
Abstract
The remarkable functional plasticity of professional antigen-presenting cells (APCs) allows the adaptive immune system to respond specifically to an incredibly diverse array of potential pathogenic insults; nonetheless, the specific molecular effectors and mechanisms that underpin this plasticity remain poorly characterized. Cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), the target of the blockbuster cancer immunotherapeutic ipilimumab, is one of the most well-known and well-studied members of the B7 superfamily and negatively regulates T cell responses by a variety of known mechanisms. Although CTLA-4 is thought to be expressed almost exclusively among lymphoid lineage hematopoietic cells, a few reports have indicated that nonlymphoid APCs can also express the CTLA-4 mRNA transcript and that transcript levels can be regulated by external stimuli. In this study, we substantially build upon these critical observations, definitively demonstrating that mature myeloid lineage dendritic cells (DC) express significant levels of intracellular CTLA-4 that they constitutively secrete in microvesicular structures. CTLA-4+ microvesicles can competitively bind B7 costimulatory molecules on bystander DC, resulting in downregulation of B7 surface expression with significant functional consequences for downstream CD8+ T-cell responses. Hence, the data indicate a previously unknown role for DC-derived CTLA-4 in immune cell functional plasticity and have significant implication for the design and implementation of immunomodulatory strategies intended to treat cancer and infectious disease.
Introduction
C
Although the mechanistic particulars by which CTLA-4 exerts its suppressive activities remain an area of substantial debate, its pattern of expression has garnered significantly less controversy. CTLA-4 is thought to exhibit a lymphoid lineage-specific pattern of expression with reports describing expression on regulatory T cells [18], activated conventional T cells [19], induced expression on B cells [20], and even a recent report of natural killer cell expression [21]. Surface staining does not generally detect CTLA-4 expression on other hematopoietic lineages. Furthermore, transgenic expression of CTLA-4 from a T-cell-specific promoter was sufficient to abrogate the lethal autoimmunity observed in CTLA-4-deficient mice, suggesting that critical functions of CTLA-4 may be primarily limited to the T-lymphoid lineage [22].
In contrast to the well-known data suggesting lymphoid specificity, there also exist a number of inconclusive reports suggesting expression of CTLA-4 in myeloid lineage hematopoietic cells, including dendritic cells (DC) [23 –27]. These sporadic data include a previous report of CTLA-4 mRNA expression from highly purified in vitro-derived myeloid DC [27]. DC are the master regulators of adaptive immunity in mammals and the only cell type capable of priming de novo T cell responses. Accordingly, definitive confirmation of CTLA-4 expression in DC with concomitant functional insight would alter the present understanding of CTLA-4 function as well as the manner by which the adaptive immune response is regulated.
In this study, we conclusively demonstrate that mature myeloid DC express intracellular CTLA-4 which is subsequently secreted into the extracellular space by means of a vesicular intermediary. DC-derived extracellular CTLA-4 competitively inhibits antibody binding of B7, and its presence negatively regulates downstream T-cell responses in vitro and antitumor immunity in vivo. The unexpected presence of functional CTLA-4 in this critical and plastic hematopoietic lineage suggests an additional level of DC control over the adaptive immune response and has significant implication for novel uses of CTLA-4 antagonist drugs.
Materials and Methods
Reagents
Western blot and coIP antibodies
αHuman CTLA-4 clone A3.6B10.G1 (Cat No. 525401; Biolegend, San Diego, CA); αHuman CTLA-4 clone BNI3 (Cat No. 555851; BD Pharmingen, San Diego, CA); αHuman CTLA-4 (Cat No. ab107198; Biolegend, Cambridge, MA); Human/Mouse αIL-12p35 (Cat No. MAB1570; R&D Systems, Minneapolis, MN); Human αIFN-γ (Cat No. ab9657; Abcam, Cambridge, MA); Protein G Plus Agarose Suspension (Cat No. IP04; Calbiochem, Billerica, MA); Human TruStain FcX™ FC block (Cat No. 422301; Biolegend); Ponceau S Solution (Cat No. 6226-79-5; Sigma-Aldrich, St. Louis, MO); Restore™ Western Blot Stripping Buffer (Cat No. 21059; Pierce, Rockford, IL); Secondary Antibodies (αGoat-HRP, αRabbit-HRP, αMouse-HRP); and β-actin (Santa Cruz, Dallas, TX).
Functional antibodies
αHuman CD3 clone UCHT1 (Cat No. 555329; BD Pharmingen) and αHuman CD28 (Cat No. 555725; BD Pharmingen).
Flow antibodies
αHuman CD11C, CD80, CD83, CD86, CD3, CD4, CD8, CD25, IFN-γ, and CTLA-4 flow antibodies (Biolegend). HIV Tetramer (HIV-pol468; ILKEPVHGV) from the Baylor College of Medicine Tetramer Core (Houston, TX).
Confocal microscopy antibodies and reagents
αCTLA-4-biotin clone BNI3 (Cat No. 555852; BD Pharmingen, San Jose, CA); Streptavidin-APC (Cat No. 554067; BD Pharmingen); Rab5 (Cat No. 108011; Mouse-Monoclonal Synaptic Systems, Goettingen, Germany); Rab11 (Cat No. 610656; BD Biosciences, San Jose, CA); Giantin (Courtesy: Dr. Rick Sifers; BCM, Houston, TX); Alexa-fluor Ms546 (Courtesy: Dr. Anna Sokac; BCM); Alexa-fluor Rb546 (Courtesy: Dr. Anna Sokac; BCM); CD3-FITC (Cat No. 555332; BD Pharmingen); CD11c clone 3.9-Alexa-fluor 488: (Cat No. 301618; Biolegend); and DAPI: SlowFade® Gold Antifade Mountant (Cat No. S36938; Molecular Probes, Grand Island, NY).
DC selection and enrichment
Human CD14 Positive Selection Kit (Cat No. 18018, EasySep; Stemcell Technologies, Vancouver, Canada) and Human Myeloid DC Enrichment Kit (Cat No. 19021, EasySep; Stemcell Technologies).
Mice
Four- to 6-week-old C57BL/6 mice and CTLA-4−/−CD28−/− double knockout mice were obtained from the Jackson Laboratory (Bar Harbor, ME). All mice were maintained in accordance with the specific IACUC requirements of Baylor College of Medicine and in accordance with animal protocol AN-1428.
