N- and IH-ECFCs (present study and [14]). This drug could, in theory, dampen SOCE by reducing the electrochemical gradient sustaining Ca2+ inflow across the plasma membrane trough the activation of the nonselective cation channel TRP Melastatin 4 (TRPM4) [65]. However, BTP-2 fully activates TRPM4 already at 2 μM, while it is effective on ECFC SOCE and proliferation only at larger doses [19]. Consistenly, 5–20 μM BTP-2 is now regarded as a rather specific compound to specifically target SOCE in a growing number of cell types [66 –68].
To confirm whether constitutive SOCE is present and larger in IH-ECFCs as related to N-ECFCs, we turned to the Mn2+-quenching technique. This methodology has widely been employed to examine Ca2+ influx in endothelial cells [33,36,45,69] and we used it to ascertain whether SOCE selectively occurs in resting IH-ECFCs. In agreement with our surmise, a basal influx of Mn2+ was a rare event in N-ECFCs, while it occurred in majority of IH-ECFCs. Moreover, the rate of Mn2+ inflow was significantly higher in the latter compared to healthy cells. Furthermore, Mn2+ entry was inhibited by BTP-2, 10 μM La3+, and Pyr6, while it was unaltered by G418, which interferes with Stim2-dependent Ca2+ influx. These data support the postulate that a constitutive SOCE is present in ECFCs and is significantly larger in IH-ECFCs. It is, therefore, feasible that the partial depletion of the InsP3-sensitive Ca2+ pool recruits Stim1, Orai1, and TRPC1 to secure SOCE activation and recharge the empty stores in resting IH-ECFCs. Nevertheless, Ca2+ influx is not sufficient to fully restore ER Ca2+ levels, thereby triggering a positive feedback loop that sustains SOCE over time. This mechanism has also been shown in brain neurons, whose ER is fully or moderately depleted even in the absence of synaptic stimulation [70]. For instance, Stim1 recruits Orai2, that is, the established neuronal SOC [40], to mediate Ca2+ inflow and maintain ER Ca2+ levels in nonstimulated mouse cerebellar Purkinje and granule cells [71,72]. Likewise, Orai1 and TRPC1 bring about a basal Ca2+ influx in MDA-MB-468 breast cancer cells [44], whereas Stim1 and Orai1 interact to support resting SOCE and proper ER Ca2+ filling in HL-1 cardiomyocytes [73] and human skeletal myoblasts [74]. The question next arises as to why constitutive SOCE is not able to fully replenish the ER Ca2+ reservoir in IH-ECFCs. We speculate that such feature is attributable to the different organization of the plasma membrane-ER Ca2+ circuit between N- and IH-ECFCs. Resting Ca2+ influx in both cell types is hard to detect by simply switching the extracellular Ca2+ concentration from low (0.0 mM) to high (1.5 mM). Conversely, it is unmasked by replacing Ca2+ with Ba2+, which cannot be captured by Ca2+-transporting systems located either on the plasma membrane or on the ER, in the reentry protocol. Consistent with the Mn2+-quenching experiments, Ba2+ entry was significantly greater in IH-ECFCs as respect to N-ECFCs. These experiments suggest that SOCs reside in close apposition with the superficial ER domains, thereby generating a local Ca2+ microdomain beneath the plasma membrane in both cell types. Entering Ca2+ is likely prevented from reaching the bulk cytosol by the concerted interaction of several Ca2+ clearing mechanisms, such as SERCA, NCX, and possibly mitochondria [75]. Earlier work conducted in unstimulated vascular endothelial cells demonstrated that, in the absence of InsP3 production [76,77], incoming Ca2+ is avidly sequestered by SERCA activity, redirected toward the subplasmalemmal space by leak channels and/or the basal activity either of InsP3Rs or ryanodine receptors (RyRs), and extruded across the plasma membrane by NCX [44,78]. In this view, it is the rate of Ca2+ leak that determines the extent of ER Ca2+ filling [79]. Therefore, one might hypothesize that ER Ca2+ leak is significantly more pronounced in IH-ECFCs compared to control cells: this would explain why ER Ca2+ content is lower and, consequently, constitutive SOCE larger in the former. Alternatively, ER Ca2+ leak might be preferentially coupled to the SERCA activity in N-ECFCs, thereby preserving [Ca2+]ER even in the absence of Ca2+ influx in N-ECFCs as earlier observed in Ea.hy926 cells [77]. These models would explain why the ER Ca2+ reservoir is not depleted after 20 min in the presence of SOC inhibitors or absence of external Ca2+. Notably, the genetic deletion of SOCE does not affect ER Ca2+ handling in N-ECFCs as demonstrated both by other research groups [18] and by ours [15,19]. It turns out that the ER of these cells may cope even with the chronic downregulation of Stim1, Orai1, and TRPC1 and do not undergo dramatic changes in its Ca2+ levels. Future work will have to experimentally probe this hypothesis and shed light on the molecular underpinnings of the ER Ca2+ leak pathways, which, along with constitutively open InsP3Rs and RyRs, might also involve translocons, pannexins, proteins related to neurodegenerative diseases (such as presenilins), and members of antiapoptotic proteins of the Bcl2 and Bcl2-associated X protein (Bax)-inhibitor-1 (BI-1) [80].
