
Abstract
All-trans retinoic acid (ATRA) is a potent inducer of osteogenic differentiation in mouse adipose-derived stromal cells (mASCs), although the underlying mechanisms responsible for its mode of action have yet to be completely elucidated. High temperature requirement protease A1 (HtrA1) is a newly recognized modulator of human multipotent stromal cell (MSC) osteogenesis and as such, may play a role in regulating ATRA-dependent osteogenic differentiation of mASCs. In this study, we assessed the influence of small interfering RNA (siRNA)-induced repression of HtrA1 production on mASC osteogenesis and examined its effects on ATRA-mediated mammalian target of rapamycin (mTOR) signaling. Inhibition of HtrA1 production in osteogenic mASCs resulted in a significant reduction of alkaline phosphatase activity and mineralized matrix formation. Western blot analyses revealed the rapid activation of Akt (Ser473) and p70S6K (Thr389) in ATRA-treated mASCs, and that levels of phosphorylated p70S6K were noticeably reduced in HtrA1-deficient mASCs. Further studies using mTOR inhibitor rapamycin and siRNA specific for the p70S6K gene Rps6kb1 confirmed ATRA-mediated mASC osteogenesis as being dependent on p70S6K activation. Finally, transfection of cells with a constitutively active rapamycin-resistant p70S6K mutant could restore the mineralizing capacity of HtrA1-deficient mASCs. These findings therefore lend further support for HtrA1 as a positive mediator of MSC osteogenesis and provide new insights into the molecular mode of action of ATRA in regulating mASC lineage commitment.
Introduction
E
ATRA's ability to influence osteoblast differentiation has been observed in several different cell systems and is considered to be largely dependent on the concentration of ATRA used. While ATRA acts to enhance osteogenesis at micromolar concentrations [1 –7, 9, 10], at nanomolar concentrations, it has been shown to inhibit both osteoblast gene expression and mineralization [11 –13]. The concentration of ATRA used to stimulate mASC osteogenesis in vitro is generally within the range of 1–5 μM, where it acts to enhance the expression of several osteogenic markers including alkaline phosphatase (Alpl) and osteopontin (Spp1), and to induce mineralization of mASC-derived osteoblasts [3,4]. In addition, ATRA's ability to direct mASCs along the osteoblast lineage in vitro has also been exploited for the purpose of enhancing mASC-induced new bone formation in vivo. Priming of mASCs with ATRA before their implantation into mouse calvarial defects resulted in accelerated bone regeneration compared with mice treated with unstimulated mASCs [14]. However, the mechanisms through which ATRA instigates its osteogenic effects in these cells remain unclear.
Findings from studies investigating the combined effects of ATRA and bone morphogenetic protein (BMP)-2 on mASC osteogenesis suggested that ATRA's primary function was to regulate BMP signaling through enhanced BMP receptor expression [1]. However, ATRA also has the ability to induce osteogenic differentiation of mASCs in the absence of exogenous BMP-2 [2 –7]. Therefore, it's likely that in addition to BMP signaling, ATRA targets other pathways critically involved in regulating mASC osteogenesis.
We have previously identified high temperature requirement protease A1 (HtrA1) as a novel mediator of human BMSC (hBMSC) differentiation, where it acts to enhance osteogenesis and subsequent mineralization by differentiating bone-forming cells [7]. Furthermore, HtrA1 expression is upregulated in mASCs in response to ATRA-containing osteogenic induction medium [7].
HtrA1 is a member of the HtrA family of serine proteases and has been linked to various biological processes by virtue of its ability to interact with numerous intracellular and extracellular substrates [15]. Tuberous sclerosis complex 2 (TSC2) was the first cytoplasmic HtrA1 substrate to be identified, and its degradation by HtrA1 was shown to result in activation of the mammalian target of rapamycin (mTOR) pathway as confirmed by alterations in the phosphorylation of downstream targets eukaryotic initiation factor 4E binding protein 1 (4E-BP1) and p70 ribosomal protein S6 kinase (p70S6K) [16]. This bears particular significance with regards to mASC osteogenesis, based on the fact that mTOR signaling plays a positive role in the osteogenic induction of several cell types including BMSCs [17 –19]. However, no studies have yet sort to investigate its involvement in mediating the osteoinductive effects of ATRA on mASCs, or whether HtrA1's ability to influence mTOR signaling plays a role in determining mASC osteogenic potential.
In this study, we investigated the role of HtrA1 in the ATRA-dependent differentiation of mASCs into mineral-forming bone cells and assessed its influence over mTOR signaling events during the course of mASC osteogenesis.
Materials and Methods
Materials
Antibodies specific for nonphosphorylated Akt, mTOR, p70S6K, 4E-BP1, and rpS6; phosphorylated Akt (Ser473), mTOR (Ser2448), p70S6K (Thr389), 4E-BP1 (Thr37/46), and rpS6 (Ser235/236) were all purchased from Cell Signaling, BioConcept. Mouse monoclonal anti-tubulin was from Sigma-Aldrich. Mouse HA-probe antibody and anti-GAPDH were from Santa Cruz Biotechnology, LabForce AG. A polyclonal anti-HtrA1 antibody was generated as previously described [20]. HRP-labeled secondary antibodies specific for mouse or rabbit IgG were purchased from Jackson ImmunoResearch. Rapamycin was purchased from Enzo Life Science. BMP-2 was kindly donated by Prof. Franz Weber (University of Zurich). ATRA, dexamethasone, and PF-4708671 were from Sigma-Aldrich. The expression plasmids pRK7-HA-S6K1-F5A-E389-R3A (Addgene plasmid # 8991) and pRK7-HA-S6K1-KR (Addgene plasmid # 8985) were kind gifts from John Blenis [21].
Isolation and culture of mASCs
Primary mASCs were isolated from SAM mice as previously described [4,5]. All animal research procedures were approved by the Animal Experimentation Committee of the Veterinary Office of the Canton of Zurich, Switzerland and followed the guidelines of the Swiss Federal Veterinary Office for the use and care of laboratory animals. Briefly, subcutaneous inguinal fat pads were removed and digested in Hepes buffer containing 0.1% collagenase A (Roche Diagnostics) and 0.2% bovine serum albumin for 40 min at 37°C. Adherent stromal cells were maintained in complete medium consisting of Dulbecco's modified eagle medium (DMEM-low glucose, with GlutaMAX) (Life Technologies), supplemented with 10% fetal bovine serum (FBS) (Bioswisstec) and antibiotics. Supernatant was replaced after 1 day with fresh complete medium and cells were used between passage 1 and 4 following initial analysis for mesenchymal and hematopoietic cell markers by flow cytometry as previously described [4].