DC preparation, enrichment, and maturation
Normal donor peripheral blood buffy coats were obtained in an anonymous, unidentifiable manner. Products were diluted 1:2 in phosphate-buffered saline (PBS; Lonza, Allendale, NJ) and centrifuged at 450 g on a Ficoll gradient (Lympholyte; Cedarlane Labs, Burligton, NC) to isolate viable white cells. Human CD14+ cells were magnetically separated from total peripheral blood mononuclear cells (PBMC) using Clinimacs CD14 beads (Miltenyi-Biotec, San Diego, CA) according to the manufacturer's instructions. CD14+ cells were cultured for 6 days in AIM-V medium (Invitrogen, Carlsbad, CA) supplemented with 10% Human AB Serum (Atlanta Biologicals, Lawrenceville, GA), 50 μg/mL streptomycin sulfate (Invitrogen), 10 μg/mL gentamicin sulfate, 2 mM
Tolerogenic DC
Buffy coat DC were prepared from adherent monocytes and incubated as previously described but were differentiated in the presence of 100 ng/mL macrophage colony-stimulating factor (M-CSF) and 10 ng/mL TGF-β (both from eBioscience, San Diego, CA). Differences from conventional DC preparations were verified by flow cytometry of CD11c, CD80, CD83, and CD86.
T-cell stimulation and analysis
PBMC were isolated from the nonadherent fraction of a Buffy Coat, resuspended in RPMI-10% fetal bovine serum (FBS), 1% anti-anti, loaded with 1 μM CFSE, and plated at 1 × 106 per well in a 96-well immunoabsorbent flat-bottom plate previously coated (24 h, 4°C) with 1 μg/mL immobilized αCD3 (clone UCHT1). The plate was incubated at 37°C, 5% CO2 for 3 days, cells were washed with PBS, replated in a 96-well round-bottom plate at 105 cells per well, and treated with αCD28 (various concentrations) for 3 days at 37°C, 5% CO2. Alternatively, PBMC were treated for 4 days with various concentrations of SEB. Cells were then analyzed by flow cytometry for CFSE content, and cultured supernatants were analyzed by western blot for IFN-γ.
siRNA transfection
CTLA-4 (mouse and human) siGenome SMART Pools and nontargeting (NT) siRNA pools were purchased from Thermo Scientific (Wilmington, DE). In brief, siRNA was reconstituted in 50 μL of siRNA buffer, and 1 μL/electroporation was prediluted in Viaspan (Barr Laboratories subsidiary of Teva Pharmaceuticals, Pomona, NY) before 1:1 addition to cells resuspended in Viaspan (20–40 × 106/mL). Cells and siRNA were incubated together on ice for 10 min before electroporation (DC—250 V, 125 μF, Ω = ∞, 4 mm cuvette; T cell—140 V, 1,000 μF, Ω = ∞, 4 mm cuvette) using a Gene Pulser Xcell Electroporator (Bio-Rad Laboratories, Hercules, CA).
Western blotting and analysis
All gel electrophoresis was performed under denaturing, reducing conditions on a 12% polyacrylamide gel with subsequent transfer to a 0.45 μm nitrocellulose membrane for antibody probing. All blocking and antibody staining steps were carried out in 5% milk, and primary antibodies were applied overnight at 4°C. Western blot chemiluminescent signal was detected using a ChemiDoc XRS digital imaging system supported by Image Lab software Version 2.0.1 (Bio-Rad Laboratories). All western blot signals were normalized by densitometry of Ponceau S (Sigma-Aldrich)-stained membranes. Contamination of supernatants with residual cell lysate or debris from cell death was controlled for by immunostaining with anti-β-actin (Santa Cruz) and additional densitometry. Densitometry was performed using ImageJ software (NIH, Bethesda, MD). All western blots are representative of at least three independent experiments.
Coimmunoprecipitation
Samples were prepared based on individual experimental approach. Either cultured media supernatant was separated from cells by centrifugation (400 g, room temperature, 5 min) or cells were lysed with various concentrations of NP-40 lysis buffer (1 h, 4°C) followed by centrifugation of debris (20 min, 4°C, 20,000 g). Samples were then precleared with naked Protein G plus beads (1 h, room temperature with rotation), followed by centrifugation of the beads (10 min, room temperature, 100 g). The remaining supernatant was then incubated with αCTLA-4-coated beads (overnight, 4°C with rotation), followed by centrifugation of the beads (10 min, room temperature, 100 g). Beads were then carefully washed 5× with either PBS or detergent, and the remaining contents of the Protein G pull-down were boiled in sodium dodecyl sulfate (SDS)- and β-mercaptoethanol-containing gel electrophoresis loading dye and subsequently analyzed by western blot.
Flow cytometry and analysis
All flow cytometric analysis was performed using an LSR II flow cytometer (BD Biosciences) and analyzed with FlowJo version 10.0.00003 for the MacIntosh (Tree Star, Inc., Ashland, OR). All flow analyses shown are representative of at least three independent experiments.
Immunofluorescence and confocal microscopy
DC were cultured and matured in a six-well plate and subsequently collected onto 12 mm round poly-
CTLA-4 RT-PCR
Loaded matured DC were resuspended in 1 mL Trizol (Life Technologies) at <1 × 107 cells per sample and total RNA was extracted according to manufacturer's instructions. RNA was treated with 1 μg/μL DNase I (Invitrogen). cDNA was synthesized from the DNase-treated RNA sample using the SuperScript™ III First-Strand Synthesis kit (Life Technologies) and amplified by PCR for 35 cycles at an annealing temperature of 55°C with CTLA-4 Fwd primer: ATGGCTTGCCTTGGATTTCAGCGGC and CTLA-4 Rev primer: TCAATTGATGGGAATAAAATAAGGCTG. Primers were designed to amplify transcripts corresponding to both soluble and membrane-bound CTLA-4 isoforms. GAPDH was amplified as a control.
In vitro coculture
DC were treated with either CTLA-4 or non-targeting (NT) siRNA for a total of 72 h and matured for a total of 48 h before coculture with autologous T cells at a ratio of 1:10 in RPMI-1640/10% FBS/1% anti-anti and incubated at 37°C in 5% atmospheric CO2. T cells were restimulated with appropriate DC on day 9, and recombinant human IL-2 at 200 IU/mL (Chiron subsidiary of Novartis, Emeryville, CA) was added to the culture on days 5, 7, 10, 12, 14, 16, 18, 20, and 22. T cells were collected at various time points for proliferative counts and flow cytometric analysis.