In addition to replenishing ER Ca2+ stores in IH-ECFCs (see above), SOCE stimulates proliferation in healthy and RCC-derived ECFCs [14,16], but not in PMF-ECFCs [20]. In N-ECFCs, the proangiogenic signal delivered by SOCE is activated by VEGF, which recruits PLCγ to deplete the InsP3-sensitive Ca2+ pool [15]. As SOCE is constitutively active in IH-ECFCs, it cannot be activated by VEGF in IH-ECFCs and is only weakly stimulated by other ER depleting stimuli, such as CPA and ATP (see above). Therefore, we focussed on the functional outcome of the constitutive SOCE in IH-ECFCs. Growth rate was significantly greater in IH-ECFCs compared to N-ECFCs. To the best of our knowledge, this is the first demonstration that tumor-derived EPCs proliferate faster than healthy cells. Moreover, this feature is compatible with the supporting role played by ECFCs during IH neovascularisation, although this hypothesis remains to be experimentally demonstrated. Importantly, the pharmacological inhibition of SOCE with BTP-2 and Pyr6 blocks IH-ECFC proliferation. The same effect was achieved by BAPTA, which buffers intracellular Ca2+ levels and prevents entering Ca2+ from activating its downstream Ca2+-sensitive decoders [15,23]. Therefore, it is possible to conclude that constitutive SOCE activation results in the aberrant IH-ECFC growth. These results are corroborated by the finding that, in the absence of extracellular growth factors, IH-ECFCs still proliferate at a higher rate compared to control cells. However, the blockade of SOCE with either BTP-2 or Pyr6 suppressed IH-ECFC proliferation, while it is ineffective in N-ECFCs. These data (1) confirm that it is constitutive SOCE that drives the excessive IH-ECFC growth and (2) further demonstrate that SOCE controls the proangiogenic activity of N-ECFCs only when it is stimulated by external mitogens, such as VEGF.
The question then arises as to the mechanistic link between SOCE and IH-ECFC proliferation. NO is required to promote EPC egression from bone marrow into peripheral blood [54] and stimulates proliferation in mature endothelial cells [26,27] and CFU-ECs [81], another EPC subtype. Moreover, eNOS is a Ca2+/calmodulin-dependent enzyme that is mainly activated by SOCE in vascular endothelial cells [28,29,82]. We found that basal NO synthesis was significantly larger in IH-ECFCs compared to N-ECFCs, thereby suggesting that constitutive eNOS activation is more sustained in the former. Moreover, NO production was abrogated by L-NAME, a selective eNOS inhibitor, BTP-2, and removal of external Ca2+: these results strongly suggest that resting SOCE drives the higher eNOS activity in IH-ECFCs. On the other hand, BAPTA did not interfere with NO levels; this finding resembles the weak effect of BAPTA on UCB-ECFC proliferation [23]. This observation, however, is not incompatible with our previous results: it suggests that eNOS is placed at a distance much shorter than 10–100 nm from the SOC pore [58,83] so that 2 h might not be enough for BAPTA to diffuse near the inner mouth of the channel and intercept the incoming Ca2+. Consistently, eNOS has been allocated to a restricted Ca2+ microdomain near Orai1 channels [55,84]. In agreement with the role of NO in ECFC growth, L-NAME inhibited N- and IH-ECFC proliferation. Overall, these data strongly corroborate the assumption that constitutive SOCE drives eNOS activation and, therefore, accelerates cell replication in IH-ECFCs compared to N-ECFCs. We do not rule out the possibility that other proangiogenic Ca2+-dependent decoders, such as NF-κB and NFAT, are recruited by the basal store-dependent Ca2+ inflow to further support IH-ECFC proliferation [15,85]. A major caveat of this study is that, because of ethical issues, we could not access age-matched controls. Therefore, healthy cells were necessarily obtained from older donors. Yet, we believe that this feature does not affect the relevance of our findings. Accordingly, the Ca2+ toolkit of IH-ECFCs, which are derived from young children, is qualitatively and quantitatively identical to N-ECFCs, which are derived from healthy subjects in their twenties. Conversely, it is strikingly different from UCB-ECFCs, which should in principle bear more similarities with neonatal cells. Accordingly, both N- and IH-ECFCs express InsP3R1, which is absent in UCB-ECFCs, but lack TRPC3, which is otherwise abundant in UCB-ECFCs [23]. In addition, there was no difference in the expression levels of all the components of the Ca2+ machinery examined in this study between N- and IH-ECFCs (see above). These observations strongly suggest that neonatal ECFCs rapidly acquire their adult Ca2+ signalosome and are, therefore, not different from their mature counterparts in terms of intracellular Ca2+ homeostasis.
In conclusion, this investigation shows that SOCE is constitutively active in IH-ECFCs, but not in healthy cells, most likely due to the chronic underfilling of the InsP3-sensitive Ca2+ pool. The resting influx of Ca2+ enhances basal NO generation and, consequently, boosts cell proliferation as respect to N-ECFCs. Such observations corroborate the notion that the Ca2+ toolkit is deranged in cancer patients and display distinct signatures depending on the tumor type [59]. Furthermore, Stim1, Orai1, and TRPC1 stand out as novel candidates to shed light on the molecular underpinnings of IH neovascularisation. It is still unknown whether ECFCs correspond to the clonal endothelial precursors, which support the early growth of IH. However, basal SOCE activation in ECFCs might now be regarded among the signalling pathways that drive the onset of such diseases. In this perspective, SOCE would provide an easy accessible and druggable target to interfere with IH neovascularisation as suggested for other solid malignancies [22,59,86 –88].
Footnotes
Acknowledgment
The present project was financially supported by the intramural funding program “Ricerca Corrente” of the IRCCS Policlinico San Matteo: RC/80520.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.