Osteogenic differentiation of mASCs
mASCs were plated at 5,000 cells/cm2 and incubated in alpha-minimum essential medium (α-MEM) (Life Technologies), supplemented with 10% FBS (Bioswisstec), 50 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate, 10 mM β-glycerophosphate, and either 5 μM ATRA or 100 nM dexamethasone and 100 ng/mL BMP-2 for up to 21 days with regular changes of medium as previously described [4]. Where indicated, PF-4708671 was also added to mASCs undergoing osteogenic differentiation to assess the influence of S6K1 inhibition on ATRA-dependent osteogenic induction. Alkaline phosphatase (ALP) activity was quantified in cell lysates using p-nitrophenylphosphate (pNPP) liquid substrate (Sigma-Aldrich) and values normalized to total protein content and reaction time as previously described [4]. Mineralization was visualized using Alizarin red and the amount of staining determined by measuring optical densities at 570 nm following extraction using 10% cetylpyridinium chloride (Sigma-Aldrich). Optical densities were then converted to micromoles (μM) of Alizarin red using a standard curve and normalized to cell number. The mean cell number was determined by automated counting of 4′,6-diamidino-2-phenylindole (DAPI) stained nuclei in at least six random fields of view. Images were captured on a Leica DMI 6000 inverted fluorescence microscope (Leica Microsystems). Image processing and nuclear counts were performed using NIH ImageJ software.
Quantitative reverse transcription PCR
Total RNA was isolated from mASCs and purified using TRIzol reagent (Invitrogen AG, Basel, Switzerland) according to the manufacturer's instructions. RNA (0.5 μg) was reverse transcribed to cDNA using Superscript II (Invitrogen AG) and random hexanucleotide primers (Promega AG). Quantification of mRNA expression was performed with TaqMan Gene Expression Assays (Applied Biosystems) specific for HtrA1 (Mm00479887), Rps6kb1 (Mm01310033), Alpl (Mm01187117), and Spp1 (Mm01611440) using the StepOnePlus Real-Time PCR System (Applied Biosystems) and values normalized to Mrps12 (Mm00488728) mRNA levels and presented as fold change according to the 2−ΔΔCT method. Each 10 μL reaction consisted of 1 × TaqMan Fast Universal PCR Master Mix (Applied Biosystems), 1 × TaqMan Gene Expression Assay, and 10 ng cDNA (based upon initial RNA concentrations). All reactions were performed in triplicate in fast optical 96-well reaction plates (Applied Biosystems) at 95°C for 20 s and 40 cycles of 95°C for 1 s and 60°C for 20 s.
Western blot analysis
Total cellular protein was extracted from mASCs using CelLytic M (Sigma-Aldrich) containing protease and phosphatase inhibitor cocktails (Sigma-Aldrich). For the analysis of HtrA1 in mASC supernatants, cells were treated for 3 days with osteogenic induction medium and then for a further 24 h in fresh FCS-free osteogenic induction medium before harvesting and concentrating supernatants 30-fold using Amicon Ultra-15, 10 kDa mwco filter units (Millipore). In each case, protein amounts were quantified using BioRad Protein Assay (BioRad). Protein samples were boiled for 5 min in loading buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 100 mM DTT, and 0.002% Bromophenol blue) and equal amounts of protein loaded onto 12% SDS-PAGE gels. Protein was then electroblotted onto PVDF membranes using the Trans-Blot Turbo blotting system (BioRad) and incubated in 5% skimmed milk, 50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% Tween 20 (TBST) for 1 h at room temperature. Membranes were then incubated overnight at 4°C with primary antibodies specific for HtrA1 or phosphorylated or nonphosphorylated Akt, mTOR, p70S6K, rpS6, and 4E-BP1. Monoclonal mouse anti-tubulin or anti-GAPDH were used to control equal protein loading of cell lysates. Coomassie blue staining was used to control equal protein loading of cell supernatants. After washing in TBST thrice for 5 min each, membranes were incubated with a HRP-conjugated anti-mouse or anti-rabbit IgG (1:10,000) for 1 h at room temperature. Following a further washing step, peroxidase activity was detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Protein levels were quantified using NIH ImageJ software. Phosphorylated and nonphosphorylated protein values were first normalized to tubulin loading control and then the phosphorylation to total protein ratio was calculated using the normalized values.
Small interfering RNA studies
Specific knock down of gene expression was performed with Silencer Select Small interfering RNA (siRNA) (Ambion, Life Technologies) specific for HtrA1 or Rps6kb1 using previously described methods [7]. Briefly, mASCs (1 × 105 cells) were transfected with 20 nM of targeted siRNA or negative control siRNA (Negative Control-1) using the NEON Transfection System (Life Technologies). Following transfection, cells were seeded in cell culture plates with fresh growth medium (without antibiotics) and incubated for 24 h at 37°C, 5% CO2. Medium was then replaced with either fresh growth medium or osteogenic differentiation medium and total RNA or protein harvested at selected time points for further analysis. The effects of siRNA mediated gene knockdown on osteogenic-induced mASC ALP activity and mineralization was determined using the ALP activity assay and Alizarin red staining respectively.
siRNA and plasmid co-transfection
mASCs were transfected with Silencer Select siRNA specific for HtrA1 or Negative Control-1 and 1 μg of mammalian expression plasmid pRK7-HA-S6K1-F5A-E389-R3A (constitutively active p70S6K), pRK7-HA-S6K1-KR (kinase inactive p70S6K), or empty control plasmid pcDNA3 using the NEON Transfection System as described above. After 24 h, cells were induced to undergo osteogenesis and mineralization quantified after 21 days using Alizarin red staining as described above.
Statistical analysis
Two-tailed unpaired Student's t-test for comparison of two groups or one-way analysis of variance (ANOVA) with Tukey's post hoc test for multiple group comparisons were performed using SPSS19.0 (SPSS, Inc.). In all cases, a P-value of <0.05 was considered statistically significant.
Results
We have previously demonstrated that HtrA1 plays a vital role in the regulation of osteogenesis in hBMSCs [7]. In the current report, we further investigated this property of HtrA1 in ATRA-stimulated mASCs and aimed to establish its role in regulating mASC osteogenesis and mASC-derived osteoblast mineralization.
HtrA1 deficiency impairs ATRA-mediated mASC osteogenic differentiation
To examine the influence of loss-of-function of HtrA1 on the osteogenic capacity of mASCs in response to treatment with osteogenic medium containing ATRA, we analyzed mineral production by mASC-derived osteoblasts, and ALP expression and enzyme activity. Analysis of HtrA1 in mASC supernatants by western blot confirmed HtrA1 protein production to be effectively reduced in osteogenic mASCs treated with siRNAs specific for HtrA1 (Fig. 1A).