In vivo B16 tumor/vaccination
Mouse bone marrow-derived DC (BMDC) were prepared as follows: BM was isolated from C57BL/6 mouse femurs and tibias, red blood cells were lysed with the ACK lysis buffer (Life Technologies) for 5 min at room temperature, and the remaining cells were resuspended in 40 mL RPMI/10%FBS/1% anti-anti and plated in a 150 mm tissue culture dish with a 20 mm grid. Media were supplemented with 20 ng/mL GM-CSF and 10 ng/mL IL-4. Media were refreshed on days 3 and 5, and immature DC were harvested on day 6. Immature BMDC were electroporated with siRNA 72 h before injection and loaded and matured 24 h before footpad injection into recipient mice. Recipient mice received 50,000 B16 cells subcutaneously on the flank 72 h before ipsilateral DC injection. Administration of DC was accompanied by peritumoral adjuvantation with 500 μg imiquimod (Sigma-Aldrich). Mice were boosted in the ipsilateral footpad on day 14 in conjunction with additional imiquimod adjuvantation. Tumors were measured by caliper every other day.
Results
Monocyte-derived DC express and secrete CTLA-4
Previous sporadic reports have suggested expression of CTLA-4 by DC or CD14+ myeloid cells under a variety of different conditions; however, conclusive characterization this phenomenon has been elusive [23
–27]. Given that DC CTLA-4 surface expression is not detectable, we sought to test the hypotheses that DC expression of CTLA-4 could be intracellular, secretory, or a combination of both possibilities. To determine whether or not DC secrete CTLA-4, we analyzed the culture medium of several different matured human DC preparations as well as that of cultured syngeneic nonadherent PBMC by western blot analysis, detecting CTLA-4 only in the culture medium of DC preparations (Fig. 1A). After verifying that CTLA-4 was not an inherent component of the culture medium itself (not shown), we further verified the identity of the presumed CTLA-4 western blot band by siRNA knockdown of CTLA-4 (also Fig. 1A) as well as by performing a CTLA-4-specific depletion of the cell culture medium using beads covalently bound to each of two different well-characterized αCTLA-4 clones (BNI3 and A3.6B10.G1). Each bead-bound antibody clone was independently able to abrogate the CTLA-4 band detected by western blot, whereas beads bound to an irrelevant isotype control antibody were not (Fig. 1B). None of the bead-bound antibodies diminished the signal of nonspecific proteins like IL-12 (also Fig. 1B). Interestingly, the CTLA-4 isoform most prominently found in the culture medium migrated just above the 37 kD molecular weight marker, the previously reported size of the full-length (flCTLA-4) isoform containing both the cytoplasmic and transmembrane domains. In contrast, CTLA-4 secretion was not detected from CD14− PBMC under native or hyperstimulatory conditions (Fig. 1A), despite significantly increased proliferation, activation, and IFN-γ release (not shown) under such conditions. Moreover, we demonstrated that our DC preparations generated by CD14 selection and subsequent CD11c enrichment were virtually devoid of CD3+ cells (Supplementary Fig. S1A; Supplementary Data are available online at

DC secrete CTLA-4.
DC CTLA-4 localizes intracellularly in a pattern distinct from that of T cell CTLA-4
Given the presence of secreted CTLA-4 attributable to DC, we presumed to find CTLA-4 on or within the DC itself. Although we were unable to routinely detect CTLA-4 on the surface of immature or mature DC, we identified CTLA-4 intracellularly by flow cytometry (Fig. 2A) and confocal microscopy (Fig. 2B). DC were observed to express significantly more CTLA-4 as they matured (Fig. 2A, B), an observation supported by RT-PCR (Fig. 2C). Indeed, upregulation of CTLA-4 expression was closely correlated with that of other maturation markers like CD80 and CD83 (Supplementary Fig. S3A). In comparison to activated T cells, the pattern of CTLA-4 localization in DC was dramatically different, dispersed throughout the inside of the cell rather than concentrated near the plasma membrane (Fig. 2D). Moreover, staining of immature tolerizing or “tolerogenic” DC generated in the presence of M-CSF and TGF-β [28] indicated a CD11c+ cell population with log-fold higher CTLA-4 expression levels than conventional DC generated with GM-CSF (Fig. 2E). Interestingly, tolerogenic DC expressed high levels of intracellular CTLA-4 whether immature or mature, whereas conventional DC appeared to upregulate intracellular CTLA-4 expression only after maturation. Furthermore, circulating CD11c+ cells harvested from healthy donors displayed comparable intracellular CTLA-4 levels with donor-matched circulating CD3+ cells, supporting in vivo physiologic relevance (Supplementary Fig. S3B, bottom panel). In addition, activation of donor PBMC with SEB led to upregulation of surface CTLA-4 on CD3+ cells, but no discernible upregulation of surface CTLA-4 on CD11c+ cells, underscoring the potential for biologically distinct roles of CTLA-4 on each different cell type (Supplementary Fig. S3B, top panel). Isotype control antibody indicated excellent staining specificity (Supplementary Fig. S4). siRNA knockdown of CTLA-4 led to a significant diminution of signal over a period of 5 days as indicated by both western blot (Supplementary Fig. S5A) and confocal microscopy (Supplementary Fig. S5C). Interestingly, CTLA-4 in DC appeared to be quite stable with nearly all diminution of signal observed between 48 and 96 h post-siRNA administration (Supplementary Fig. S5B). This contrasted significantly with the stability of T-cell CTLA-4, the knockdown of which was >90% within 24 h of siRNA administration (Supplementary Fig. S1B).
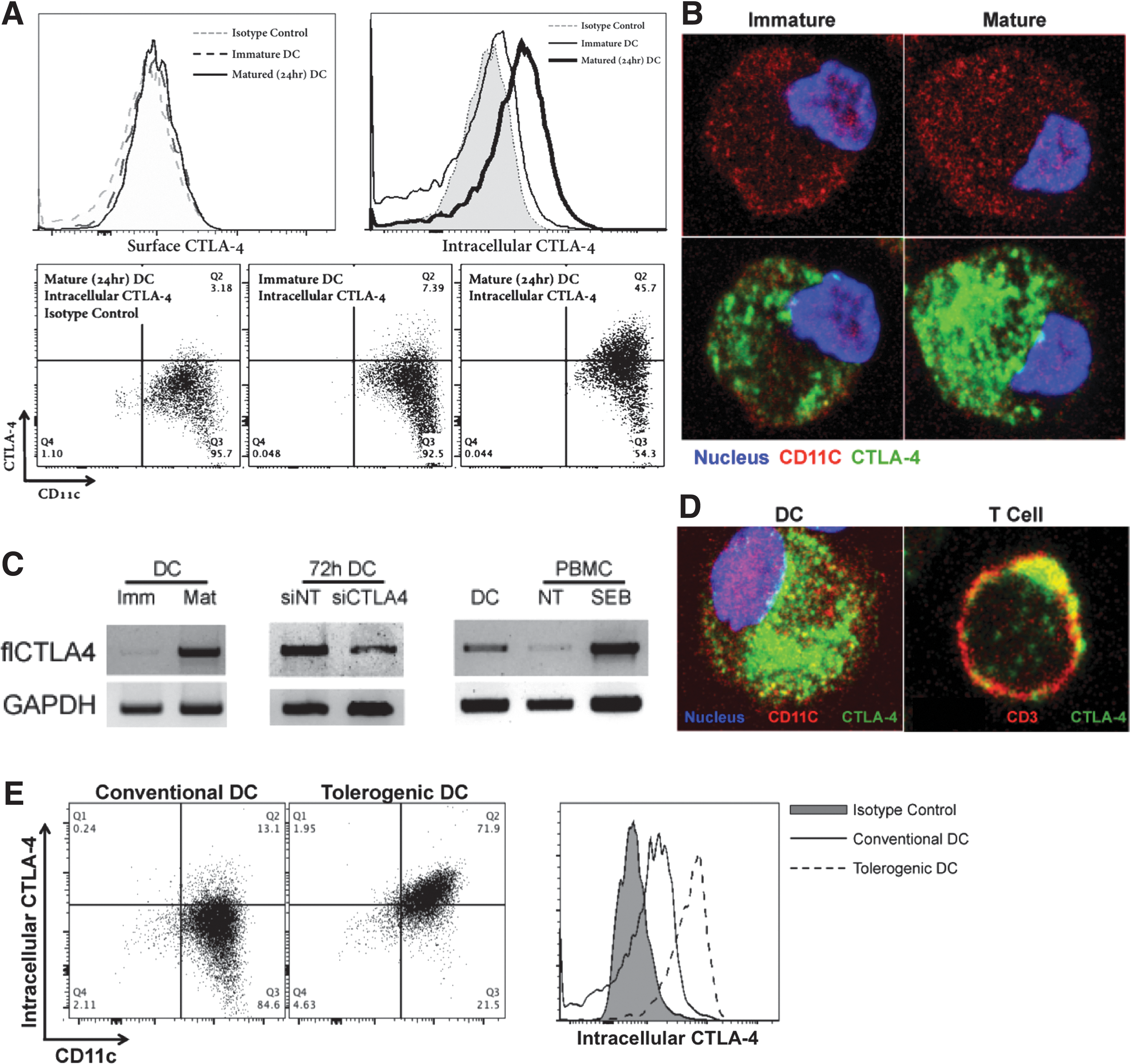
DC possess intracellular CTLA-4. Following CD14-selection, DC-differentiation, and CD11c enrichment, DC were analyzed for intracellular CTLA-4. CD11c+ DC were shown to possess intracellular CTLA-4 by
Full-length DC CTLA-4 is predominantly secreted within extracellular microvesicles
Despite detection of a CTLA-4 isoform the appropriate size of soluble CTLA-4 (i.e., expressed without the transmembrane domain, data not shown), this was not the predominant isoform of CTLA-4 detected in DC culture medium. Rather, the vast majority of detected CLTA-4 corresponded in predicted size to the flCTLA-4 isoform. DC have been reported to communicate with other cells through the directed secretion of extracellular vesicles (EV) containing numerous ligands, receptors, and other molecules [29]. Since EV possess lipid membranes, it would be feasible for flCTLA-4 to be secreted by means of DC EV release. If this were the case, then depletion of EV by coIP should also deplete CTLA-4 from the culture supernatants. LAMP-3 (lysosomal-associated membrane protein 3) or CD63 is an endosomal marker and is also one of the most abundant proteins found on the surface of circulating EV [30]. CD63 coIP of DC culture supernatant almost completely abrogated the CTLA-4 signal previously seen by western blot (Fig. 3A), indicating that removal of CD63+ EV from the DC culture media was sufficient to also remove observed flCTLA-4. Partial lysis of the EV fraction before CD63 coIP restored some flCTLA-4 signal, presumably because lysis of EV lipid membranes freed some CTLA-4 from CD63-containing vesicles. Lysis in the absence of CD63 coIP did not affect the amount of CTLA-4 detected in the media, eliminating the possibilities that the CD63 antibody nonspecifically removed CTLA-4 or that the lysis procedure interfered with western blot detection. To further confirm that the flCTLA-4 observed in the extracellular milieu was localized within CD63+ EV, supernatants were lysed with increasing concentrations of NP-40 lysis buffer for 1 h, depleted of remaining EV by CD63 coIP, and analyzed for remaining CTLA-4 content by western blot. As shown, increasing concentrations of lysis buffer lead to a more intense flCTLA-4 signal by western blot, on par with that of supernatants not depleted of EV by CD63 coIP (Fig. 3A).