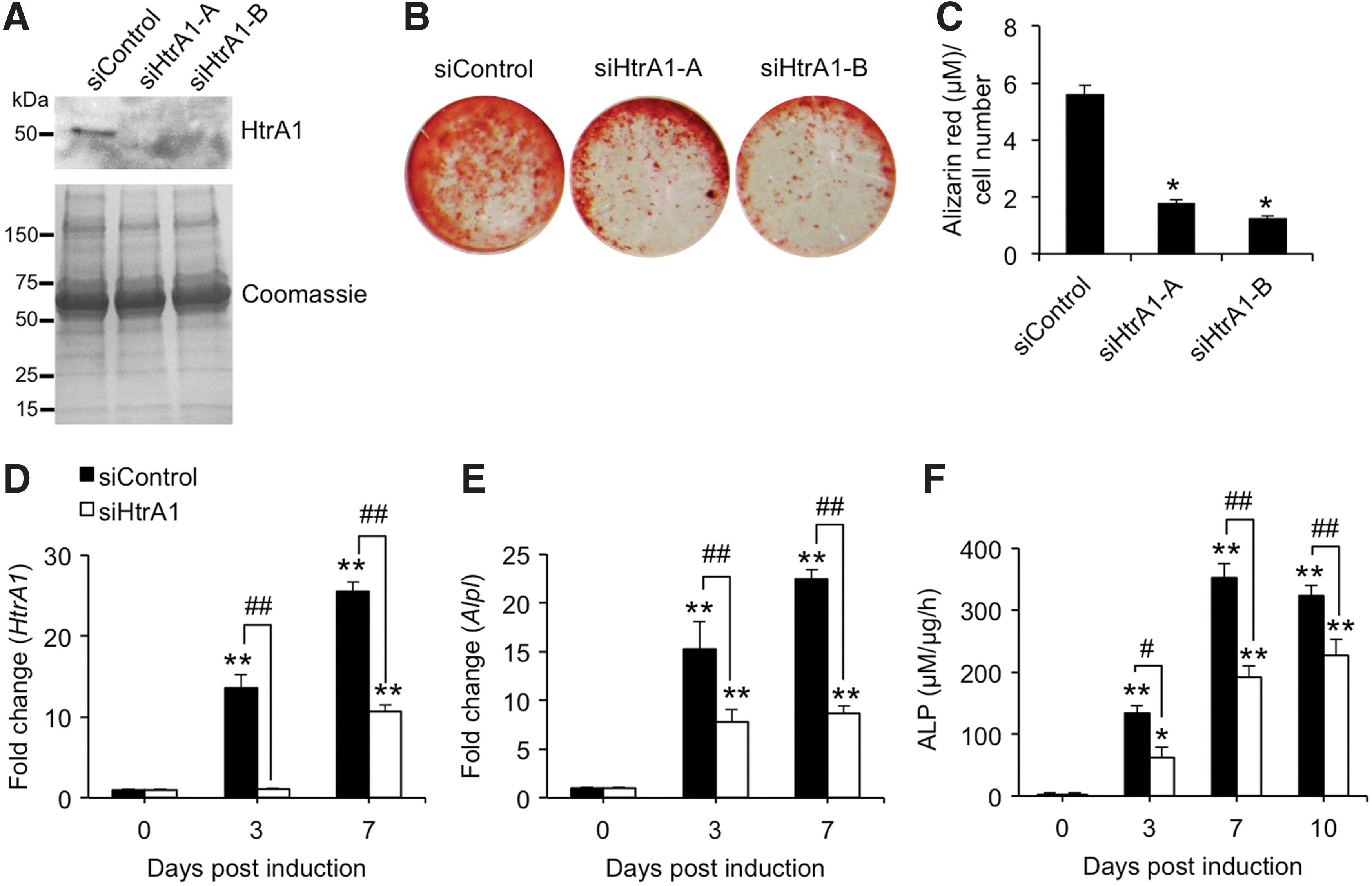
Effect of HtrA1 knockdown on osteogenic induction of mASCs. mASCs were pretreated with control siRNA (siControl) or two selected siRNAs targeting HtrA1 (siHtrA1-A and siHtrA1-B) for 24 h and induced to undergo osteogenesis for up to 10 days.
We next assessed the influence of loss-of-function of HtrA1 on the osteogenic potential of mASCs. mASCs treated with control siRNA underwent efficient osteoblastogenesis and mineralization following stimulation with osteogenic medium for 10 days, as determined by Alizarin red staining (Fig. 1B). However, Alizarin red staining of osteogenic-induced mASCs in which HtrA1 had previously been depleted was noticeably reduced (Fig. 1B). Similarly, HtrA1 deficiency also resulted in significant reductions in Alizarin red staining in mASCs induced to undergo osteogenic differentiation in response to BMP-2 treatment (Supplementary Fig. S1; Supplementary Data are available online at
In accordance with our previous findings [7], ATRA-mediated osteogenic induction of mASCs resulted in significant increases in both HtrA1 (Fig. 1D) and Alpl (Fig. 1E) expression levels in a time-dependent manner. Similarly, ALP enzyme activity was also significantly enhanced in response to osteogenic induction at all time points tested (Fig. 1F). As expected, HtrA1 knockdown of mASCs significantly suppressed HtrA1 expression in osteogenic mASCs over the course of the study (Fig. 1D). ATRA-mediated increases in Alpl expression levels were also significantly impaired in HtrA1-deficient mASCs (Fig. 1E) and were accompanied by significant decreases in ALP enzymatic activity at all time points tested (Fig. 1F). These findings therefore demonstrate a functional role for HtrA1 in regulating ATRA-mediated mASC osteogenesis and in the generation of a mineralized matrix by mASC-derived osteoblasts.
HtrA1 deficiency impairs ATRA-mediated p70S6K activation in mASCs
Having demonstrated HtrA1 to be a necessary component for efficient mASC osteogenesis, we next considered its potential mode of action. Based on HtrA1's previously reported role in the activation of mTOR targets p70S6K and 4E-BP1 in tumor cell lines [16], we assessed the possible influence of HtrA1 deficiency on mTOR signaling in ATRA-stimulated mASCs. As no studies have yet sought to determine the effects of ATRA-mediated osteogenic induction on mTOR signaling in mASCs, we initially performed a series of western blot analyses to ascertain the activation status of kinases located both upstream and downstream of mTOR. Phosphorylation levels of Akt (Ser473), mTOR (Ser2448), p70S6K (Thr389), and 4E-BP1 (Thr37/46) were assessed in mASCs over the course of 2 h following ATRA-mediated osteogenic induction. A noticeable increase in Akt and p70S6K phosphorylation was already evident in osteogenic-induced mASCs after only 10 min and had reduced to basal levels by 1 h (Fig. 2A). However, basal levels of phosphorylated mTOR were only minimally affected after 10 min, and 4E-BP1 remained relatively unchanged at all time points tested. We next proceeded to investigate the influence of HtrA1 silencing on Akt/mTOR/p70S6K/4E-BP1 phosphorylation in differentiating mASCs. Western blot analysis of cell lysates from HtrA1-deficient mASCs revealed no reduction in phospho-Akt levels, and only minor reductions in phospho-mTOR and–4EBP1 levels compared with siControl (Fig. 2B). By comparison, however, HtrA1-deficient mASCs demonstrated marked reductions in the levels of phosphorylated p70S6K and rpS6. These results are therefore suggestive of ATRA-mediated p70S6K activation as being a potential HtrA1 target and a means by which it could influence ATRA-dependent mASC osteogenesis.