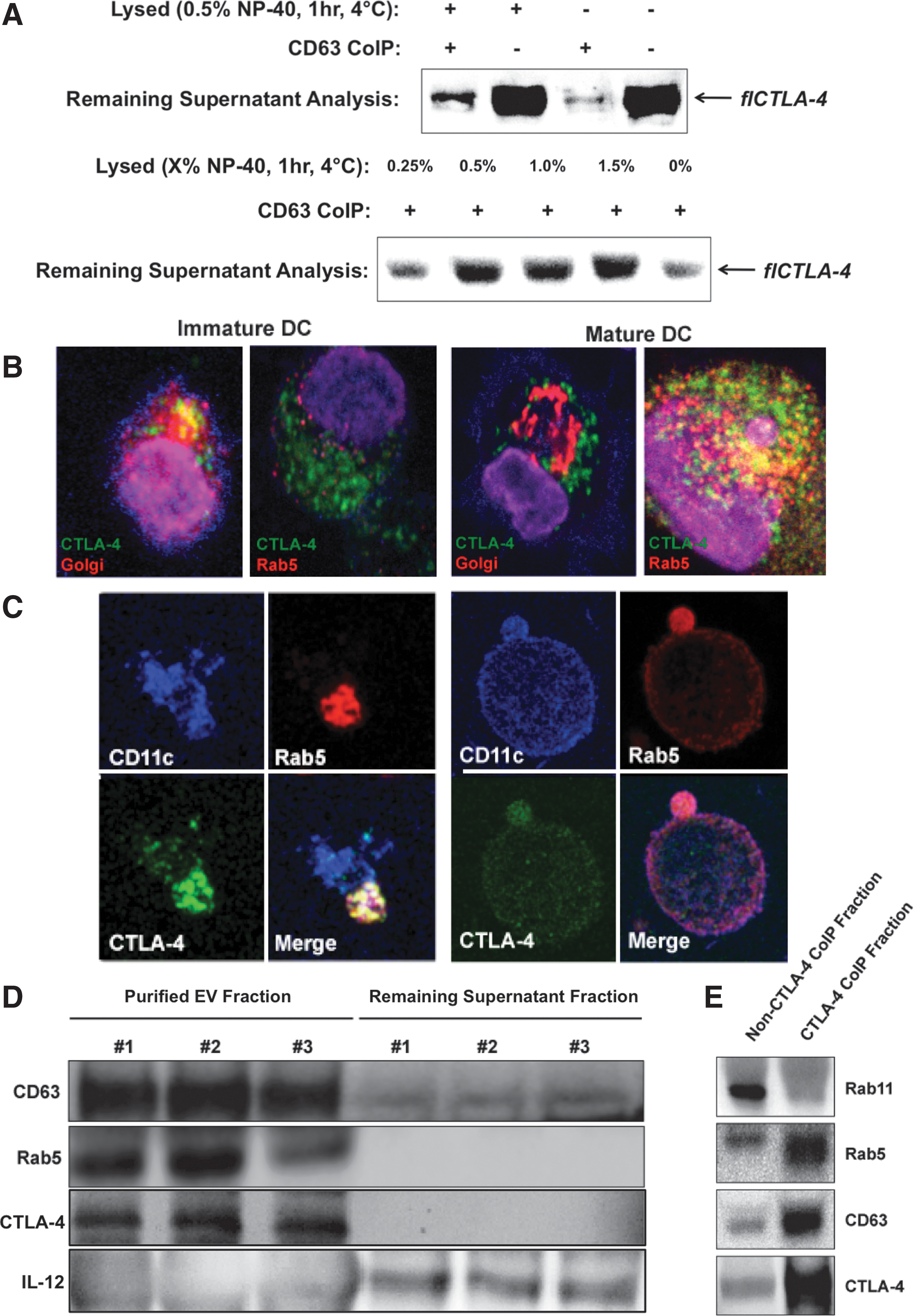
DC secrete full-length CTLA-4 packaged within microvesicular structures.
The presence of extracellular CTLA-4 in secreted vesicles should correspond with the presence of intracellular CTLA-4 colocalized with components of the secretory machinery. Confocal microscopy indicated good colocalization of intracellular CTLA-4 within the Golgi apparatus of immature DC (Fig. 3B). Upon maturation, CTLA-4 colocalization migrated from the Golgi to Rab5, a small GTPase known to be a master regulator of endosome biogenesis and a marker that identifies secretory endosomes (Fig. 3B, C) [31]. Rab11, a marker of recycling endosomes [32], was not observed to colocalize with CTLA-4 to any significant degree (not shown). Colocalization with the Golgi in immature DC or with Rab5 in mature DC was mutually exclusive. Confocal data indicated that CTLA-4 colocalization with Rab5 could occur in a highly polarized manner (Fig. 3C, left panel) within the cytoplasm as well as within secretory export vesicles in the process of budding (Fig. 3C, right panel). EV 30–120 nm in size were purified from DC supernatants derived from three independent biological samples using the Total Exosome Isolation Kit, and efficiency of the EV isolation protocol was analyzed by comparing the remaining supernatant to the EV fraction for the presence of CD63. While the soluble supernatant fraction continued to contain secreted proteins such as IL-12, Rab5 and CTLA-4 were localized exclusively within the EV fraction (Fig. 3D). CTLA-4 coIP of 30–120 nm EV and subsequent analysis indicated that CTLA-4 colocalized extracellularly with Rab5, but not Rab11, similar to what was observed intracellularly by confocal microscopy (Fig. 3E). Taken together, the data indicate that flCTLA-4 is packaged for secretion in immature DC, becomes associated with the active secretory machinery upon maturation, and is ultimately secreted into the extracellular environment within intact microvesicles.
Microvesicular CTLA-4 mediates uptake of CTLA+ EV and regulates B7 surface expression
To ascertain functional significance of DC-secreted microvesicles, we first sought to determine if such vesicles could be internalized. To ascertain this, DC were labeled with CFSE. Figure 4A demonstrates that CFSE uptake by DC was relatively uniform throughout the cell and also colocalized significantly with CTLA-4+ endosomes. CFSE-labeled DC were then cultured for 48 h during which time CFSE+ EV were secreted into the culture supernatant. These CFSE+ supernatants were then harvested and added to unlabeled DC onto which CFSE+ EV could subsequently be shown to bind (Fig. 4B) and ultimately be internalized (Fig. 4C), a process that could be followed over time by both confocal microcopy (Fig. 4B, C) and flow cytometry (i.e., Fig. 4D and Supplementary Fig. S7A). Preclearance of supernatants with beads conjugated to anti-CTLA-4 clone BNI3 could reduce the uptake of CFSE-labeled EV by unlabeled DC in a manner dependent upon the concentration of bead-conjugated BNI3 antibody (Fig. 4D and Supplementary Fig. S7A). Because CFSE+ EV uptake could easily be monitored and quantitated by flow cytometry, the consequences of such uptake on B7 surface expression could be monitored as well. As indicated in Fig. 4E, DC that became CFSE+ exhibited log-fold lower levels of CD80 and CD86 surface expression with little to no change observed in the expression of other surface markers such as CD11c. The process of B7 diminution was time dependent. Although no uptake or diminution of B7 signal was observed after 3 h of incubation, after 6 h of incubation in CFSE-labeled microvesicles, 12%–13% of CFSE+ DC were B7 “low” (in comparison to 5%–6% of CFSE− DC); and, after 12 h of incubation, 65%–75% of CFSE+ DC were B7 “low” (in comparison to 15% of CFSE− DC). Staining of B7 for 20 min at 4°C in the presence of 0.1% sodium azide (i.e., conditions under which B7 receptor downregulation could not occur) in 100% DC supernatant indicated the presence of a titratable factor in the supernatant that reduced the amount of antibody binding to B7 without reducing the amount of antibody binding to CD11c (Supplementary Figs. S6A and S7B). As with previous experiments, this factor could be removed by preclearance with beads conjugated to anti-CTLA-4 BNI3 (Supplementary Figs. S6B and S7C). To further characterize the potential dependency of EV uptake upon CTLA-4, DC were first treated with either NT siRNA or CTLA-4-specific siRNA before CFSE labeling and DC maturation. As shown in Fig. 5A and B, after 9 h of incubation in CFSE-labeled supernatants derived from CTLA-4 siRNA-treated DC, recipient DC remained predominantly CFSE−, whereas recipient DC treated with CFSE-labeled supernatants derived from NT siRNA-treated DC became CFSE+, suggesting that EV uptake was dependent upon CTLA-4/B7 interaction.