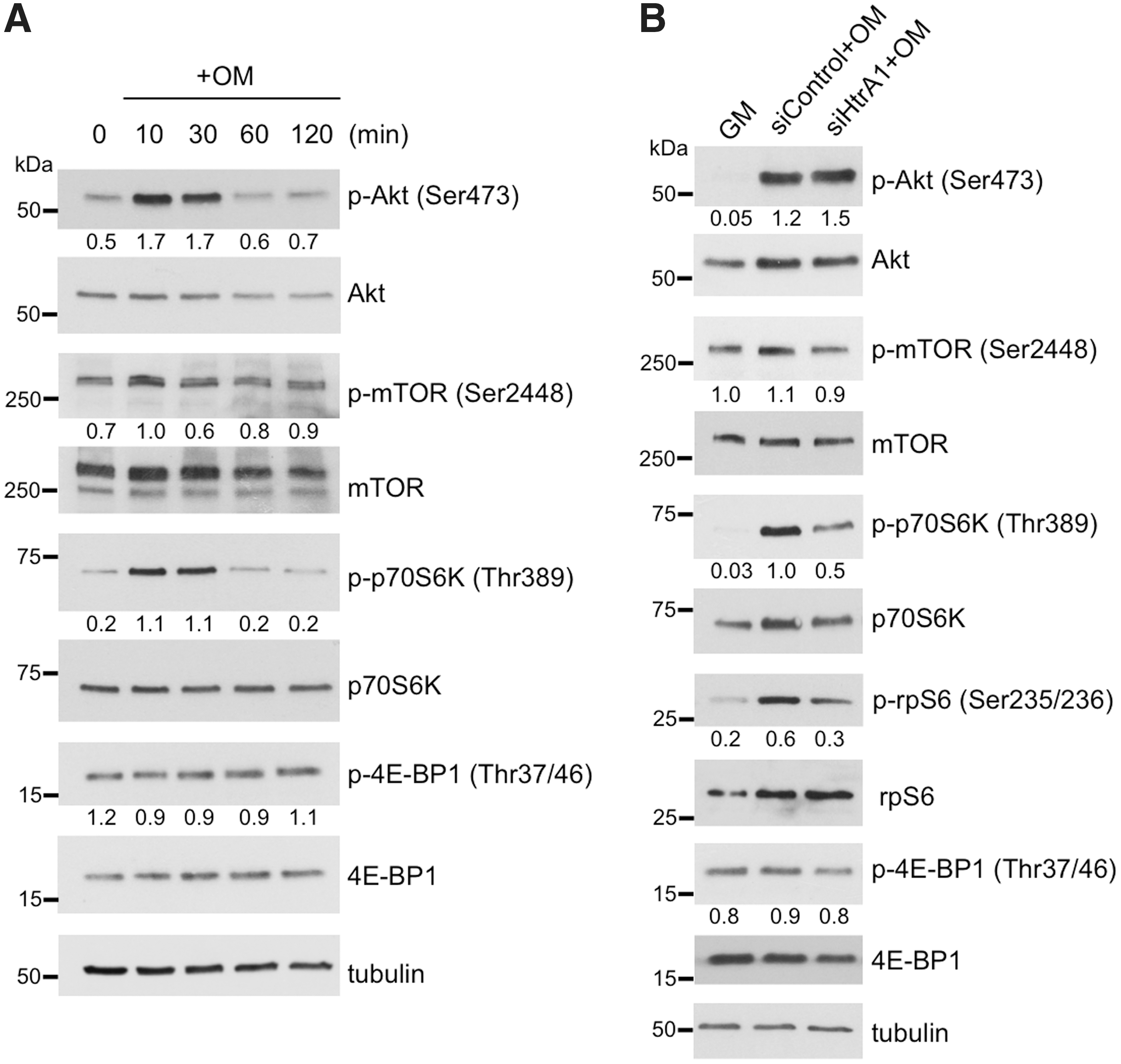
Western blot analysis of kinase activity in mASCs in response to ATRA-mediated osteogenic induction.
Loss-of-function of p70S6K impairs mASC osteogenesis
To address the functional relevance of p70S6K activation in the context of mASC osteogenesis, we next assessed the effects of p70S6K inhibition on ALP expression and matrix mineralization in mASCs undergoing ATRA-mediated osteogenic differentiation. Short-term treatment of mASCs with the mTOR inhibitor rapamycin before osteogenic induction markedly reduced mTOR phosphorylation and completely abolished p70S6K phosphorylation, while Akt phosphorylation levels remained unaffected (Fig. 3A). Next, we evaluated the effects of long-term exposure of mASCs to rapamycin with regards to their ability to differentiate into mineralizing osteoblasts. Alizarin red staining of osteogenic mASCs was significantly reduced by rapamycin treatment in a concentration-dependent manner (Fig. 3B, C), thus confirming that mTOR signaling was required for mASC-derived osteoblastogenesis.
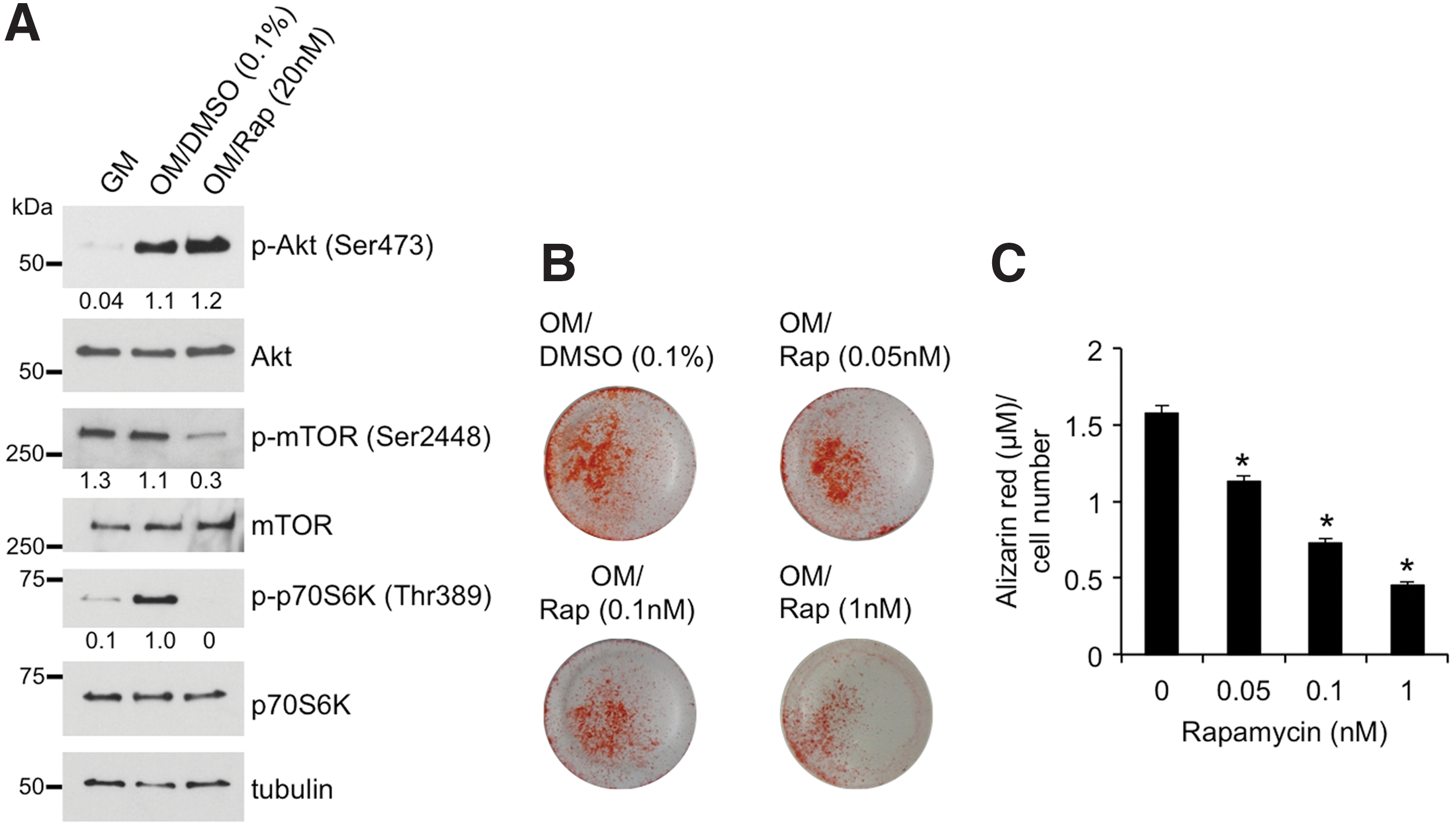
Effect of rapamycin on osteogenic induction of mASCs.
Further investigations employing siRNA-mediated knockdown of the p70S6K gene Rps6kb1 were also performed to assess the effects of specifically inhibiting p70S6K activity on mASC osteogenesis. p70S6K protein and activity levels were noticeably reduced in Rps6kb1-deficient mASCs after short-term osteogenic induction (Fig. 4A). We next investigated the effects of Rps6kb1 knockdown on mASC-derived osteoblast mineralization using Alizarin red staining. A marked reduction in Alizarin red staining was observed in Rps6kb1-deficient mASCs after 14 days of culture in osteogenic medium (Fig. 4B). Further quantitative analysis of extracted Alizarin red stain revealed the mineralizing capabilities of p70S6K-deficient mASC-derived osteoblasts to be significantly impaired compared with siControl (P < 0.001) (Fig. 4C). In support of these findings, significant reductions in Alizarin red staining of mASC-derived osteoblasts were also observed in cultures treated with the specific S6K1 inhibitor PF-4708671 (Supplementary Fig. S2). Gene expression analyses of mASCs undergoing osteogenesis confirmed efficient Rps6kb1 knockdown throughout the course of the study and revealed a significant reduction in Rps6kb1 gene expression at day 14 in siControl in response to osteogenic induction (Fig. 4D). Furthermore, the expression levels of osteogenic markers Alpl (Fig. 4E) and osteopontin (Spp1) (Fig. 4F) in mASCs undergoing osteogenesis were significantly reduced in p70S6K-deficient cells. These results clearly identify p70S6K activation as being a necessary requirement for efficient osteogenic differentiation of mASCs and provide a potential means through which HtrA1 may regulate ATRA-mediated mASC osteogenesis.
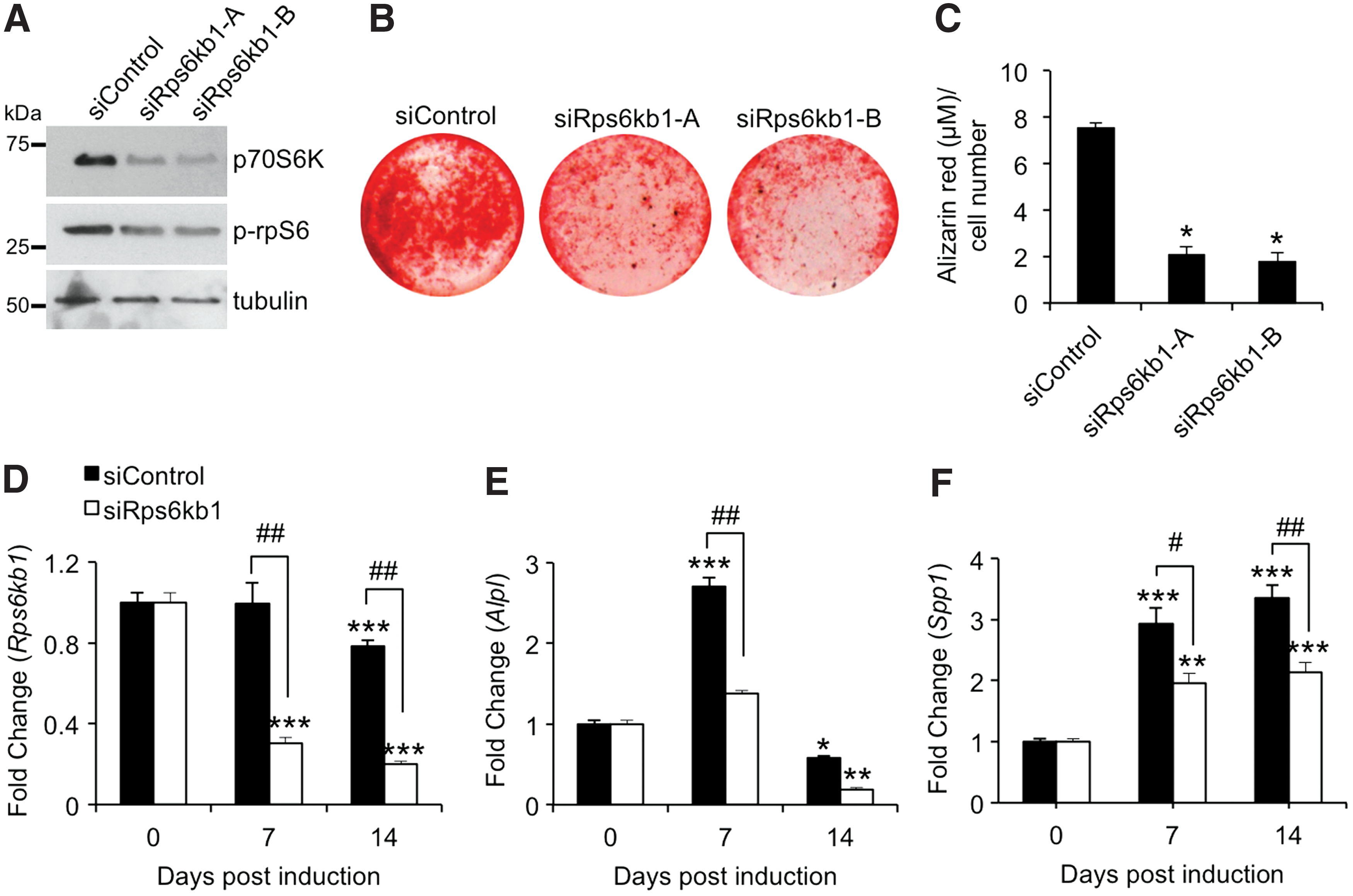
Effect of Rps6kb1 knockdown on osteogenic induction of mASCs. mASCs were pretreated with control siRNA (siControl) or two selected siRNAs targeting Rps6kb1 (siRps6kb1-A and siRps6kb1-B) for 24 h and induced to undergo osteogenesis for up to 14 days.