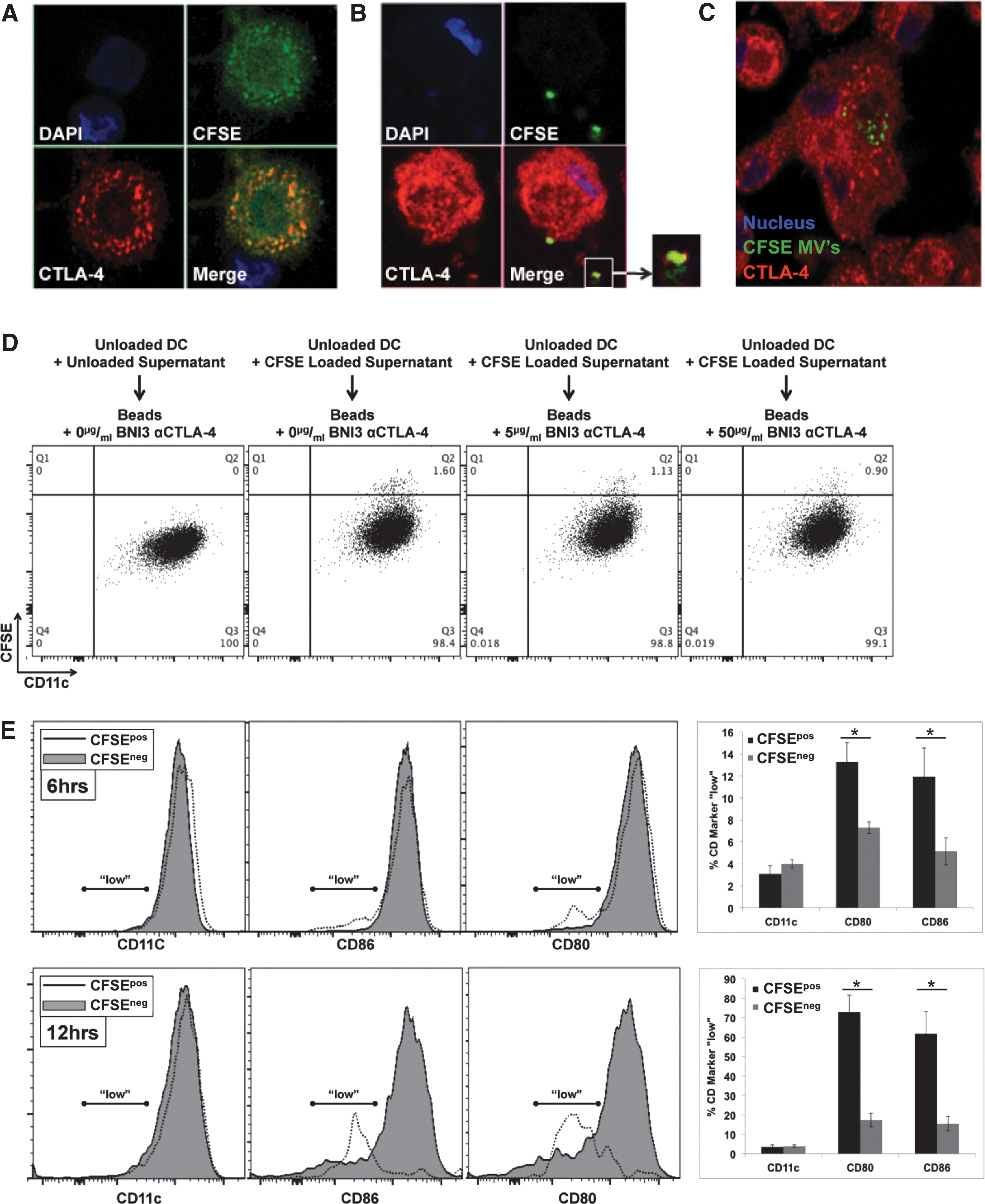
DC-derived EV are internalized by DC in an autocrine/paracrine manner mediated by EV surface CTLA-4.
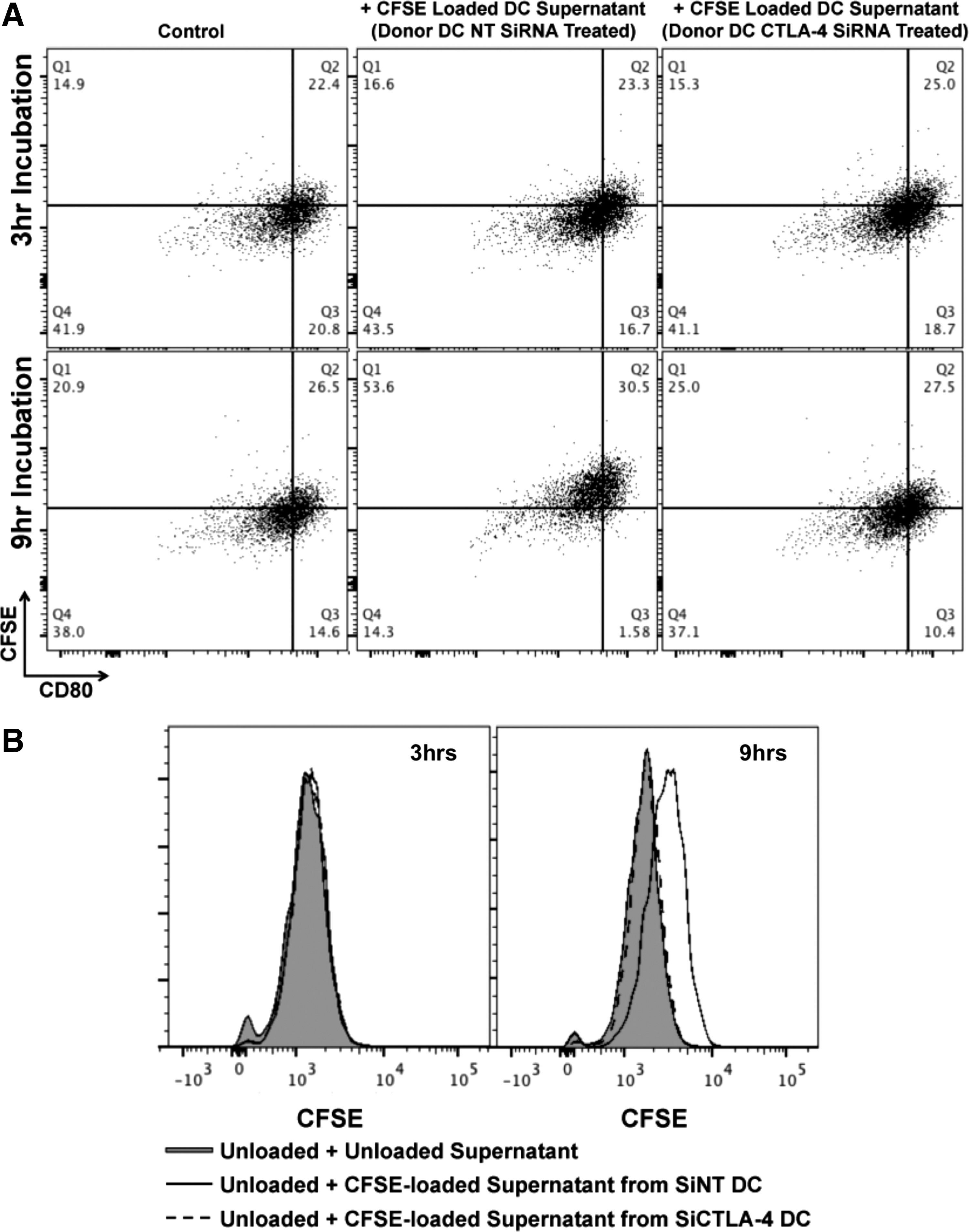
siRNA knockdown of CTLA-4 in CFSE-loaded DC diminishes uptake of CFSE-loaded EV by unlabeled recipient DC.
DC CTLA-4 is functional and suppressive
Since there are almost no reports on the existence and/or function of DC CTLA-4, we next assayed whether or not DC CTLA-4 was functional relative to the canonical understanding of CTLA-4 biology. To test whether DC-secreted CTLA-4 negatively regulates T-cell activation, human PBMC were cocultured with DC treated with either NT siRNA or CTLA-4 siRNA (for 72 h before initiation of coculture). Although the CD8:CD4 ratios were similar at early time points (e.g., day 5), the CD8+CD25+IFN-γ+ fraction was much greater when PBMC were cultured with DC lacking CTLA-4 (Fig. 6A, left panel). Following the early trend, by day 25, T cells cultured with CTLA-4-deficient DC consistently exhibited a highly significant increase in the CD8:CD4 ratio with a near doubling of the percentage of CD8+ cells (range +31% to +305%, mean =+88%, n = 8, P = 0.01) (Fig. 6A). In addition, the percentage of CD4+CD25+Foxp3+ tregs was significantly less when DC lacked CTLA-4 (Fig. 6A, right panel). Similarly, incubation of total splenocytes in siRNA-treated DC culture supernatants possessing differing amounts of CTLA-4+ microvesicles demonstrated that subsequent proliferation of CD8+CD25+ cells was dependent upon low supernatant CTLA-4 content as well as proportional to the concentration of CTLA-4 in the supernatant (Fig. 6B).

Knockdown of DC CTLA-4 enhances the TH1 response and antitumor immunity.
To test the physiological relevance of DC CTLA-4 in vivo, a B16 melanoma DC vaccine study was conducted in which the only variable altered was the addition of either NT or CTLA-4 siRNA 48 h before electroporation of DC with B16 mRNA. Following electroporation, DC were matured for 24 h and injected into recipient mice with preestablished palpable B16 tumors. Mice that received the CTLA-4 siRNA DC vaccine exhibited significantly delayed tumor growth (Fig. 6C), decreased metastasis, and increased survival (Fig. 6D). Given the association of DC CTLA-4 knockdown with enhanced production of CD8+IFN-γ+ cells and augmented antitumor immunity, we sought to determine if TH polarization might play a role in DC CTLA-4 release. Maturing DC were incubated in either