Restoration of HtrA1-deficient mASC osteogenesis using a constitutively active p70S6K mutant
In view of the fact that mASC osteogenesis is dependent on both HtrA1 and p70S6K, and that loss-of-function of HtrA1 impairs p70S6K activation, we sought to determine whether transfection of mASCs with a constitutively active p70S6K mutant could relieve the detrimental effects of HtrA1 deficiencies on mASC osteogenesis. mASCs were transfected with either an empty plasmid, or plasmids encoding a rapamycin-resistant constitutively active (p70S6KCA) or kinase inactive (p70S6KKI) HA-tagged p70S6K [21]. The results shown in Fig. 5A are representative of the controls used to confirm that the plasmids encoding p70S6KCA and p70S6KKI were performing as expected. Both protein products were labeled with an HA-tag, and so plasmid-mediated protein expression could be accurately and specifically detected using an anti-HA antibody. As shown in the top lane of Fig. 5A, HA-labeled protein is evident in cells treated with plasmids encoding p70S6KCA and p70S6KKI as expected, but no signal is detected in cells treated with empty plasmid as no HA-labeled protein has been produced. Cells transfected with empty plasmid therefore serve as an additional control to confirm Western blot specificity and the robustness of the cell transfection system used.
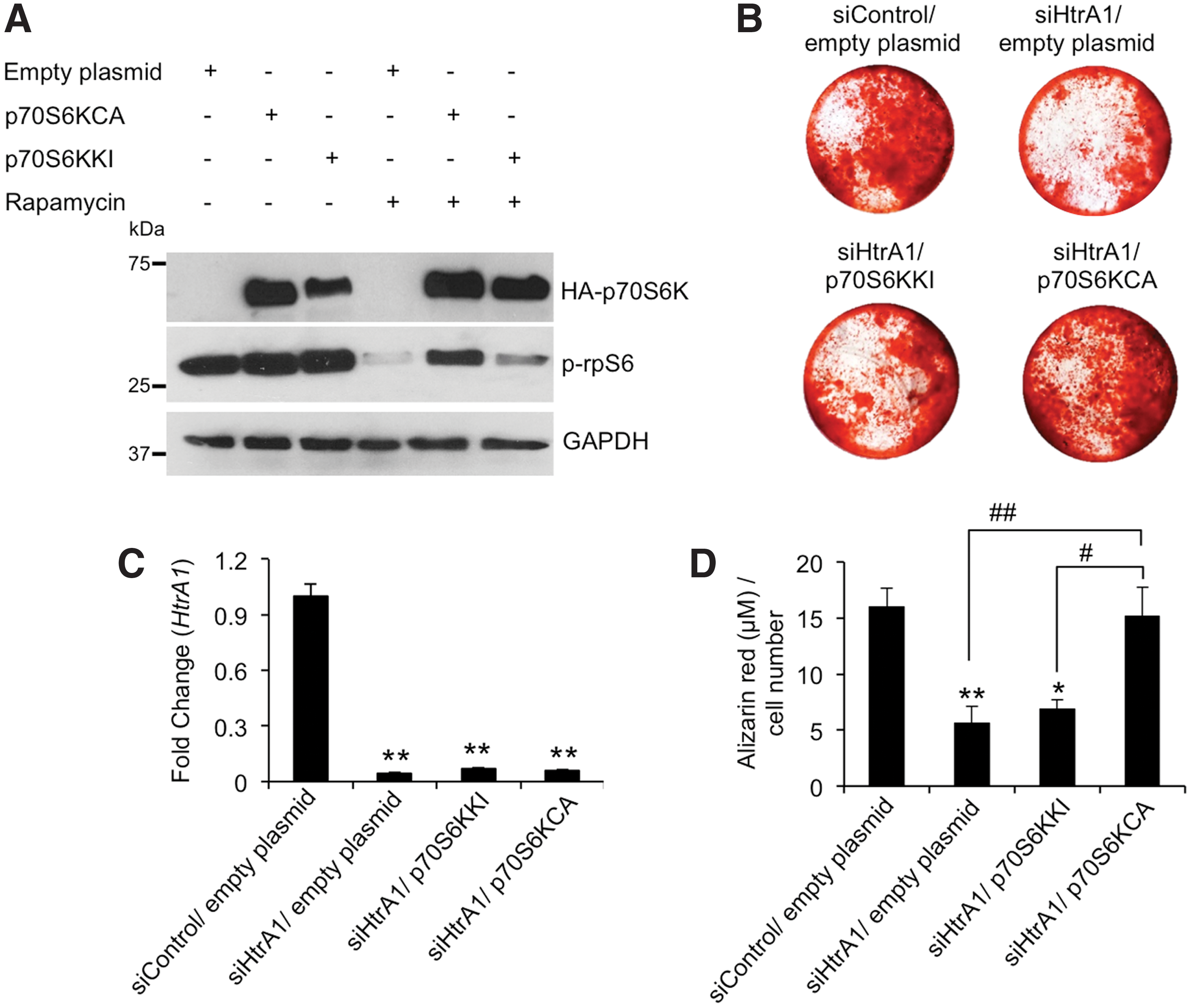
Influence of p70S6K activity on siHtrA1-mediated suppression of mASC osteogenesis.
The p70S6K substrate rpS6 is phosphorylated by activated p70S6K and therefore represents a useful means by which to visualize p70S6K activation by western blot. As such, inhibition of p70S6K activity by rapamycin treatment is expected to result in reduced phosphorylated rpS6 (p-rpS6) levels. Indeed, upon rapamycin treatment of mASCs transfected with empty plasmid or plasmid encoding p70S6KKI, we observed a noticeable reduction in p-rpS6 levels in cells. This would be expected as neither the empty plasmid nor the kinase inactive p70S6KKI can generate active, rapamycin-resistant p70S6K. However, cells expressing the rapamycin-resistant, active P70S6KCA protein can still phosphorylate rpS6 even in the presence of rapamycin. Therefore, these results are confirmation that mASCs, when transfected with plasmid DNA, can produce the relevant p70S6K proteins and that they are either active (p70S6KCA) or inactive (p70S6KKI). Quantitative reverse transcription PCR confirmed that siRNA mediated reduction of HtrA1 mRNA expression was unaffected in cells co-transfected with plasmid DNA (Fig. 5B).
Next, we transfected HtrA1-deficient mASCs with empty plasmid, p70S6KCA or p70S6KKI and assessed their ability to influence mASC-derived osteoblast formation through quantification of Alizarin red staining after 21 days. The assumption was that reduced p70S6K activity in HtrA1-deficient mASCs could be compensated for through expression of the constitutively active p70S6K (p70S6KCA), and thereby restore osteogenic potential. Indeed, as shown in Fig. 5C and D, Alizarin red staining was restored in HtrA1-deficient mASCs transfected with p70S6KCA. The kinase inactive (KI) p70S6K (p70S6KKI) was included to control as accurately as possible for the introduction of plasmid DNA into the cells and expression of p70S6K protein. It differs with respect to p70S6KCA in that it is not active, and so should not compensate for the reductions in p70S6K activity in HtrA1-deficient mASCs. This was indeed the case, as shown in Fig. 5C and D where Alizarin red staining could not be restored in HtrA1-deficient mASCs transfected with p70S6KKI. Empty plasmid was also included as an additional control, and it demonstrated no influence over mASC osteogenesis as expected. Taken together, these findings identify p70S6K as being of critical importance in ATRA-mediated mASC osteogenesis and that activation of p70S6K is reliant, at least in part, on the actions of HtrA1.