high-dose IL-12 to induce TH1 polarization or SEB [33] to induce TH2 polarization, and CTLA-4 release was quantitated by western blot analysis of DC culture supernatants. As shown in Fig. 6E, TH1 polarization of DC resulted in a near-complete abrogation of CTLA-4 secretion, whereas TH2 polarization resulted in an increased CTLA-4 secretion in a dose-dependent manner. In aggregate, the data suggest that DC CTLA-4 serves a clear functional purpose in the regulation of CD8+ CTL activity with a concomitant physiologic consequence in tumor immunity.
Discussion
The remarkable functional plasticity of professional APCs that permits a specific response to a diverse array of pathogens is well-documented; nonetheless, many of the specific molecular effectors and mechanisms that underpin this plasticity remain poorly characterized. Although discovered more than a quarter century ago [34], immunoglobulin superfamily member CTLA-4 continues to be recognized as one of the most critical negative regulatory molecules governing the adaptive immune response. Given such a prominent history, it is surprising that significant expression of CTLA-4 in a vital immune cell subset remains uncharacterized. Because transgenic CTLA-4 expression from the proximal Lck promoter (i.e., CD3-specific) is known to prevent the fatal autoimmune phenotype of knockout animals (Masteller et al. [22]), there has presumably been little impetus to look for critical functions of CTLA-4 in other nonobvious cell types. It has also been reported that CTLA-4−/−-mixed BM chimeras can be rescued by T-cell sufficient, but not T-cell deficient CTLA-4+/+ BM or CTLA-4+/+ T cells expressing a fixed TCR repertoire (Friedline et al. [35]). The present data may be reconciled with these published observations by means of one of the following hypothesis, namely: (1) that supraphysiologic CD3+ T-cell expression of CTLA-4 in Masteller was sufficient to mask CTLA-4 deficiency in other cell types; (2) that the absence of DC-expressed CTLA-4 is not sufficient to mediate fatal autoimmunity or significant lymphoproliferation within the 2–3 month time frames analyzed by Masteller and Friedline; or (3) that DC-expressed CTLA-4 becomes important for downregulation of T-cell function only after significant infection, inflammation, and DC maturation. Characterization of CD11c-Cre/CTLA-4flox/flox conditional knockout mice generated by our group will ultimately resolve this issue in a conclusive manner.