Discussion
mASCs represent a readily available source of osteoprogenitor cells, which unlike mBMSCs, have the advantage of being able to sustain a high level of osteogenic differentiation potential with age and under conditions of low bone quality [4, 22 –25]. Subsequently, mASCs are fast becoming the preferred choice for stem cell-based approaches in bone tissue engineering [26,27]. Certainly, results from our previous studies have confirmed that mASCs harvested from SAMP6 mice, a model for senile osteoporosis, have the capability of increasing bone quality when re-injected back into SAMP6 tibia [5]. However, despite their widespread usage, the underlying mechanisms through which mASC osteogenic differentiation is controlled remains incompletely understood.
In the current report, we identify the serine protease HtrA1 as being a positive regulator of ATRA-induced mASC osteogenesis and mASC-derived osteoblast mineralization. Furthermore, we provide evidence, which supports p70S6K as playing a role in mediating the pro-osteogenic effects of HtrA1 in mASCs in response to ATRA.
Mammalian HtrA1 was originally identified by Zumbrunn and Trueb [28] and has since been implicated in numerous biological processes and diseases [15,29]. Findings from our own studies have revealed HtrA1 to be a potent modulator of hBMSC multipotency as evidenced by its ability to inhibit adipogenesis and stimulate osteogenesis [7]. However, several studies also exist in which HtrA1 has been classified as a negative regulator of osteogenesis both in vitro and in vivo [30,31]. Clearly therefore, further investigations are required to clarify HtrA1's function in osteogenesis and to ascertain its potential mechanism of action. Here, we provide further evidence in support of HtrA1's role as a positive regulator of multipotent stromal cell osteogenesis.
HtrA1 is classified as a secreted serine protease and as such, its influence over cellular processes is largely thought to be due to its extracellular actions [7,20,32]. However, it is also equally likely that HtrA1 instigates many of its effects intracellularly. Indeed, HtrA1 has been shown to interact with and functionally regulate several intracellular substrates including tubulin [33], proTGFβ1 [34], tau [35], X-linked inhibitor of apoptosis protein (XIAP) [36], and TSC2 [16]. In the context of the present study, HtrA1's regulatory influence over TSC2 activity holds particular relevance given the importance of mTOR signaling in stem cell multipotency [17 –19].
The mTOR protein makes up the catalytic subunit of two separate complexes, namely mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [37]. mTORC1 functions to control cell growth and protein synthesis through phosphorylation of 4E-BP1 and p70S6K, and it has been implicated in osteoblast differentiation [17,38 –40]. mTORC1 is negatively regulated by the GTPase-activating protein TSC1/2 complex and as such, relies on the actions of Akt for its activation through phosphorylation of TSC2 [41]. Although several studies have demonstrated activation of the Akt/mTOR signaling pathway in response to ATRA [42 –44], no investigations have yet been undertaken to examine Akt/mTOR activation in ATRA-stimulated mASCs or to evaluate its consequences for their commitment toward osteoblasts.
Our findings have demonstrated that the Akt/mTOR/p70S6K pathway is rapidly activated in mASCs in response to ATRA. Similar rapid increases in several other kinase cascades have previously been demonstrated in various cell systems in response to ATRA [45 –48]. Such effects are considered to be independent of the classical genomic effects of ATRA, and these are instead regulated through atypical, nongenomic events possibly through interactions with membrane-associated retinoic acid receptors [49]. The ability of the mTOR inhibitor rapamycin to completely suppress ATRA-mediated p70S6K activation in these cells suggests that despite the minimal increases in phospho-mTOR (Ser2448), ATRA-mediated p70S6K activation in mASCs is rapamycin sensitive and as such, reliant on mTOR signaling.
It is important to note that although p70S6K is well recognized as a downstream target of mTOR, phosphorylation of mTOR at Ser2448 is also considered to represent a feedback signal from p70S6K [50]. If indeed the case, then ATRA-mediated p70S6K activation in mASCs may possibly be reliant on increases in mTOR activity through the phosphorylation of sites other than Ser2448. Certainly, mTOR has been reported to have several potential phosphorylation sites whose functions remain largely undefined [51]. As with rapamycin treatment, loss-of-function of HtrA1 also resulted in reductions in mTOR and p70S6K phosphorylation in the absence of any changes in phospho-Akt. However, in contrast to rapamycin, the removal of HtrA1 was unable to completely abolish mTOR or p70S6K phosphorylation.
Although p70S6K activity is considered to be of paramount importance in determining mASC adipocyte lineage commitment [52], its functional role in mASC osteogenesis has not yet been established. Our findings from studies using the mTOR inhibitor rapamycin, along with siRNA-dependent inhibition of Rps6kb1 gene expression, confirmed that the mTOR/p70S6K signaling pathway was indeed an essential requirement for efficient mASC osteogenesis and mASC-derived osteoblast mineralization. As far as we are aware, this is the first report to demonstrate such a role for mTOR/p70S6K in ATRA-mediated mASC osteogenesis. Therefore, these studies identified both HtrA1 and mTOR/p70S6K as being important regulators of ATRA-mediated mASC osteogenesis.
However, it was still unclear as to whether HtrA1's ability to regulate p70S6K phosphorylation in response to ATRA was directly related to its pro-osteogenic effects. We therefore performed a study in which we introduced plasmids encoding DNA for either constitutively active or kinase inactive mutants of p70S6K into HtrA1-deficient mASCs in an attempt to rescue osteoblastogenesis. Indeed, our results revealed that the mineralizing capacity of HtrA1-deficient mASC-derived osteoblasts could be fully restored when cells were engineered to express the constitutively active p70S6K mutant.
These findings therefore identify p70S6K activation as a regulatory target of HtrA1 and an important event in mediating HtrA1's pro-osteogenic effects in ATRA-stimulated mASCs. However, based on the fact that mTOR phosphorylation at Ser2448 may be the result of feedback regulation by p70S6K, further investigations are needed to determine whether HtrA1 can directly influence mTOR activation. Certainly, we would anticipate that if HtrA1 were acting to regulate p70S6K activity through its interaction with TSC2 [16], then reductions in mTOR activity would be apparent [53]. However, the suggestion that the TSC-complex may in fact regulate p70S6K independently of mTOR [54], may offer an additional means by which HtrA1 could activate p70S6K without the need for alterations in mTOR activity.
Alternatively, HtrA1 may act to regulate p70S6K phosphorylation through mTOR, but in a TSC2-independent manner. Certainly, mTOR is not solely reliant on TSC2 inhibition for its activation as confirmed by studies in which Akt was shown to activate mTOR by relieving the inhibitory effects of proline-rich Akt/PKB substrate 40 kDa (PRAS40) on mTORC1 [55], and more recently, through its ability to promote mTORC1 phosphorylation at Ser1415 via the actions of IkB kinase alpha (IKKα) [56]. Further studies are therefore required to ascertain the involvement of TSC2 and mTOR in mediating the effects of HtrA1 on p70S6K phosphorylation in osteogenic mASCs.
Despite us having now demonstrated HtrA1 to be a positive regulator of both mASC and hBMSC [7] osteogenesis, its role in mediating bone formation remains highly controversial. This is highlighted by findings from previous studies in which HtrA1 was deemed to be a negative regulator of osteogenesis in stromal cells derived from long-term bone marrow cultures [31] and mouse 2T3 osteoblasts [57]. By contrast however, a more recent report has identified HtrA1 as being a necessary requirement for the osteogenic differentiation of periodontal ligament cells [58]. One possible explanation for such discrepancies may lie in the fact that in each of these studies, a different cell culture system was used. If indeed the case, this would imply that HtrA1 acts to mediate cell-specific responses to osteogenic stimuli, the result of which may impart an inhibitory or enhancing effect on osteogenic induction.
Interestingly, our new findings also demonstrate that HtrA1's involvement in regulating mASC osteogenesis extends beyond it's ability to influence ATRA-mediated osteogenic induction as evidenced by significant reductions in mineral formation in HtrA1-deficient mASC cultures stimulated with BMP-2.
To try and further elucidate HtrA1's role in regulating bone formation, investigations have also been conducted in mice with a targeted gene deletion of HtrA1 [30]. However, although HtrA1-deficient mice displayed significant increases in several bone parameters, the influence of such changes on bone quality and strength remain to be determined. Furthermore, no studies were undertaken to address the possible involvement of compensatory mechanisms in HtrA1-deficient mice, such as the upregulation of other members of the HtrA family (eg, HtrA3 and HtrA4), which may also have an influence on bone formation. Clearly, more in-depth investigations are required to reconcile these conflicting studies and thereby help clarify HtrA1's role as a modulator of bone formation.
In summary, we have identified p70S6K as an important regulator of mASC osteogenesis, being activated in response to ATRA via pathways involving mTOR and HtrA1 (Fig. 6). As such, it is proposed that HtrA1 represents a newly identified positive regulator of ATRA-mediated mASC osteogenesis and mASC-derived osteoblast mineralization.
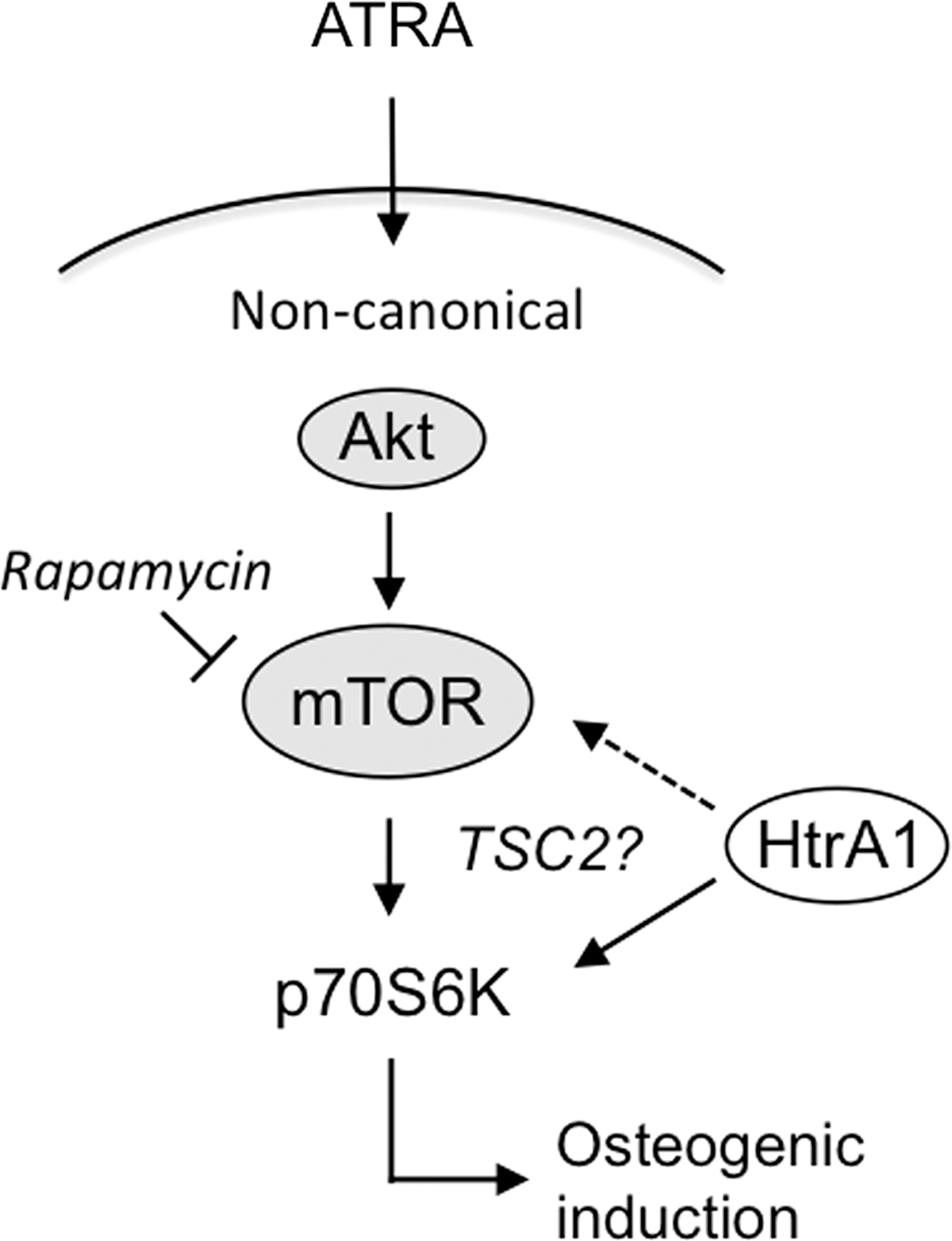
Model of ATRA-induced mASC osteogenesis. Based on our findings, we propose that ATRA drives osteoblast commitment of mASCs through activation of p70S6K in a noncanonical and nongenomic manner. This appears not only to be dependent on rapamycin-sensitive signaling pathways, but also on the actions of HtrA1. However, further investigations are required to determine HtrA1's mechanism of action and to clarify the potential involvement of the HtrA1 substrate TSC2 in mediating its effects on p70S6K activity and mASC osteogenesis. ATRA, all-trans retinoic acid; TSC2, tuberous sclerosis complex 2.
Footnotes
Acknowledgments
SG and GF were supported by SNSF grants 31003A_134935 and 31003A_156313. AM and ANT were supported by the Uniscientia Foundation and Forschungskredit University of Zurich. CLF was supported by the Whitaker International Program.
Author Disclosure Statement
The authors declare that they have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.