Nearly all previous reports characterizing CTLA-4 expression and/or function have indicated an expression pattern limited to lymphoid lineage hematopoietic cells or to pathological settings such as cancer [23,36,37]. Although a few sporadic reports previously hinted at myeloid lineage (i.e., DC) CTLA-4 expression, in normal immune homeostasis such reports were rare and largely inconclusive [24 –26]. In this study, we present for the first time a thorough and conclusive characterization of DC CTLA-4 expression and function. T-cell expressed CTLA-4 is thought to downmodulate T-cell activity by one of two different mechanisms. The cell extrinsic function of CTLA-4 on either conventional or regulatory T cells inhibits binding of CD28 to B7 by a variety of mechanisms and may also downmodulate APC functionality by generating a negative regulatory signal through B7 ligation. In contrast, induced expression of CTLA-4 on the surface of conventional T cells appears to counterbalance activation in a cell-intrinsic manner by negative regulatory signaling at the synapse [7,8,11,12]. Our data suggest that DC-expressed and -secreted CTLA-4 could function in a manner most similar to the former, that is, by binding to APC-expressed B7 in an autocrine and paracrine manner that interferes with CD28 engagement.
In addition, we show that bystander DC which internalize CTLA-4 containing secreted microvesicles exhibit a log-fold decrease of B7 surface expression over time. While binding of B7 by CTLA-4 might mediate internalization and destruction of B7 by a mechanism loosely analogous to transendocytosis [8], it is also possible that uptake of vesicular intermediates serves to deliver regulatory miRNAs that degrade the stability of B7 mRNA transcripts [38 –41]. In addition, the interaction that mediates CTLA-4+ microvesicle uptake was not fully addressed by this work. Additional studies will determine which mechanism(s) might account for the loss of B7 upon uptake of microvesicle-bound CTLA-4 as well as whether or not uptake of CTLA-4+ microvesicles by bystander DC is dependent upon direct interaction between CTLA-4 and B7.
This study does not specifically address questions pertaining to potential regulation of DC CTLA-4 secretion. Might DC CTLA-4 secretion be a constitutive process meant to self-limit, elevate the threshold, or ensure high-level specificity of T-cell activation? Or alternatively, might DC-secreted CTLA-4 be subject to positive and negative regulation in response to external stimuli? siRNA ablation of DC-expressed CTLA-4 significantly enhanced the generation of CD8+ T cells in in vitro priming experiments, suggesting that CTLA-4 could be downregulated in response to environmental cues associated with viral or other intracellular-type infections. Accordingly, TH1 polarization of DC by incubation in IL-12 completely abrogated DC CTLA-4 secretion. Given these data, the cues that physiologically regulate DC CTLA-4 secretion might potentially include certain Toll-like receptor (TLR) agonists, NOD-like receptor agonists, RIG-I-like receptor agonists, and type I and/or type III interferons known to be associated with TH1 polarization [42 –45]. DC culture in the presence of TLR agonists such as poly(I:C) (TLR-3), LPS (TLR-4), flagellin (TLR-5), imiquimod (TLR-7), and CPG-ODN (TLR-9) did not impact CTLA-4 secretion (data not shown), suggesting that innate pattern recognition alone might not completely explain the regulation of DC CTLA-4 secretion. Future studies will determine whether discernible regulation of CTLA-4 might depend upon other types of signals or certain combinations of pattern recognition and interferon signaling.
Ipilimumab (anti-CTLA-4) has been administered as an anticancer therapy on the theory that its mechanism of action relies primarily upon activation of preexisting antitumor T cells by inhibition of the regulatory T-cell response or blockade of negative regulatory signaling in conventional effector T cells [46,47]. More recent data suggest that ipilimumab might also deplete the regulatory T-cell subset in an Fc-dependent manner through ADCC [47,48]. The present study suggests that ipilimumab might provide additional anticancer efficacy through a fourth mechanism of action if administered in conjunction with immune adjuvantation and antigen-specific vaccination.
The data indicate that blockade of DC-secreted CTLA-4 during active immune priming might enhance CD8+ T-cell responses and lead to better outcomes. Indeed, in a recent randomized trial in which ipilimumab was compared to ipilimumab+GVAX (allogeneic irradiated cancer cells transduced with GM-CSF) in heavily pretreated patients with advanced pancreatic ductal adenocarcinoma, patients assigned to the ipilimumab+GVAX vaccine arm exhibited significantly greater median OS (5.7 months vs. 3.6 months) and 1 year OS (27% vs. 7%). Most importantly, 2 of 15 patients in the ipilimumab+GVAX arm experienced long-term disease stabilization and were alive at 30 months, whereas all 15 patients receiving ipilimumab only had died by 17 months posttreatment [49]. While the tumor studies outlined in this study were performed with CTLA-4 siRNA, future studies using a murine equivalent of ipilimumab [50] should be instrumental toward the clarification of this point.
In summary, we demonstrate conclusively that DC express CTLA-4, a critical regulator of T-cell immunity. We show that CTLA-4 expression is upregulated upon DC maturation, CTLA-4 protein in mature DC colocalizes with secretory endosomes, and CTLA-4 is secreted from the DC in microvesicles. We also provide significant insight into the manner by which DC CTLA-4 is likely to function by demonstrating that (1) CTLA-4-containing microvesicles competitively inhibit antibody binding to B7; (2) uptake of CTLA-4-containing microvesicles by DC leads to downregulation of B7 surface expression; and (3) ablation of DC-expressed CTLA-4 by siRNA upregulates proliferation and activation of CD8+ T cells. We further demonstrate the physiologic relevance of these findings in an in vivo model system of cancer. These data firmly establish the validity of nonlymphoid CTLA-4 expression, provide significant functional insights into molecular effector mechanisms of APC plasticity, and should open up a new area of study in CTLA-4 biology and regulation of the adaptive immune response.
Footnotes
Acknowledgments
The authors gratefully acknowledge the contributions of Dr. Anna M. Sokac who assisted with confocal microscopy. This project was also supported, in part, by funding from Alex's Lemonade Stand Childhood Cancer Foundation (to W.K.D.) as well as by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (AI036211, CA125123, and RR024574) and the expert assistance of Joel M. Sederstrom. The authors are also deeply indebted to the Faust family for its outstanding leadership and unwavering commitment to pediatric cancer research in the Texas Medical Center.
Author Disclosure Statement
W.K.D. and M.M.H. own shares of Diakonos Research, Ltd. All other authors declare no potential conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.